
Abstract
Oxidative injury contributes to cellular damage during and after cerebral ischemia. However, the downstream catabolic pathways of damaged cellular components in neurons are largely unknown. In the current study, the authors examined the formation of oxidized proteins and their active degradation by the proteasome. In near-pure rat primary cortical neurons, it was found that protein-bound carbonyls as markers for oxidized proteins are increased after oxygen-glucose deprivation (OGD). During and after OGD, degradation of proteins metabolically radiolabeled before OGD increases two-to threefold compared with the normal protein turnover. Proteolysis after reoxygenation was attenuated by the presence of dimethylthiourea, a radical scavenger, and was blocked by lactacystin, a specific proteasome inhibitor. Lactacystin also increased the amount of protein carbonyls formed. In contrast, the activity of the proteasome complex itself after OGD was not different from sham-washed controls. The authors suggest that oxygen-glucose deprivation increases free radicals, which, in turn, oxidize proteins that are recognized and actively degraded by the proteasome complex. This protease itself is relatively resistant against oxidative injury. The authors conclude that the proteasome may be an active part of the cellular defense system against oxidative stress after cerebral ischemia.
Keywords
Oxidative damage of cellular components is an important component of neuronal injury in cerebral ischemia (Chan, 1996). Numerous studies showed increased markers of oxidative injury after ischemia and conversely reduced damage when antioxidants are increased (Keller et al., 1998; Love, 1999). Oxidative damage occurs also physiologically as a consequence of electron flux during oxidative phosphorylation. In addition to lipid peroxidation (Yoshino et al., 1997) and nucleic acid damage, proteins, because of their abundance, are main targets of free radicals like the superoxide anion, nitric oxide, or other oxidants. Numerous reports showed damage to neuronal proteins after ischemia (Minger et al., 1998). Neurons possess many proteases and chaperones that may prevent the accumulation of oxidized and nonfunctional proteins, which contribute to neuronal death (Oliver et al., 1990). In other cell types, other than tasks in growth, differentiation, and signal transduction, the proteasomal system is responsible for the degradation of the bulk of oxidized proteins within the cytosolic and the nuclear compartment. This system consists of several regulatory components influencing the activity of the 20S “core” proteasome (DeMartino and Slaughter, 1999). The 20S proteasome is able to recognize and degrade oxidized proteins specifically (Grune et al., 1997). It can be assumed that in neurons failure to degrade damaged proteins or protein aggregates contributes to secondary damage not only after stroke but also in neurodegenerative diseases.
However, the adaptive events downstream of oxidative damage and the fate of oxidatively damaged proteins are largely unknown. In the current study, in an established in vitro model of central nervous system hypoxia-reoxygenation, the authors measured the accumulation of oxidized proteins and the proteolysis of new synthesized proteins.
MATERIALS AND METHODS
Cell culture
Cortical neurons were prepared from whole cerebral cortices of fetal Wistar rats (E16–17) as described previously (Shimohama et al., 1993). After removing the meninges, the tissue was minced and digested with trypsin followed by mechanical dissociation. Cells were seeded in 24 well plates (250 to 350,000/well) and grown in Neurobasal Medium with B-27 (Gibco, Rockville, MD, U.S.A.) where glial proliferation is slowed down without addition of cytosin/arabinosin. Eight-five to 90% of cells stained positive for neuronal specific enolase (NSE; DAKO, Hamburg), 10% to 15% of cells were positive for glial fibrillary acidic protein (GFAP), suggesting the presence of astroglial cells.
Oxygen–glucose deprivation
Cells were placed in a humidified and temperated (36.5°C ± 0.3°C) hypoxia chamber at a Po2 of 0 to 4 mm Hg (5% CO2/95% N2) measured by a polarographic electrode (Licox Po2, GMS, Kiel) as described previously (Bruer et al., 1997). Experiments were performed after 8 to 10 days in vitro as described previously. Culture medium was washed out three times with phosphate-buffered saline and replaced by a deoxygenated balanced salt solution containing (in mmol/L): Na+ 143.8, K+ 5.5, Ca2+ 1.8, Mg2+ 0.8, Cl− 125.3, HCO3− 26.2, H2SO42− 1.0, SO42− 0.8, glycine 0.01 at pH 7.4. The same solution with 25 mmol/L d-glucose under normoxia served as control. Oxygen–glucose deprivation then was started by placing the cells in the hypoxia chamber and terminated by returning the stored culture medium and returning the cells to the normoxic incubator.
Quantitation of cell damage
Cell injury was assessed 24 hours after OGD by phase contrast microscopy or by measuring supernatant lactate dehydrogenase release (LDH), which corresponds to the number of damaged neurons (Koh and Choi, 1987). Lactate dehydrogenase release activity was quantitated by the linear OD decline of NADH at 340 nm at excess pyruvate calculated against a standard LDH sample. The amount of LDH in sister cultures, washed with the same salt solution containing oxygen and 20 mmol/L d-glucose, was subtracted as background. Oxygen-glucose deprivation values were normalized to 100%.
DNA laddering
DNA was extracted using a modified commercial Easy-DNA Kit (Invitrogen BV, Netherlands). Two micrograms DNA was separated on a 2% agarose gel and stained by ethidium bromide. DNA fragments were analyzed using a fluorescence imager (Typhoon, Pharmacia Amersham Biotech, Freiburg, Germany).
Measurement of overall proteolysis
Protein degradation or proteolysis is assessed by measuring trichloracetic-soluble amino acids and small peptides that were prior constituents of larger proteins that were degraded by intracellular proteases) from a prelabeled protein pool. Proteolysis was measured after metabolically labeling the cellular protein pool with [35S]-methionine in glutamate-free and methionine-free minimal essential medium. After 16 hours of incubation at 37°C, the labeling mixture was removed and the cells were washed. The degradation of metabolically labeled proteins was quantified after addition of an equal volume of 20% trichloroacetic acid, and scintillation counting was performed of the counts in the supernatant after centrifugation at 14,000×g for 10 minutes. Proteolysis was calculated as follows: % degradation = (trichloracetic-soluble activity − background) × 100/(total activity − background). After removal of nonincorporated label, the trichloracetic-soluble amount of [35S]-methionine released into the supernatant correlates to the rate of protein breakdown (Sitte et al., 1998).
Protein carbonyl measurement
The protein carbonyl content was determined in cell lysates (4 mg/mL) by a dinitrophenylhydrazine-based ELISA as described (Buss et al., 1997; Sitte et al., 1998). The proteins were absorbed on microtiter plates (Immunosorb) overnight. The detection system used was an anti-dinitrophenyl-rabbit-IgG-antiserum (Sigma, Deisenhofen, Germany) as primary antibody and a monoclonal anti-rabbit-IgG-antibody peroxidase conjugated (Sigma, Deisenhofen, Germany) as secondary antibody, developed with o-phenylenediamine. Lactacystin was dissolved in balanced salt solution.
Activity of the proteasome
The proteolytic activity of the proteasome can be measured by its chymotrypsin-like activity (Grune et al., 1995). The degradation of the fluoropeptide suc-LLVY-MCA was measured after addition of the substrate to cell lysates. Cells were washed and then lysed in 1 mmol/L dithiothreitol during vigorous shaking for 1 hour at 4°C. Nonlysed cells, membranes, and nuclei were removed by centrifugation at 14,000×g for 30 minutes. The supernatant was incubated in a buffer containing 50 mmol/L Tris-HCl (pH 7.8), 20 mmol/L KCl, 0.5 mmol/L Mg-acetate, and 1 mmol/L dithiothreitol. After a 1-hour incubation with 200 μmol/L of the fluorogenic peptide suc-LLVY-MCA, the proteolysis was terminated by addition of an equal volume of ice-cold ethanol. Measurements were performed at 380 nm excitation and 440 nm emission after addition of 0.125 mol/L sodium borate (pH 9.0) using free MCA as standard.
Statistics
Statistical difference between groups was calculated using a one-way analysis of variance with Bonferroni post hoc comparison. * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001.
RESULTS
Protein carbonyl content is increased after oxygen-glucose deprivation
First, the authors tested whether OGD is capable of oxidizing proteins. Previous studies have shown that approximately 180 minutes of OGD causes significant damage to neurons (Bruer et al., 1997). In the current study, the authors examined the effect of 150 minutes of OGD, which causes less severe neuronal damage, but likely involves free radical processes during hypoxia/reoxygenation. Protein carbonyl contents after OGD are shown in Fig. 1. In the control group there was an increase of carbonyls over time. In contrast, in the OGD group there was a transient increase 3 hours after OGD to approximately 140% compared with control. Twenty-four hours after OGD, a decrease of the protein carbonyl level was measured, so that the control and the OGD protein carbonyl concentration did not differ significantly. Similar results were obtained when the duration of OGD was only 75 minutes (data not shown).
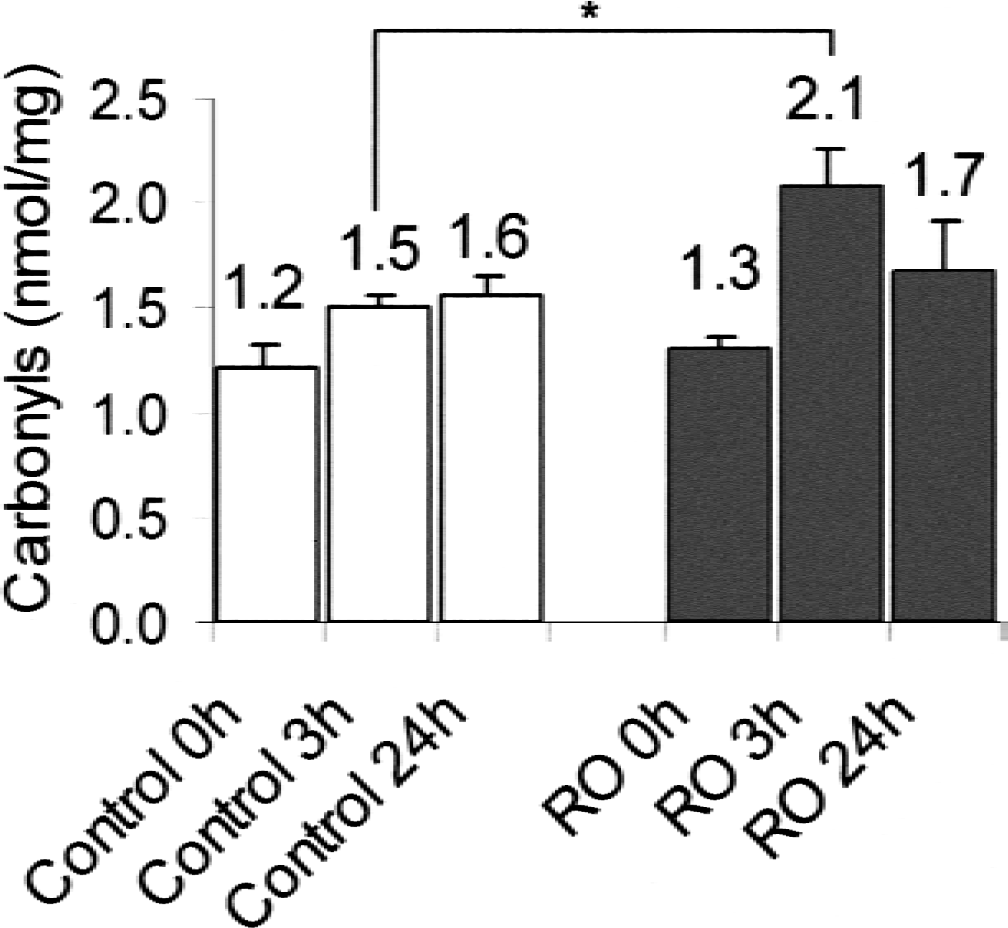
Protein carbonyls after oxygen-glucose deprivation (OGD). Near-pure neuronal cells at 8 to 10 days in vitro were either treated with oxygenated balanced salt solution with 20 mmol/L glucose (control) or were exposed to OGD (deoxygenated balanced salt solution without glucose) (Bruer et al., 1997) for 150 minutes. Immediately after OGD (RO: reoxygenation), 0, 3, and 24 hours after returning the cells to their conditioned culture medium, protein carbonyls were measured as described and data are expressed as carbonyls/mg cellular protein to correct for cell loss after OGD. Neurons were collected, washed, and frozen in liquid nitrogen until analysis. Protein carbonyl determination was performed as described. Means + SD are displayed, four cultures per condition. Data are from three independent primary cultures. * P < 0.05 by one-way analysis of variance followed by Bonferroni post hoc comparison.
Degradation of metabolically radiolabeled proteins is enhanced during and after oxygen-glucose deprivation
The effect of 150 minutes of OGD on the breakdown and appearance of proteins that were synthesized during the labeling period is shown in Fig. 2. Already during OGD there was a rapid increase of protein degradation (approximately 3-fold at 150 minutes; 2.4-fold at 26 hours) compared with control, indicating enhanced protein degradation. After OGD, during reoxygenation, the protein degradation slowed down markedly. The protein breakdown in control cultures represents the normal protein turnover in neuronal cells indicating the breakdown of metabolically radiolabeled proteins. The inset of Fig. 2 demonstrates the normalized protein breakdown after termination of OGD. It is clear that the degradation of proteins in neurons after OGD is still enhanced in comparison with the control. This indicates an enhanced protein breakdown in comparison with the normal protein turnover.
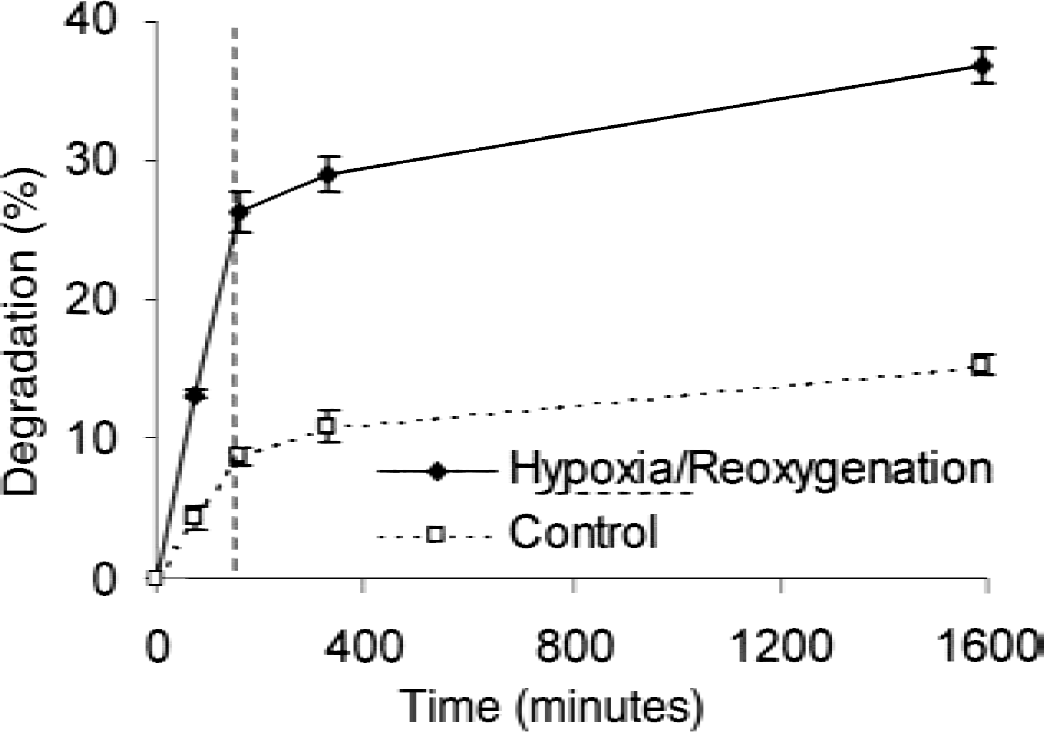
Degradation of metabolically radiolabeled proteins during and after oxygen-glucose deprivation (OGD). Twenty-four hours before each experiment cells were radiolabeled with 35S methionine/cysteine in glutamate and methionine-free minimum essential medium (MEM) medium. Neuronal cells were exposed to OGD or balanced salt solution as control at time point zero for 150 minutes. After the indicated time points, OGD proteolysis was measured by analysis of the acid-soluble radioactivity. The percent degradation was calculated by: (acid-soluble count − background)/(total radioactivity − background). n = 4 cultures per condition; 3 independent experiments. Dashed line indicates end of OGD.
Proteolysis is attenuated by scavenging of free radicals with DMTU and is blocked by lactacystin, a proteasome inhibitor
Because the protein turnover after OGD was enhanced, the authors' hypothesis was that OGD-oxidized nonfunctional proteins are selectively degraded. It is known from earlier studies that free radicals and other oxidants are formed after OGD and that the proteasomal system is responsible for the selective recognition and degradation of oxidized proteins. Therefore, the authors tried to block free radical formation using dimethylthiourea (DMTU; 100 μmol/L), an hydroxyl radical scavenger (Pieper et al., 1996), and measured the effect of this compound on protein degradation after OGD (Fig. 3, right panel). Previous studies have shown an important role of the proteasome in the breakdown of proteins after oxidative damage in various cell types (Grune et al., 1995). To further elucidate the type of protease involved in protein degradation in the authors' neuronal cell system, the authors tested whether nontoxic doses of lactacystin (5 μmol/L), a specific proteasome inhibitor (Fenteany and Schreiber, 1998), affected proteolysis after OGD. Because lactacystin has been shown to inhibit the proteasome in neuronal cells and induces neuronal death (Qiu et al., 2000), the authors first determined lactacystin toxicity in their system (Fig. 3A). Neuronal cells were incubated with lactacystin at 1, 5, 10, and 20 μmol/L. Lactate dehydrogenase in the supernatant was measured after 24 hours. As described in Materials and Methods, 20 μmol/L lactacystin caused a significant increase of LDH efflux as a marker for neuronal membrane leakage. Doses up to 10 μmol/L lactacystin did not cause increased apoptosis compared with control, as indicated by DNA-laddering (Fig. 3C). Therefore, 5 μmol/L lactacystin was chosen as safe for further experiments. Figure 3B shows that DMTU was able to prevent the increased protein turnover after OGD by 48%, indicating involvement of free radical processes during enhanced proteolysis. Lactacystin was able to prevent the increase in protein degradation after OGD, without influencing the normal protein turnover substantially. This is in accordance with previous results indicating that a small proteasomal activity remaining after the usage of lactacystin is able to cover the normal protein turnover, but insufficient to degrade oxidized proteins.
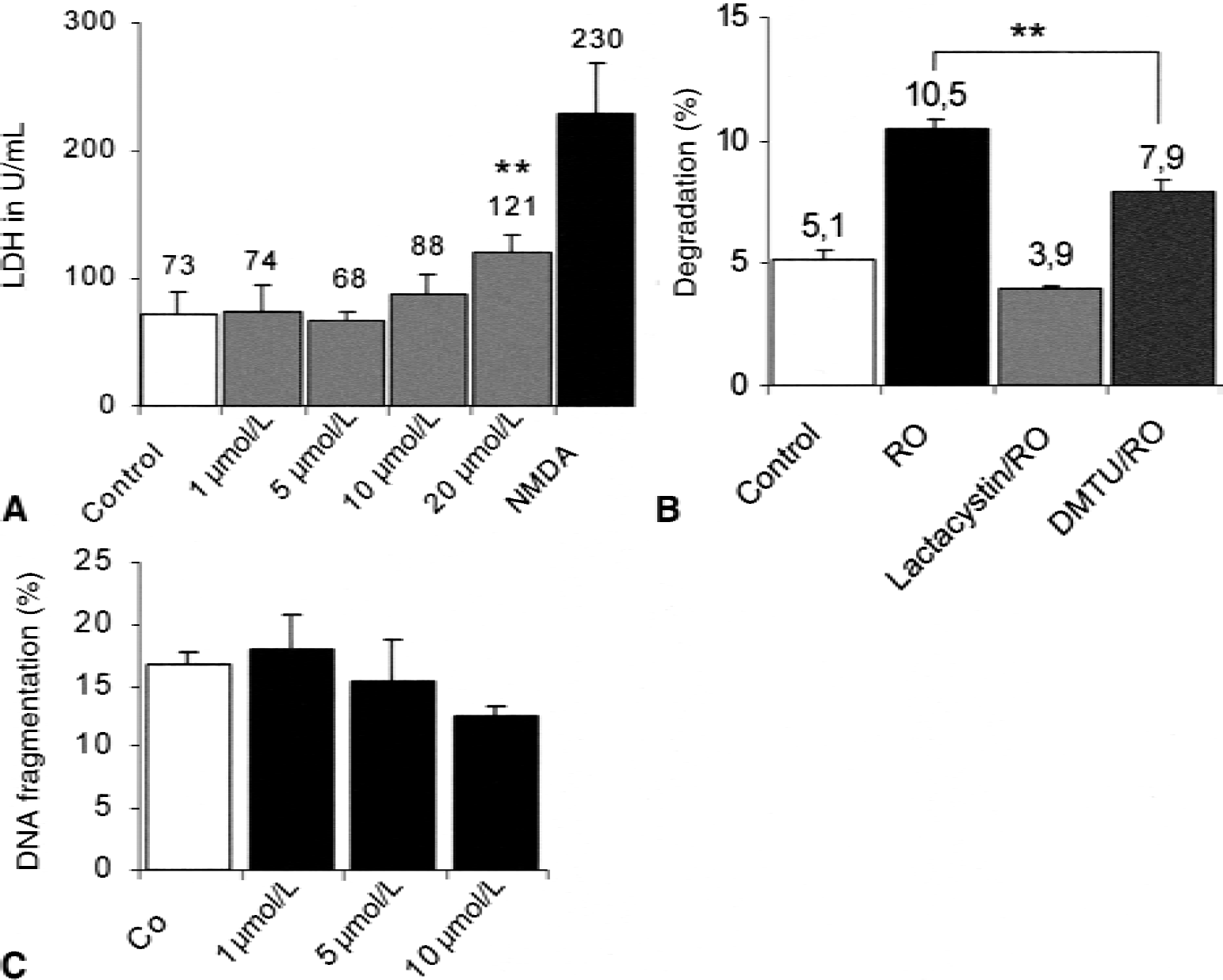
Proteolysis is attenuated after treatment with dimethylthiourea (DMTU) or nontoxic doses of lactacystin.
Proteasome inhibition increases protein carbonyl content
The authors' experiments suggested an involvement of the proteasomal system in the increased protein turnover after OGD. The authors next tested whether proteasome removes oxidized proteins after OGD. To test this hypothesis, cells were treated with or without lactacystin in the reoxygenation phase after OGD and protein-bound carbonyls were determined 24 hours later. As shown before, OGD/reoxygenation increases protein carbonyl content (Fig. 4). The proteasome inhibition by lactacystin after OGD was accompanied by a higher level of protein carbonyls 24 hours after (Fig. 4), indicating the role of the proteasomal system in the removal of oxidized proteins in neuronal cells.
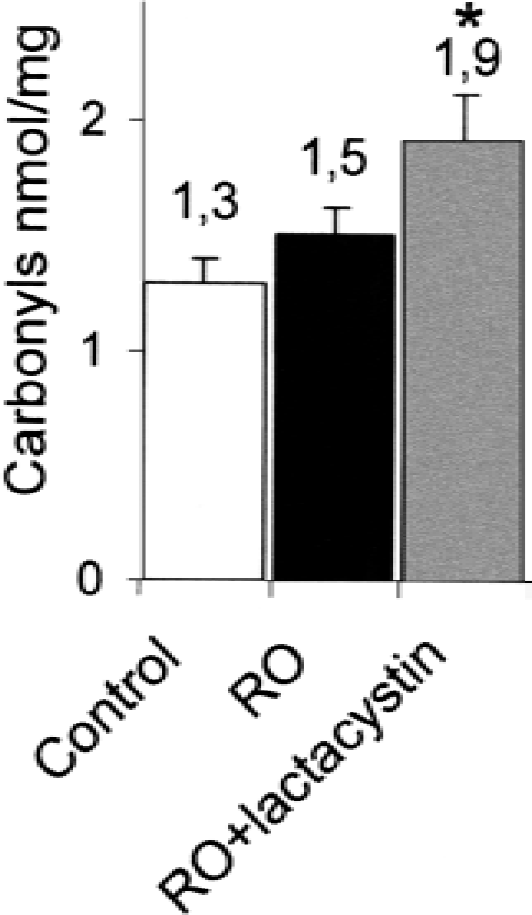
Oxidized proteins are increased by proteasome inhibition. Rat cortical neurons on 24 well plates were subjected to 150 minutes of OGD and then returned after 24 hours to the normoxic incubator (RO: reoxygenation) with or without lactacystin (5 μmol/L). Protein carbonyls (ELISA) were determined after 24 hours in cell lysates (anti-dinitrophenyl-rabbit-IgG-antiserum as primary antibody, monoclonal anti-rabbit-IgG as secondary antibody) and normalized by protein content. * P < 0.05 versus RO in one-way analysis of variance with Bonferroni post hoc comparison. Data are means + SD from n = 8 from 2 independent experiments.
Proteasomal activity is reversibly reduced after oxygen-glucose deprivation
The authors next tested whether the activity of the proteasomal system itself is altered after OGD. There is a rapid decrease of proteasomal activity after medium withdrawal and washing procedures in OGD cells as well as in sham-washed sister cultures that received balanced salts with glucose and O2, but not in unwashed control cultures (Fig. 5).
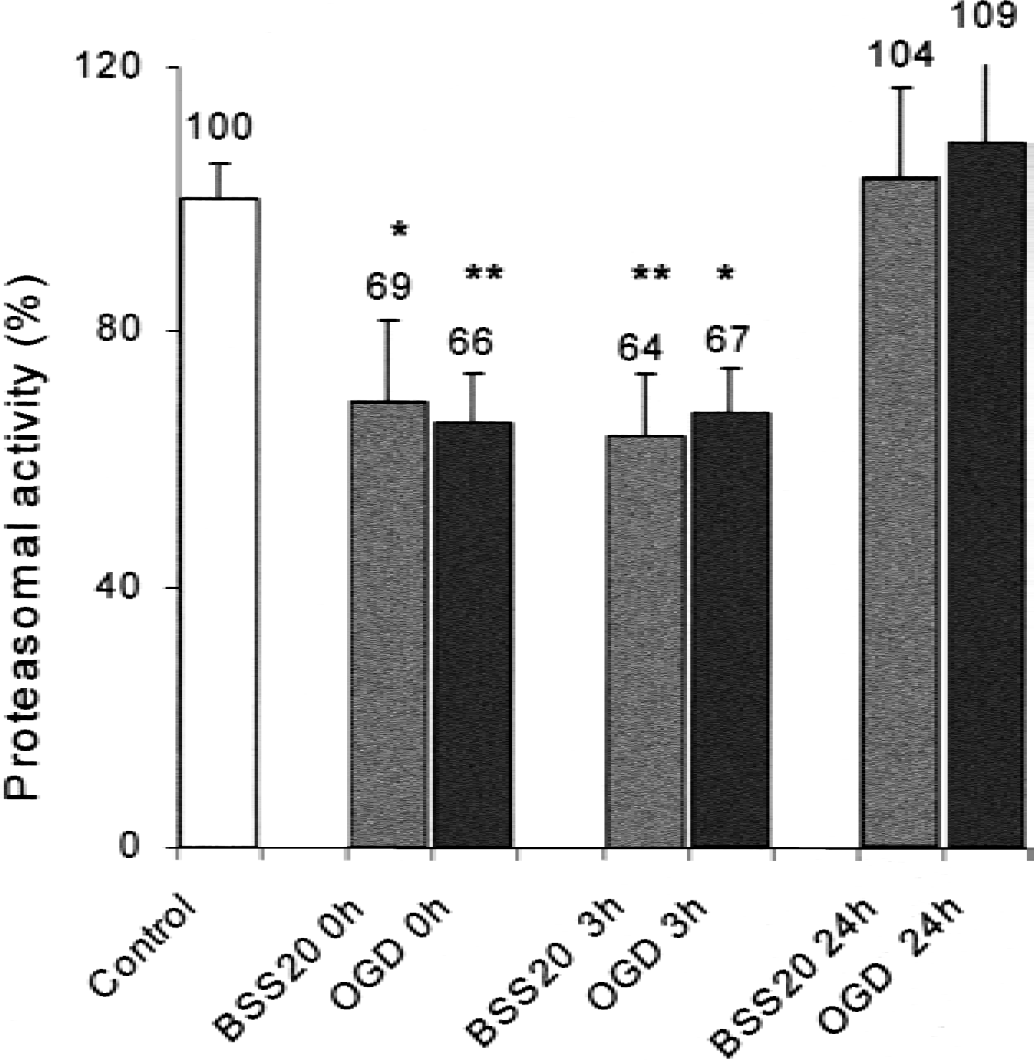
Proteasomal activity was determined in lysed cells and analyzed by the cleavage of the proteasomal substrate fluoropeptide suc-LLVY-MCA. Proteasomal activity of unwashed sister cultures served as control and values were normalized to percent of control. Proteasomal activity was determined after washing in balanced salt solution (“sham wash”: BSS20, with 20 mmol/L glucose and O2) or balanced salt solution without glucose or O2 (OGD: oxygen-glucose deprivation). 0h represents values immediately after washing, 3h represents proteasomal activity 30 minutes after 2.5 hours OGD where cells had their stored regular culture medium again. There was a rapid decrease in proteasomal activity immediately after beginning of OGD in both groups and no further decrease after the end of OGD (3h) in the early reoxygenation phase. Twenty-four hours (24h) later, proteasomal activity in both sham wash and OGD returned to baseline of unwashed sister cultures. * P < 0.05, ** P < 0.01 compared with control, as determined by analysis of variance. Data are means + SD; four conditions per well.
Consistent with a relative resistance of the proteasome against oxidative stress, 150 minutes of OGD does not change proteasomal activity between the groups 3 hours (30 minutes after reoxygenation) and 24 hours after beginning of OGD (Reinheckel et al., 1998). The results were unchanged when cells were subjected to OGD for only 75 minutes (data not shown). Therefore, the authors concluded that the differences between sham-washed control and the OGD treatment in the protein turnover is rather caused by the increased amount of available substrates, in this case oxidized proteins, and not to changes in the proteasomal activity itself.
DISCUSSION
The authors' principal finding is that after oxygen-glucose deprivation proteins in neuronal-enriched cultures are oxidatively damaged, as assessed by protein-bound carbonyls. This accumulation of oxidized proteins is followed by enhanced proteolysis. This proteolysis is probably mediated by the proteasome complex and not by other proteases, because this proteolysis was completely blocked after incubation with a proteasomal inhibitor.
The proteasomal system is a nonlysosomal proteolytic machinery consisting of the barrel-like central 20S proteasome, the dumbbell-shaped 26S proteasome, and several regulators. Numerous proteins are marked for degradation by covalent attachment of ubiquitin. The 26S proteasome has been implicated in diverse cellular functions like development, metabolism, cell cycle control, or control of transcription factors (DeMartino and Slaughter, 1999). Inhibition of the proteasome induces apoptosis in rat primary neurons and glioma cells (Qiu et al., 2000; Kitagawa et al., 1999), possibly by increasing ADP-ribosylation (Keller and Markesbery, 2000), but might also block apoptosis (Sadoul et al., 1996). In addition, proteasome also has a key role in recognizing and degrading proteins after oxidative damage in diverse cell types (Grune et al., 1995, 1996; Sitte et al., 1998).
The authors found a rapid decline of proteasomal activity after beginning of OGD, possibly because of rapid adenosine triphosphate depletion. Surprisingly, this decline also was observed in sham-washed cells that received glucose and oxygen. This finding currently cannot be explained. However, the fact that proteasomal activity was not lower in OGD is consistent with the hypothesis that the proteasome itself is relatively resistant to oxidative stress (Reinheckel et al., 1998). However, other authors have found decreased proteasomal activity after cerebral ischemia with reperfusion in mice (Keller and Markesbery, 2000) and in other models (Okada et al., 1999). These differences are probably because of different models, assays to determine proteasomal activity, and the severity of oxidative stress as well as the presence of other protective molecules like heat shock proteins (Conconi et al., 1998).
Whether predominantly neurons or astroglial cells are damaged by oxidized proteins after OGD has not been examined in this study. Oxidative damage has been suggested to occur in cerebral ischemia and reperfusion and has been attributed to play a major role in neuronal damage. However, because of their short-lived nature and technical limitations, direct assessment of free radicals and associated damage to cellular constituents has been limited. Protein oxidation has been measured by diverse methods as methionine sulfoxide formation, protein surface hydrophobicity, or dityrosine formation. Oxidative damage to proteins may occur as alterations of the protein backbone or amino acid side-chains. Aliphatic side chains are oxidized to alcohols, hydroperoxides, and carbonyl compounds. Because carbonyl compounds are not abundant in normal proteins, they are good markers for the overall oxidative damage to proteins, especially for in vitro studies (Davies et al., 1999). In the current study, a carbonyl-ELISA was used, based on the 2,4-dinitrophenylhydrazine assay, which is applicable for tissue culture and has been shown to correlate sufficiently with other measures of oxidative damage, like glutathione or the lipid peroxidation product malondialdehyde (Pantke et al., 1999). In the current study, protein carbonyls were increased at the end of hypoxia and remained elevated after reperfusion, suggesting that, in this model, free radicals are produced during hypoxia and the reoxygenation period.
Besides damage to nucleic acids and lipid peroxidation, proteins are main targets of free radicals after cerebral ischemia (Oliver et al., 1990). Protein damage may lead to inactivation of enzymes and ion channels resulting in functional and structural damage of neurons. Oxidative damage to proteins occurs during physiologic conditions as a consequence of oxidative phosphorylation and other processes. In ischemia–reperfusion injury, oxygen free radicals (mainly superoxide and hydroxyl radicals) are produced by several pathways: oxidative phosphorylation, by the xanthine oxidase, the Fenton reaction with iron, arachidonic acid catabolism, or secondary to the calcium-dependent nitric oxide synthase. Several studies have examined protein metabolism after cerebral ischemia. New protein synthesis is disturbed after ischemia, much like other endergonic processes. Oxidative damage in the brain leads to structural alteration of membrane-associated proteins (Hall et al., 1995). Also, the activity of rate-limiting enzymes in the protein synthesis process like initiation factor 2 is reduced after ischemia (Hu and Wieloch, 1993). In contrast, little is known about the fate of oxidized proteins or physiologic proteolytic responses after ischemic stress in neurons. The current results stress the importance of the proteasome after oxidative injury/oxygen-glucose deprivation in neuronal cultures.
Currently, the role of the proteasome in neuronal death is not entirely clear. Proteasomal inhibition has been shown to induce apoptosis in rat primary neuronal culture (Qiu et al., 2000), but inhibited NGF-induced apoptosis in sympathetic neurons (Sadoul et al., 1996). In a rat model of focal cerebral ischemia, proteasomal inhibition reduces infarct volume, probably by antiinflammatory actions (Phillips et al., 2000).
The authors conclude that oxidized proteins after hypoxia are not only targets of free radical–induced damage, but also are subjected to active degradation by physiologic proteases like the proteasome complex. Among other antioxidative systems, the proteasome may be an active part of the cellular defense against oxidative stress after cerebral ischemia.