
Abstract
A modern understanding of how cerebral cortical lesions develop after acute brain injury is based on Aristides Leão’s historic discoveries of spreading depression and asphyxial/anoxic depolarization. Treated as separate entities for decades, we now appreciate that these events define a continuum of spreading mass depolarizations, a concept that is central to understanding their pathologic effects. Within minutes of acute severe ischemia, the onset of persistent depolarization triggers the breakdown of ion homeostasis and development of cytotoxic edema. These persistent changes are diagnosed as diffusion restriction in magnetic resonance imaging and define the ischemic core. In delayed lesion growth, transient spreading depolarizations arise spontaneously in the ischemic penumbra and induce further persistent depolarization and excitotoxic damage, progressively expanding the ischemic core. The causal role of these waves in lesion development has been proven by real-time monitoring of electrophysiology, blood flow, and cytotoxic edema. The spreading depolarization continuum further applies to other models of acute cortical lesions, suggesting that it is a universal principle of cortical lesion development. These pathophysiologic concepts establish a working hypothesis for translation to human disease, where complex patterns of depolarizations are observed in acute brain injury and appear to mediate and signal ongoing secondary damage.
Keywords
Introduction
The most important clinical conditions causing acute brain damage are cerebrovascular disease (ischemic and hemorrhagic stroke), head trauma, cardiac arrest, and asphyxiation-hypoxia. These neurologic emergencies affect people of all ages and backgrounds, and cardiac arrest and stroke in particular are the top two causes of mortality globally. In survivors, permanent brain damage often leads to severe disability and loss of independence. Thus, these conditions have been a major priority for public research funding and for pharmaceutical and medical device companies to improve prevention, diagnostics, and treatment. In particular, the concept of neuroprotection emerged from advances in understanding the fundamental pathophysiology of these conditions, spawning hope that new therapeutic approaches could limit the extent of damage, and therefore, the burden of morbidity and mortality. 1
This issue of JCBFM highlights a central mechanism in the development of acute brain injury that until recently has scarcely been recognized. The fundamental mechanism was discovered 70 years ago by the Brazilian physiologist, Aristides Leão, who described what is commonly known as “spreading depression” of cortical activity. 2 Beginning in the 1980s, it was increasingly accepted that this mechanism was relevant to acute brain injuries, particularly those resulting from ischemic and hemorrhagic stroke, as well as trauma. Research on spreading depression and its variants gained further momentum when they were convincingly demonstrated in the human brain, 3 occurring with high incidence in each of these diseases. 4 The reader is referred to excellent reviews of spreading depression, covering topics of biophysics and cellular mechanisms,5,6 vasculature and hemodynamics,7,8 and relevance to human disease.4,8–10
Definitions of spreading depolarization and spreading depression.
In this article, we review the history and scientific rationale that establishes the spreading depolarization continuum. On the basis of this understanding, we discuss the role of the initial spreading depolarization in early damage from severe ischemia and then summarize the conclusive evidence that subsequent, spontaneous waves of spreading depolarizations cause secondary lesion growth in focal ischemia. We then review application of the same concepts to other models of acute cortical lesions, including subarachnoid hemorrhage (SAH) and traumatic brain injury (TBI), and conclude with brief comments on clinical translation. Due to space limitations, we focus on acute neuronal cell death in adult cerebral cortex; white matter, developmental changes, specialized structures or cell types, subcortical or other gray matter, graded insults (e.g., concussion or hypoglycemia), and treatment implications are beyond the article’s scope. Collectively, the spreading depolarization continuum emerges as a conceptual and practical framework that is essential to understanding the full spectrum of clinical neurologic conditions resulting in acute brain damage.
Foundations of the spreading depolarization continuum
Mass depolarization is a fundamental response of cerebral cortical regions subjected to severe ischemia. This was first shown by Leão in 1947 when he described a negative “slow voltage variation” in cortical recordings that occurred 2–5 min after interruption of the cerebral circulation and persisted as long as the arteries were occluded.
11
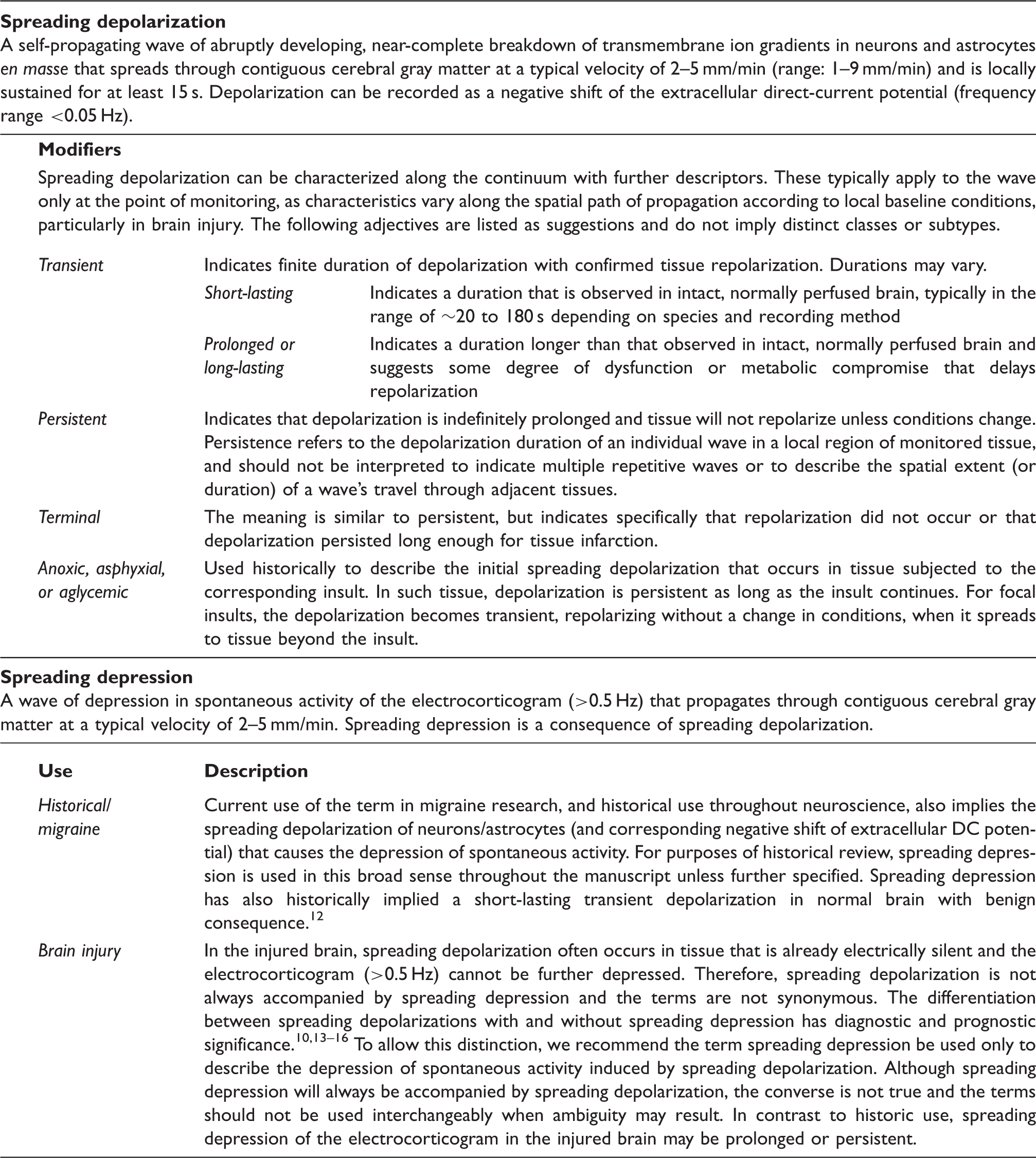
He noted that this slow voltage variation—a negative shift in extracellular direct current (DC) potential—was similar in several respects to the slow voltage variation that accompanied the spreading depression of spontaneous cortical activity: in amplitude, in abrupt rate of development, and in the spreading, nonsimultaneous onset across the cortex. He further observed that recovery of the negative DC shift of spreading depression in normally perfused cortex could be delayed indefinitely by sudden occlusion of the cerebral arteries. Thus, Leão speculated that in the spreading depression of activity, “some change of the same nature as one resulting from prolonged interruption of the circulation occurs in the cerebral cortex” (Figure 1).
Direct-current potential shifts in spreading depression and after ischemia are of the same nature. In all traces, negative is up, amplitude divisions are 3 mV, and time divisions are 1 min. (a) The original tracing published by Leão showing the initially negative slow voltage variation that accompanies spreading depression.
11
Spreading depression was elicited by application of a tetanic current to the cortex at time marked “S.” Inset shows recording and stimulation schematic. (b) In the same article, he showed a similar but persistent negative slow voltage variation several minutes after clamping (denoted by * in the trace) of the common carotid and basilar arteries. (c) The similar nature of these phenomena was further demonstrated by inducing spreading depression with electrical stimulation as in (a) and then clamping the arteries for 1 min (from * to #) as soon as the slow voltage variation of spreading depression was observed. In this case, the negative slow voltage variation of spreading depression was prolonged. Subsequent studies showed that both spreading depression and the response to ischemia are spreading mass depolarizations that are dependent on energy supply for reversal. In the experiment shown in (c), we further find a prediction of how spreading depolarization causes infarct growth: if the depolarization itself, rather than an investigator’s hand, can worsen ischemia, the tissue should remain depolarized. Adapted from Leão
11
with permission.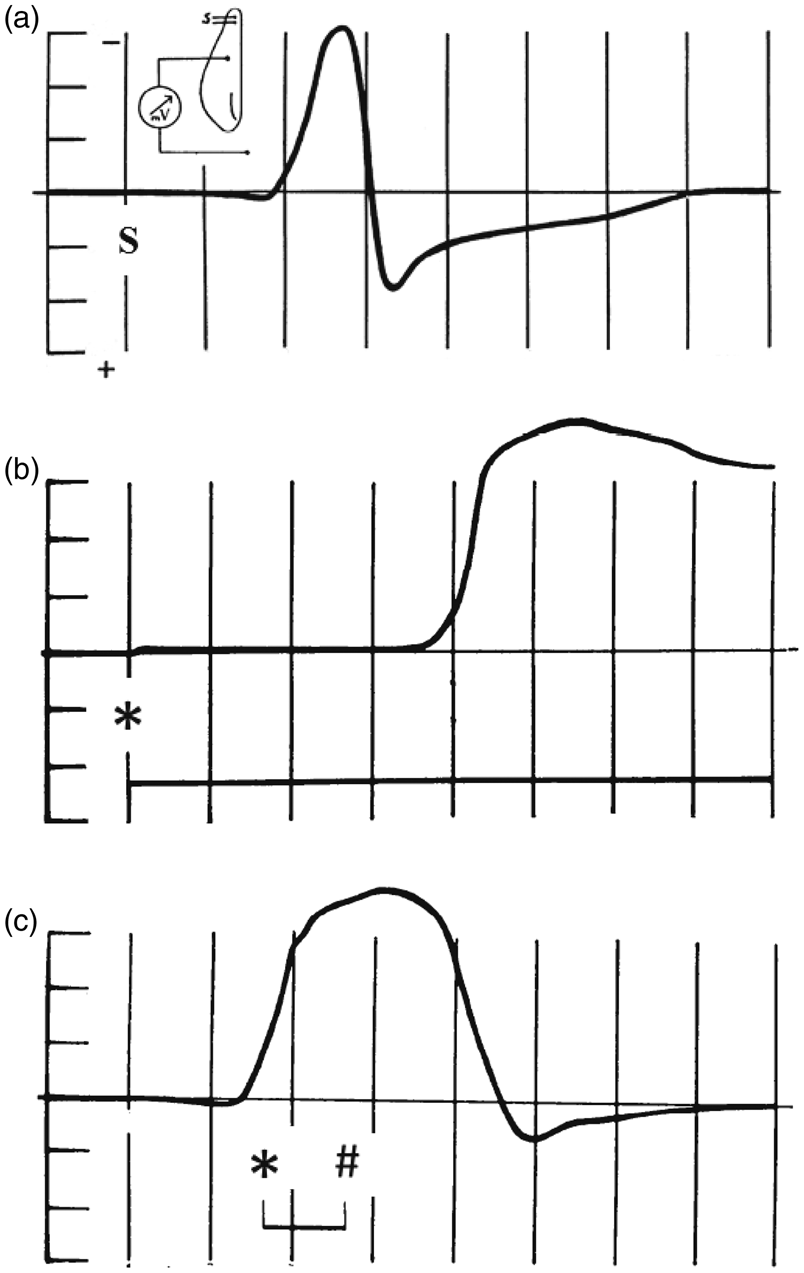
In a striking manner, this single study established two poles of a continuum of a fundamental pathological response of the cerebral gray matter to injurious stimuli. In the following years, indirect evidence suggested that the nature of the response, in the case of spreading depression, was a profound depolarization of cortical neurons that blocked neuronal discharges, resulting in suppression of the electrocorticogram.17,18 With intracellular recordings, Collewijn and van Harreveld 19 then confirmed that the DC potential shifts both of spreading depression and following cerebral circulatory arrest were indeed “of the same nature,” corresponding in both cases to an abrupt loss of neuronal membrane potential to nearly 0 mV. They concluded that “these asphyxial changes are identical with the corresponding features of (spreading depression) … and are caused by a common mechanism.”
Ironically, van Harreveld resisted this conclusion prior to his intracellular recordings20,21 since it had been shown that Leão’s spreading depression was not asphyxial, but on the contrary provoked a vasodilation and increase in blood oxygenation above the normal resting levels.22,23 This is interesting as an illustration of the schism in thought that developed concerning spreading depression on one hand and “asphyxial depolarization” on the other, despite evidence for their identity. In the decades after Leão’s publications, spreading depression was investigated primarily as a phenomenon provoked in the otherwise healthy brain, in accordance with its original description. 2 This was encouraged by early identification of its possible clinical relevance in migraine aura. Karl Lashley’s mapping of the neurologic deficits during aura—spreading scotomas in the visual field—predicted a neural correlate that closely matched the speed of Leão’s spreading cortical depression.2,24,25 Spreading depression was thus considered clinically relevant only in migraine, a disease which, except in rare cases, does not cause permanent neuronal damage.26,27 In agreement with the clinical condition, Nedergaard and Hansen 28 later demonstrated that even multiple repetitive waves of spreading depression do not cause damage to the normally perfused brain. Thus, contemporaneous studies on ischemic brain injury and asphyxial (later called anoxic) 29 depolarization were conducted under a framework quite distinct from those on spreading depression, and the two phenomena were considered separate entities. Hansen notes in 1985, for instance, that “although the two abnormalities resemble each other and may have common explanations, the common denominator is not evident.” 30
It was in this context that spreading depression-like events were first observed to occur spontaneously in animal models of focal cerebral ischemia,31–33 a breakthrough discovery that now implicated spreading depression in the pathophysiology of acute brain injury. As evidence accumulated that these depolarization waves might augment injury in the context of an existing insult, new terminology emerged to differentiate them from spreading depression, a term with a benign connotation. These included peri-infarct, peri-focal, or peri-lesional depolarization, hypoxic spreading depression, ischemic depolarization, and spreading depression-like depolarization.5,34,35 As a further matter, such waves were often evident only as DC shifts in electrically silent tissue, which precluded the possibility for the depolarization to induce spreading depression of the electrocorticogram. Thus, as the effects of these waves in injured brain were investigated, confused by proliferating terminology, a re-formulation of pathophysiologic concepts was needed.
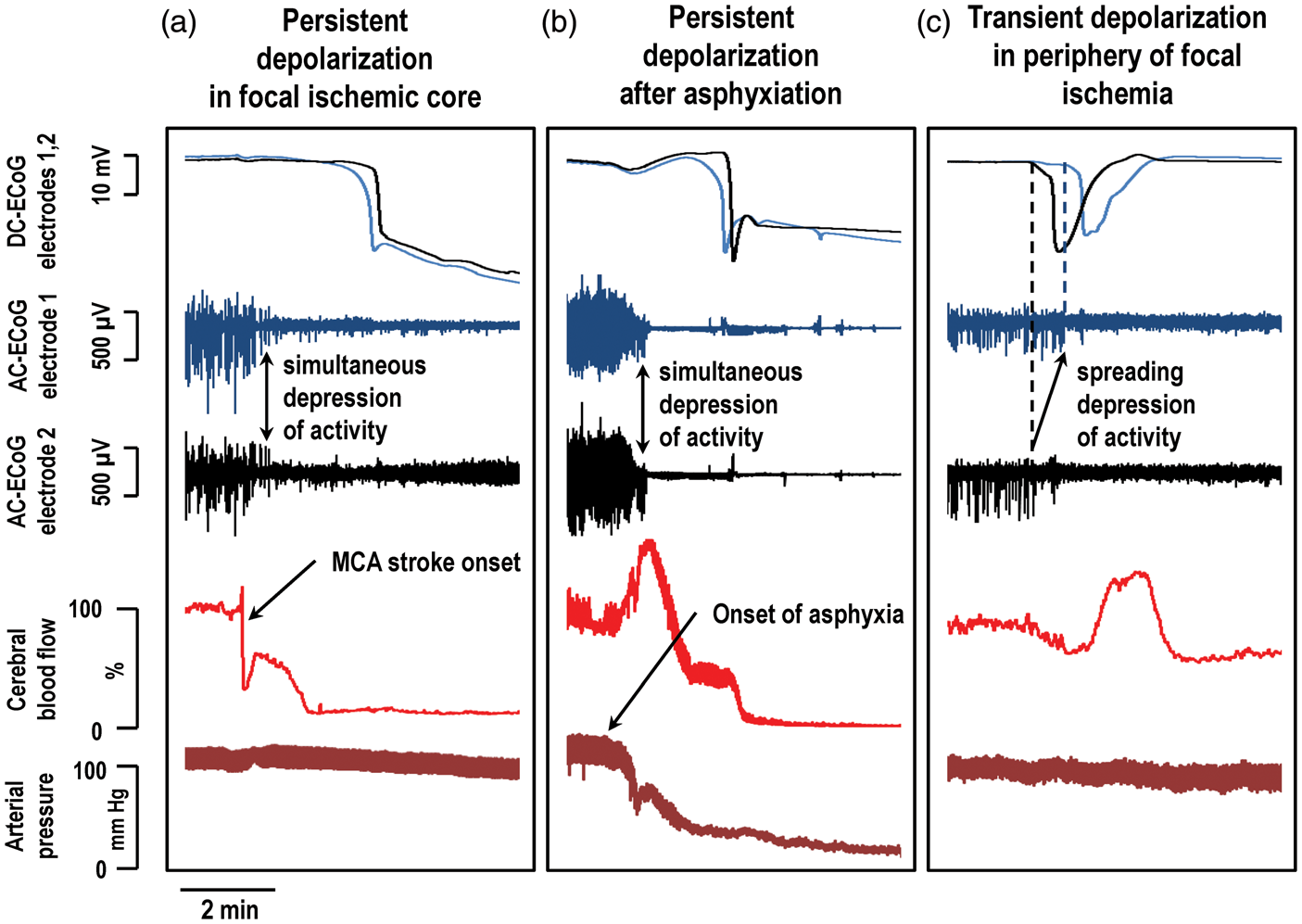
The first hint of this new concept was provided by studies that, for the first time, demonstrated a true continuum between spreading depression and anoxic depolarization in focal cerebral ischemia. Recording from multiple electrodes across the cerebral cortex after middle cerebral artery occlusion (MCAO), these studies revealed persistent depolarization in the ischemic core, spreading depression in the periphery, and intermediate-duration depolarizations in the intervening penumbra.36–39 Most notably, the first depolarization that erupts in the core propagates into the normal periphery as a single wave, but with gradually changing characteristics and durations (Figure 2(a)). In the ischemic core, this persistent depolarization is preceded by simultaneously developing suppression of the electrocorticogram (Figure 3(a)), identical to Leão’s asphyxial depolarization in global ischemia (Figure 3(b)).
11
When the same wave spreads to normally perfused cortex with spontaneous electrical activity, however, it induces spreading depression of the electrocorticogram (Figure 3(c)). The continuity of such singular waves may have been the “common denominator”
30
to finally unite prior descriptions of spreading depression and persistent asphyxial depolarization.
Persistent depolarization defines the ischemic core and causes initial and delayed secondary infarct development. Schematic diagrams illustrate various zones of focal ischemia, such as middle cerebral artery occlusion (MCAO), defined by electrophysiologic function and regional cerebral blood flow (rCBF). Direct current (DC) potential recordings (black) of spreading depolarizations and rCBF (red) are shown below for the three monitoring locations indicated by stars. The diagram in (a) depicts the status of ischemic zones after passage of the initial spreading depolarization (Time A). Within seconds of ischemic onset, a broad area with rCBF <15–23 ml/100 g/min simultaneously develops (in a nonspreading manner) complete suppression of the electrocorticogram (electrical silence). After 2–5 min, spreading depolarization then develops, as shown for instance by the cross-hairs, in a more central zone where rCBF is <5–10 ml/100 g/min. Here, the depolarization is persistent and will be terminal if perfusion is not restored. The zone of persistent depolarization and rCBF <5–10 ml/100 g/min is defined as the ischemic core, and the zone just beyond the core with electrical silence is the classical (inner) penumbra. The initial depolarization spreads through penumbra (arrows) and into normally perfused tissue as a transient wave with decreasing duration determined by local rCBF. (b) In the subsequent course of minutes, hours, or even days, spreading depolarizations arise repeatedly in penumbral hot zones as a result of local energy supply-demand mismatch. These hot zones may be located in the outer penumbra (oligemic tissue with preserved spontaneous activity) or in the inner penumbra. In the former case, as illustrated, the depolarization propagates not only outward, but also inward toward the ischemic core, inducing persistent depolarization and further perfusion decrease in the innermost penumbra (recording location 2). Thus, the ischemic core and region of developing infarction increase in a step-wise manner after the passage of each spreading depolarization (Time B). Note that spreading depolarizations that are prolonged or recur in high numbers can have the same cumulative effect as a single persistent depolarization, reaching the threshold duration of ionic derangement and metabolic impairment that commits neurons to cell death. The spreading depolarization continuum unites anoxic (persistent) depolarization and spreading depression. (a) After middle cerebral artery occlusion, simultaneous suppression of the electrocorticogram (AC-ECoG, 0.5–70 Hz) develops within seconds in the ischemic core and classical penumbra. This is followed ∼2 min later by persistent depolarization that develops in a spreading manner, as revealed by a negative DC shift of the cortical potential (DC-ECoG). (b) The same sequence of events occurs after asphyxiation, as shown in global ischemia by Leão.
11
(c) In focal ischemia, the initial spreading depolarization propagates into the normally perfused periphery, where it induces spreading depression of spontaneous AC-ECoG activity.
2
Here, asphyxiation was induced by reducing inhaled oxygen to 0% and focal ischemia was induced by injecting a blood clot near the origin of the middle cerebral artery.
69
ECoG was recorded from two glass micropipette electrodes separated by 2 mm and cerebral blood flow was monitored by laser Doppler flowmetry.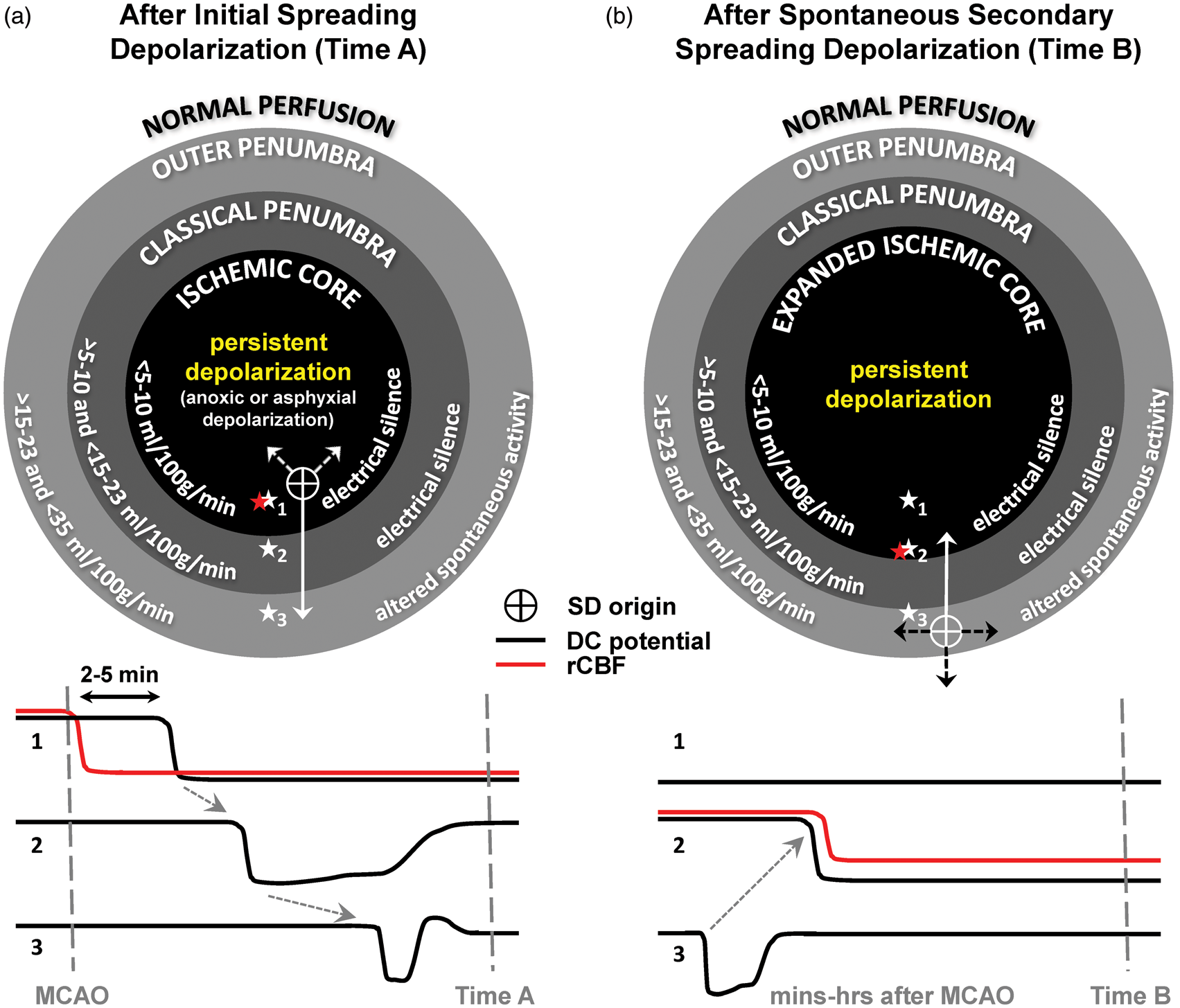
While these concepts and their relation to lesion development are reviewed in detail below, of relevance here is that the essential phenomenon of this spreading depolarization is unchanged in its defining features as it transitions through the continuum from persistent depolarization in severely ischemic tissue to spreading depression in normal brain,
5
including (a) degree of cellular depolarization and changes in principal ion concentrations (Figure 4), (b) release of neurotransmitters, (c) structural changes of cytotoxic edema and dendritic beading, (d) intrinsic optical signal, (e) abrupt onset of extracellular DC shift, and (f) slow speed of spread (1–8 mm/min). Thus, it has become clear that in the context of brain injury, Leão’s spreading depression and asphyxial depolarization are two ends of the same spectrum, and are indeed “of the same nature.”
11
The reader is referred to comprehensive reviews of differences that arise along the spreading depolarization continuum, and how the continuum may explain diverse clinical correlates.6,9,26,40 In particular, we note that there are some differences between spreading depolarizations evoked by noxious stimuli in otherwise intact cortex, and those triggered by ischemia. These include preceding tissue changes, such as gradual acidification and rising [K+]e in ischemia, and thus the pharmacology of initiating mechanisms.5,6,41 These differences, including the nature of the precipitating insult, also exist on a continuum.
9
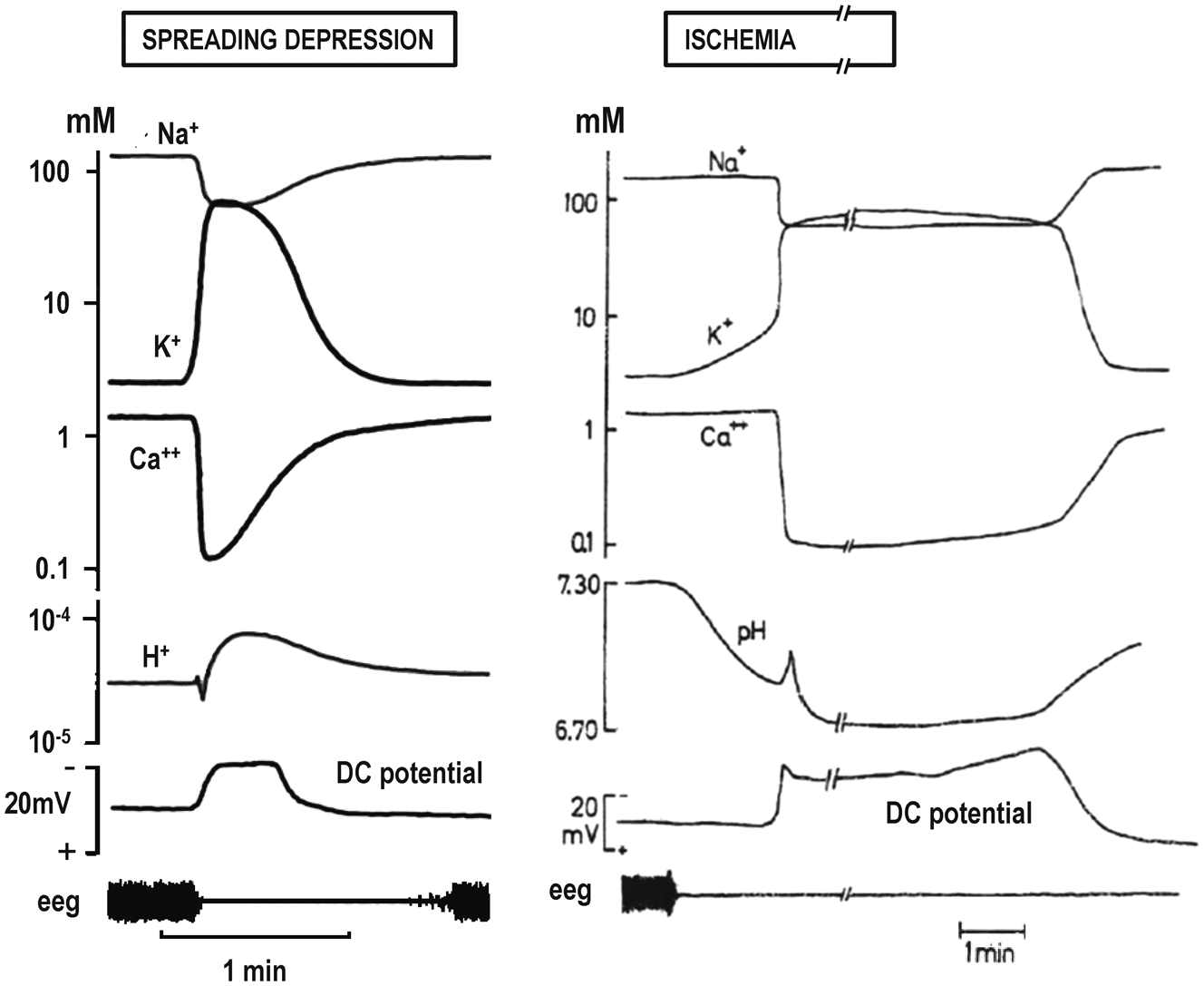
Changes in extracellular ion concentrations in spreading depression and asphyxial depolarization. Extracellular ion concentrations change to a similar degree and with similar time course during spreading depression (historical use, Table 1) in the normal brain and during the spreading depolarization that develops after asphyxiation. In the latter case, depolarization persists until cerebral perfusion is restored. A notable exception between the two cases is the gradual rise in [K+]e and decline of pH prior to the abrupt shifts during asphyxial depolarization. Adapted from Hansen and Lauritzen.
41
Reflecting a new conceptualization of brain injury depolarizations, the term “spreading depolarization” (SD) is now used as a generic name for all variants of mass tissue depolarization along the continuum originally described by Leão (Table 1). While the term he coined in his 1944 article, spreading depression,2,42 has been widely used for decades, it was unknown at that time that the underlying mechanism, reflected in the slow voltage variation, was mass neuronal/astrocytic depolarization. Nor could Leão have appreciated the extent to which he was correct in speculating that the slow voltage variation after cerebral circulatory arrest was the same essential phenomenon underlying spreading depression. In hindsight, therefore, we consider that Leão’s contribution was discovery of the full continuum of SDs, not only the subset that induces spreading depression of the electrocorticogram. While a subtle shift in terminology may seem a disservice to Leão, we rather consider that it more fully honors his rightful legacy, while also installing a neutral and uniquely descriptive term that may prove durable in growth of the field.
Spreading depolarization initiates neuronal injury in global and focal cerebral ischemia
In the sudden onset of severe cerebral ischemia, as occurs in cardiac arrest, asphyxiation, or ischemic stroke, the first electrophysiologic consequence is the suppression of spontaneous activity, reflected in the 0.5–70 Hz band of the electrocorticogram, which develops simultaneously in widespread regions 10–20 s after reduction of blood flow below the critical threshold of 15–23 ml/100 g/min (Figures 2(a) and 3(a) (b).31,43,44 Loss of synaptic activity results from neuronal hyperpolarization and adenosine-mediated suppression of vesicular transmitter release, which reduce energy consumption as a survival response to ischemia.45–48 If blood flow is reduced below 5–10 ml/100 g/min, the widespread depression of activity is followed, after a delay of 2–5 min, by mass tissue depolarization (Figures 2(a) and 3(a) and (b)).11,19,20,49 This depolarization, observed as a negative DC shift of extracellular potential, develops first focally and then spreads through contiguous cortex at 3.3–6.8 mm/min.50–55 In some cases, the apparent propagation velocity of this initial SD developing in severely ischemic cortex may be greater than SDs observed in normally perfused cortex. This occurs mainly because other SD foci develop in adjacent ischemic regions before they are engulfed by the initial wave 55 ; the full extent of severely ischemic tissue is then rapidly depolarized by convergence of waves from multiple focal origins.
In both global and focal ischemia, this initial depolarization is persistent in regions where blood flow is below the 5–10 ml/100 g/min threshold (Figure 2(a)). It has sometimes been described as “terminal depolarization,” 29 which is accurate when blood flow is not restored, since depolarization then persists until the tissue becomes infarcted (Table 1). As a generic term, however, “terminal depolarization” fails to recognize that, depending on time, the initial persistent depolarization in severe ischemia is a reversible phenomenon.53,56,57 This was first demonstrated by Leão, who found that the negative DC shift could be reversed by restoring perfusion after ischemia up to 12 min in duration. 11 There is no precise threshold duration beyond which mass depolarization is irreversible, since recovery can occur in a graded or partial manner. Importantly, reversal of the negative DC shift does not indicate that neurons will survive.
As noted above, the initial persistent depolarization that erupts after severe ischemic onset has historically been termed asphyxial or anoxic depolarization, with implications of conditions and consequences that are distinct from SD.20,29,58 However, specific definitions of “anoxic depolarization” prove to be elusive and are all misleading in some important respect, which only further illustrates the nature of the SD continuum. First, hypoxia-anoxia during SD is not unique to cases of proximal arterial occlusion, but occurs to some extent in normal tissue59–61 and more severely when SD induces the profound microvascular constriction known as spreading ischemia, as discussed below.62–64 Second, SDs can be triggered by decreased perfusion not only after initial occlusive ischemia, but also by hypotensive transients in the ischemic penumbra as a secondary injury process occurring hours after the initial ictus. 65 Third, not all SDs induced by initial occlusive ischemia are persistent, and persistent depolarization is not unique to this condition. For instance, SDs occurring within minutes of microembolization are short and transient.66,67 On the other hand, spontaneous SDs occurring hours after more severe focal ischemia can induce persistent depolarization in the inner penumbra, as reviewed below.36,68–71 Finally, the term “anoxic” is misleading since it is not only the lack of oxygen, but also of glucose and blood flow that are essential in inducing and maintaining tissue depolarization. Therefore, here we use the term “persistent depolarization” to describe indefinitely prolonged depolarizations under conditions of severe ischemia (Table 1). “Terminal depolarization” is used more specifically to describe persistent depolarization that endures until tissue becomes infarcted.
As discussed in the following, it is the initial persistent depolarization that triggers a loss of ion homeostasis and cytotoxic edema, and therefore marks the onset of structural injury and countdown to irreversible damage, in both global ischemia and the core of focal ischemia. Delayed lesion expansion in focal ischemia occurs in a similar fashion, triggered by secondary spontaneous SDs that induce persistent/terminal depolarization and excitotoxic injury in the penumbra. Definitions of the ischemic core and penumbra used here are provided in Figure 2 and explained below.
Spreading depolarization triggers the loss of ion homeostasis and release of neurotransmitters after ischemic onset
The sequence of pathophysiologic events following severe ischemia has been well characterized and generally involves the rise of [K+]e, depolarization of presynaptic terminals, excessive extracellular accumulation of neurotransmitters, activation of N-methyl-D-aspartate (NMDA) receptors, a general loss of ion homeostasis (Ca2+, K+, Na+, H+, Cl−, HCO3−) with attendant rise of intracellular Ca2+, and the onset of cytotoxic edema.72–74 In the general literature, the central role of SD in these processes is rarely acknowledged. This again may be due to the historical view of asphyxial/anoxic depolarization as distinct from Leão’s actively propagating phenomena. When the time course of the extreme ionic changes in ischemia were described, investigators noted the similarity to ionic changes during spreading depression (Figure 4).33,41,75–78 However, since only SD in normal brain (i.e., spreading depression, “a pathological event without pathology”) 75 was considered, it was difficult to reconcile these transient changes without damage to the persistent ionic changes associated with anoxic-ischemic damage. Hence, the concepts of the ischemic penumbra and neuroprotection developed initially, and persist largely today, without consideration of the SD continuum.31,43,44 Marshall was an exception to this trend in describing asphyxial depolarization as asphyxial SD. 79
Understanding now that persistent DC shifts (i.e., anoxic depolarization) are SDs, there is unequivocal evidence that SD is the network/systems process that triggers the major changes of the ischemic pathophysiologic cascade. In various in vivo models including hypoglycemia, respiratory paralysis, and ischemia, [K+]e first rises slowly over 2–4 min until it reaches 9–11 mM, a “ceiling level”
80
in the cortex which is only exceeded when SD occurs. Thus, the next phase is a sharp rise of [K+]e to a new equilibrium up to 75 mM, which coincides with the negative DC shift that signals the onset of persistent depolarization (Figure 4(b)).30,49,77 In the same simultaneous event, there is a sharp decline in extracellular Ca2+ ([Ca2+]e) from 1.3 to <0.1 mM, [Cl−]e from 150 to 95 mM, and [Na+]e from 150 to 60 mM.9,30,33,78,81–83 These shifts develop in seconds, while prior to depolarization there is little change in these ions. The same results have been confirmed with imaging studies of global ischemia in vivo and oxygen-glucose deprivation in vitro: after onset of hypoxia-ischemia, the steep rapid rise in intracellular Ca2+ occurs only as a consequence of persistent depolarization (Figure 5).53,84–86 By comparison, the time course of glutamate changes in relation to the onset of persistent depolarization has been more difficult to resolve in vivo since microdialysis techniques have limited resolution. However, several studies suggested a close association, with glutamate increasing at the time of depolarization or thereafter.87–89 Recent use of amperometric techniques confirmed that a steep rise in glutamate after arterial occlusion develops simultaneous with depolarization onset, and does not precede it,
69
as for SD in normal cortex.
90
Spreading depolarizations evoked in hippocampal slices by oxygen-glucose deprivation and by high K+. CA1 neurons were loaded with Ca2+ indicator fura-6F. The first image panel shows raw 380 nm flourescence and subsequent panels are pseudocolor images that represent [Ca2+]i. (a) Following oxygen-glucose deprivation, there is no increase in Ca2+ in ∼11.5 min prior to onset of spreading depolarization (SD). After SD, there is a large irrecoverable Ca2+ increase (∼24 µM) that originates in the soma and progresses toward apical dendrites, resulting in rapid neuronal injury. (b) SD evoked by high K+, by contrast, produces a transient Ca2+ elevation in distal dendrites that propagates toward, but never fully involves the soma, and [Ca2+]i returns to basal levels in <2 min without neuronal injury. SD propagation rates are similar in the two conditions. Initiation and propagation of SD evoked by high K+, but not by oxygen-glucose deprivation, is dependent on the intracellular Ca2+ influx and can be prevented by Ca2+ removal from the bath, illustrating a mechanistic difference that arises along the continuum. Reproduced from Dietz et al.
85
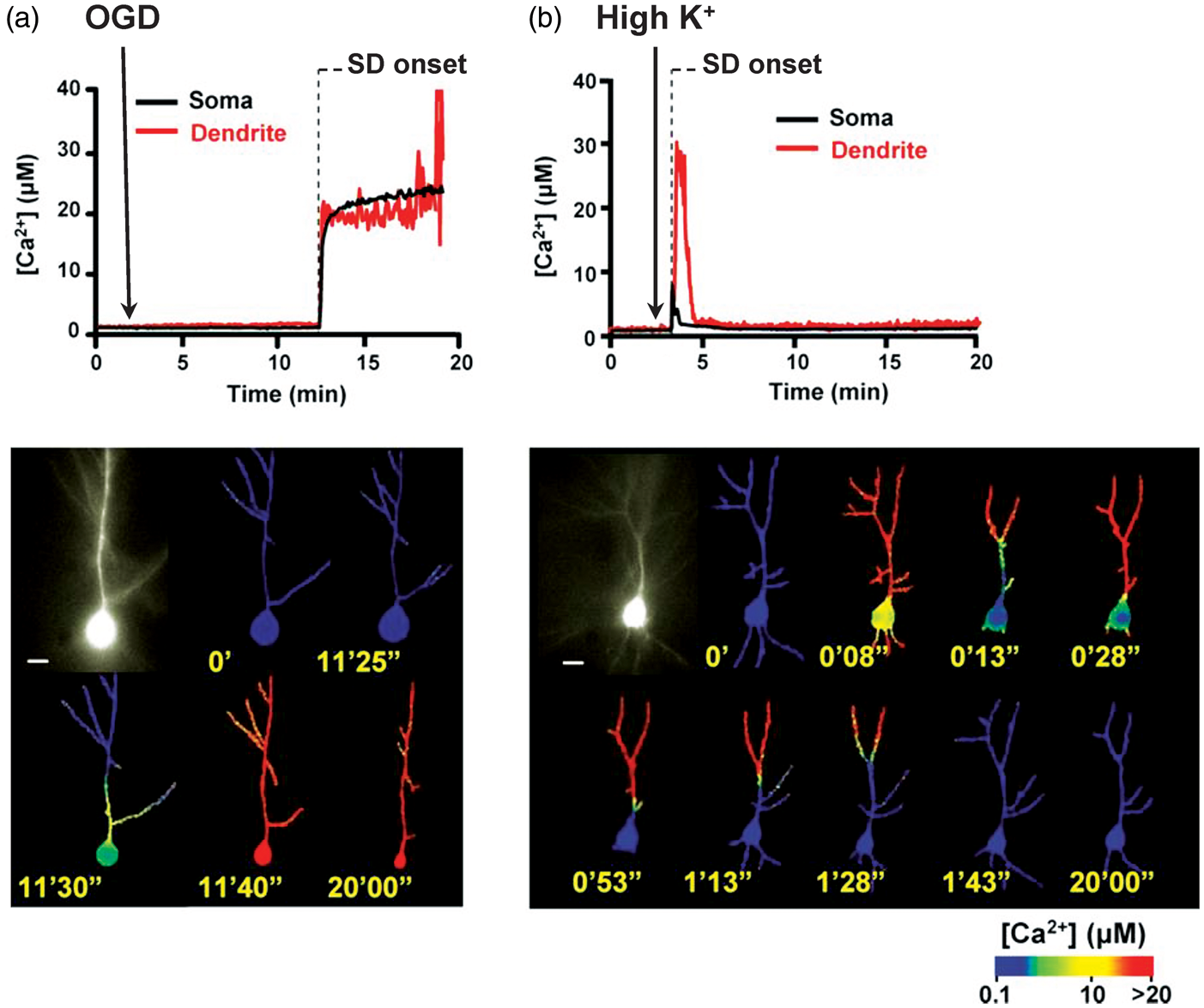
The extreme ionic loading that occurs during the initial persistent depolarization wave might be sufficient to kill neurons simply as a consequence of the increased metabolic demand coupled with the lack of substrates to fuel ATP-dependent ion pumps. That is, even though many of the mechanisms of excitotoxic injury are active at this end of the SD continuum, including profound glutamate accumulation and Ca2+ loading, it appears NMDA-dependent Ca2+ pathways may not be required for injury. In some global ischemia and in vitro models, for instance, prevention of Ca2+ loading or antagonism of NMDA receptors fails to protect against the initial structural injury induced by persistent depolarization.53,55,91
Spreading depolarization causes cytotoxic edema and characteristic changes in imaging diagnostics of ischemic stroke
Persistent depolarization following severe ischemia is also an important mechanism to recognize since it induces the structural changes, known as cytotoxic edema, that are the basis of clinical stroke diagnosis by diffusion-weighted imaging. Disruption of the physiological ion gradients during SD results in intracellular water accumulation and abrupt neuronal and astroglial swelling. Neuronal cytotoxic edema involves a plasma membrane reorganization that gives dendrites the appearance of beads on a string, termed dendritic beading (Figure 6(a) and (b)). Neuronal and astroglial swelling was first recognized as an abrupt increase in cortical tissue impedance that accompanied the “asphyxial depolarization,”92,93 a change also observed for SD in normal brain.
79
These results provided evidence of water and chloride uptake into apical dendrites,94,95 and dendritic swelling was visualized using electron microscopy.
96
Cytotoxic edema following persistent depolarization of severely ischemic cortex thus results in a 50–65% shrinkage of the extracellular volume, similar to that during SD in normal brain.57,97–99
Cytotoxic edema consequent to persistent depolarization is the basis of diffusion lesions in clinical imaging of stroke. (a) and (b) show two-photon images of apical dendrites in superficial layers of mouse cerebral cortex following bilateral common carotid artery occlusion (BCCAO) at times shown in DC potential recording above. The recording shows the onset of spreading depolarization in the imaging field 4 min 30 sec after BCCAO. Normal dendritic morphology with spines, which are sites of excitatory synaptic transmission, is observed for the first minutes after BCCAO, but spines disappear and dendrites become beaded after the tissue depolarizes. Image in (b) is taken 26 sec after depolarization onset. (c) and (d) show diffusion-weighted MRI scans from patients with ischemic stroke and aneurysmal subarachnoid hemorrhage. The scan in (c) was taken within 48 h of cardioembolic stroke in the right middle and anterior cerebral artery territory of a 41-year old woman with initial NIH stroke scale of 16. The scan in (d) was obtained 3 days following rupture of a basilar tip aneurysm in a 57-year old man who presented as Hunt-Hess grade 4 and modified Fisher grade 2. At this time, transcranial Doppler showed moderate-severe vasospasm in the right and mild-moderate vasospasm in the left middle cerebral arteries. Hyperintense regions reflect decreases in apparent diffusion coefficient caused by cytotoxic edema, dendritic beading and restricted intracellular diffusion of water. Two-photon imaging studies suggest that these changes reflect the occurrence of persistent tissue depolarization.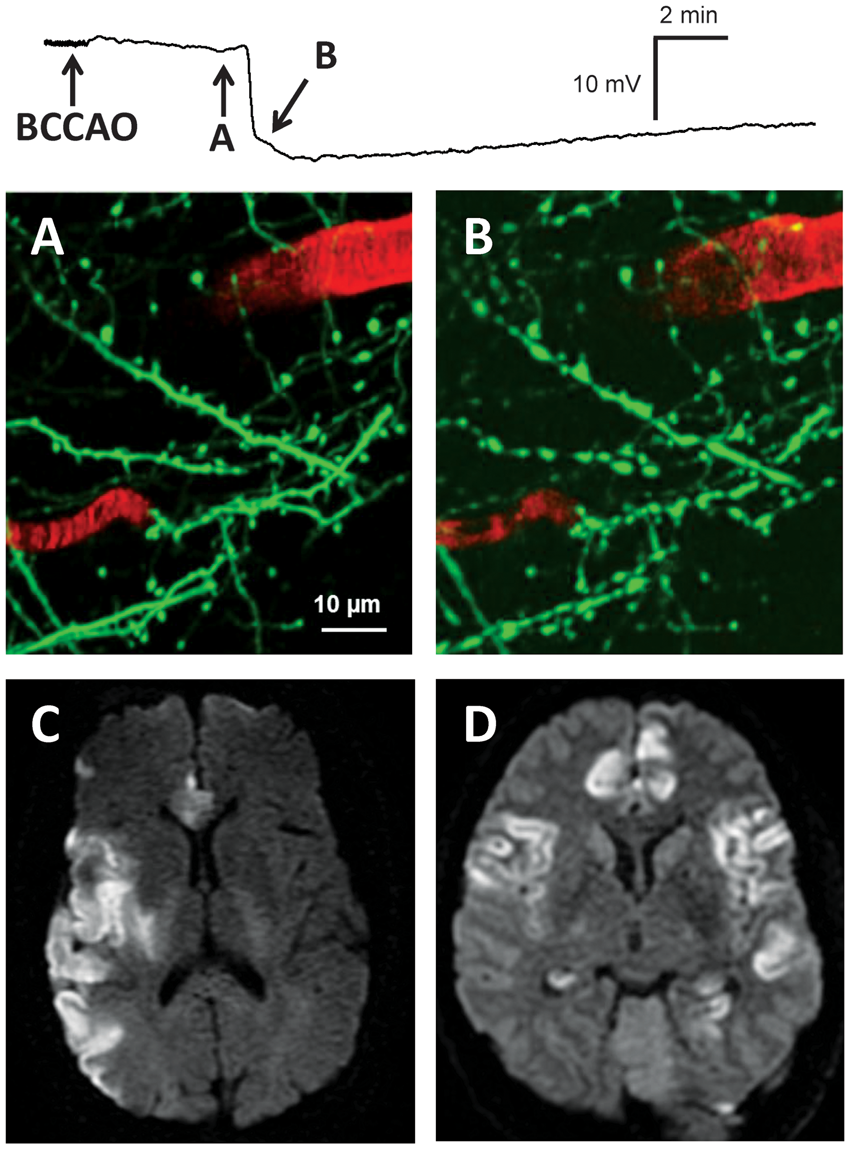
These structural changes can be visualized by magnetic resonance imaging (MRI) of restricted water diffusion in cerebral gray matter. Diffusion MRI techniques are a clinical standard in the diagnosis of ischemic stroke, since they have high sensitivity and specificity and characteristic changes are visible within minutes of stroke onset (Figure 6(c) and (d)). 100 The decrease in diffusion coefficient reflects restricted intracellular movement of water following edema onset and the development of beaded neurite morphology. 101 Importantly, the imaging changes occur abruptly and simultaneously (within the resolution of MRI sequences) with the negative DC shift of persistent depolarization.98,102 SD is sufficient to induce the imaging changes since diffusion restriction is observed as a propagating pattern even during SD in normally perfused cortex. 103 Some have disputed whether diffusion restriction after stroke is a consequence of depolarization,104,105 but this question was conclusively resolved with two-photon real-time imaging of global ischemia in the mouse.53,106 About 2–3 min after ischemia onset, prior to any structural changes to cellular processes, persistent depolarization developed with its attendant spreading wave of increased intracellular Ca2+. This was followed, ∼6 s later, by abrupt beading of dendrites and astroglial swelling that progressed in a spreading pattern after the SD wavefront (Figure 6(a) and (b)). These structural changes persist as long as depolarization is maintained and mark the onset of permanent damage to synaptic circuitry. 107
While it has been assumed that neurons swell during SD because osmotically obligated water follows Na+, Cl−, and Ca2+ influx, pyramidal neurons do not express functional aquaporins and are largely water-impermeable under acute osmotic stress. 108 Recent evidence rather suggests that water is carried by select chloride-coupled neuronal cotransporters as a consequence of the altered electrochemical gradients during SD. 109
Persistent depolarization defines the core of severe focal ischemia
The work of Lindsay Symon and colleagues was the first to suggest a connection between persistent depolarization and permanent damage from severe focal ischemia. An early study suggested that damage from MCAO was confined to regions with the lowest post-occlusive blood flow (<10 ml/100 g/min). 110 To investigate what changes occur at this threshold, they examined the effects of progressive ischemia on cortical physiology by combining MCAO with controlled steps of exsanguination (hypotension).31,43 They found that [K+]e levels changed steeply according to a flow threshold of 8–11 ml/100 g/min, with [K+]e remaining <10 mM above this range, but rising persistently to a range of 30–90 mM below the threshold. Of note, sharp but transient rises in [K+]e and decreases in [Ca2+]e were also observed at higher levels of flow.31,33,111 A similarity to spreading depression was noted for the transients, but SD was not invoked to explain the sharp, persistent increases, which were attributed generically to ion pump or membrane failure. This membrane failure, marked by the persistent [K+]e increase, was proposed as the sign of impending infarction. Subsequent work has confirmed that the [K+]e elevation (and [Ca2+]e depletion) in the core of focal ischemia is triggered and maintained by persistent depolarization, as for global ischemia reviewed above.111,112
The main legacy of this work was not the role of SD in focal ischemia, however, but rather the classical concept of the ischemic penumbra (Figure 2(a)). The threshold for the steep rise of [K+]e (persistent depolarization) and consequent structural damage was considerably lower than that for functional failure (suppression of electrocorticogram), identified as ∼20 ml/100 g/min. This suggested the idea of a penumbra between the dual thresholds, where neurons remain structurally intact but functionally inactive.43,44,113 Thus, the inner boundary of the ischemic penumbra was defined as the spatial limit of persistent depolarization, which defines the ischemic core (Figure 2(a)).
Three determinants of neuronal damage: depolarization, impaired energy supply, and time
The threshold duration for persistent mass depolarization to cause permanent neuronal injury has been termed the commitment point. 40 The commitment point is not a universal value but depends strongly on the model and the level of residual perfusion, developmental stage and cell populations, and also the marker of damage chosen.114–116 Threshold durations have been reported as short as 5 min for selective neuronal injury and as long as 60 min for pannecrosis.117,118 In another noteworthy study of global ischemia, it was shown that 15 min of persistent depolarization, but not 10 min, is sufficient to cause massive neuronal necrosis. 119 Interestingly, animals that did not develop SD, despite severe ischemia at 10% baseline blood flow for 20 min, exhibited minimal or no histologic damage. Thus, depolarization appears to be a requirement for severe acute neuronal damage to develop from ischemic insults of these durations.107,119,120 Critically, however, ischemia is also required. While persistent depolarization will eventually cause infarction even in healthy brain, 28 1 h of persistent depolarization resulted in cortical infarction only when accompanied by severe ischemia. 121 Thus, as discussed below, it appears that there are three key determinants for lesion development: depolarization, impaired energy supply, and a minimum duration of each.
It is noteworthy that the minimum durations for depolarizations to cause neuronal cell death appear to be shorter than the maximum durations for reversibility of depolarization. In focal ischemia, for instance, reversal of persistent depolarization in the ischemic core has been observed even after 3 h. 68 Importantly, this means that reversal of the negative DC shift (i.e., at least partial repolarization) is not an indicator that tissue will survive, as the commitment point for cell death may have already been crossed. 68
Lesion expansion in the focal ischemic penumbra
In comparison to global ischemia, focal ischemia presents a more complex scenario in which a gradient of blood flow and impaired metabolism exists from the ischemic core, through the classical penumbra, and into the normally perfused periphery. Following ischemic onset, SD first develops in the core, where it is persistent and thus the only wave to affect this region.36,37,54 The initial SD is not restricted to the core, however, but propagates outward in a radial pattern through ipsilateral cortex, traversing tissue with a gradient of increasing blood flow. In these regions, the wave transitions to a transient event with prolonged duration in the penumbra and short duration in non-ischemic cortex (Figures 2(a) and 3). Different depolarization durations are determined by residual perfusion, and thus reflect the perfusion gradient. 122 With the passage of time, additional SDs arise due to energy supply–demand mismatch in the classical penumbra,67,123 or even in more peripheral cortex with impaired metabolism but preserved excitability (Figure 2(b)). 65 These waves can propagate in diverse patterns such as spreading radially or cycling through the penumbra around the ischemic core, sometimes repeatedly, following the path of greatest susceptibility.54,68,123,124
The causal role of secondary spreading depolarizations in ischemic lesion expansion
The hypothesis 113 that these spontaneous, secondary SDs are a causal mechanism of secondary injury was suggested by an association between final infarct volume and the burden of transient SDs observed in the ischemic periphery/penumbra—burden being measured either in SD count or cumulative depolarization duration.34,36,65,68 Moreover, the time course of SD activity through 24 h post-ischemia matches the kinetics of lesion growth: both exhibit an early (<2 h) and delayed (8–18 h) phase. 125 Therapeutic studies further supported this hypothesis, as interventions that reduced the frequency of spontaneous SDs also reduced infarct volumes. These included both physiologic (hyperoxia 126 and hypothermia 127 ) and pharmacologic (glutamate receptor antagonists125,128–131 and gap junction blocker132,133) therapies. Nevertheless, a fair critique of this cumulative evidence is that these studies did not prove a causal role of SD in infarct growth, since neuroprotection from treatments may be achieved through other pathways that cannot be excluded, and SD may be an epiphenomenon of lesion growth, arising as a consequence but not contributing causally to the process.
Evidence for the role of spreading depolarizations in penumbral expansion of focal ischemic lesions.

SD: spreading depolarization; DW-MRI: diffusion-weighted magnetic resonance imaging; NADH: nicotinamide adenine dinucleotide.
The study by Busch et al. also provided a second line of direct evidence. 134 A series of diffusion-weighted MRIs (DWI) were obtained every 15 min after stroke in synchrony with remote SD induction. They found that the persistent diffusion lesion grew in a stepwise fashion through 2 h post-stroke, in synchrony with the invasion of SD into the penumbra, thereby directly confirming the effect of SD inferred from final lesion volumes. In particular, SDs caused a transient increase in DWI signal intensity throughout the ipsilateral cortex, but a small rim around the existing lesion did not recover, becoming indistinguishable from the lesion core. Similar results were obtained in a separate study using NADH (nicotinamide adenine dinucleotide) fluorescence imaging. 68 These experiments not only directly visualized a causal role of SD in lesion growth, but also suggested how this is effected: by inducing terminal depolarization in the inner penumbra, thus expanding the zone of the terminally depolarized core (Figure 2(b)).
This conclusion is supported by electrophysiological studies that have shown terminal depolarizations develop in a delayed fashion as a consequence of the spread of spontaneous SDs.36,68,69,70,71 Surprisingly, these delayed terminal depolarizations are observed not only in the classical penumbra, where metabolism is more severely impaired, but also in tissue with preserved spontaneous activity. 69 That propagating SDs can induce terminal depolarization under conditions of metabolic compromise is confirmed in single neurons 137 and in in vivo models of partial global ischemia. 51 Together, these studies provide a third demonstration of SD’s causal effect, since it is the propagating wave that induces terminal depolarization, regardless of how or where the SD was remotely initiated.
How can an invading SD cause terminal depolarization in tissue where baseline blood flow was previously adequate to maintain membrane integrity? It is possible that the blood flow requirement for restoration of membrane potentials is higher than that for maintenance. However, it is also known that the SD wave itself induces complex perfusion changes that affect the tissue’s capacity to repolarize.7,138,139 While SD in uninjured cortex evokes a spreading hyperemia followed by oligemia, 140 SD in the penumbra elicits a blunted, absent or inverse hemodynamic response, known as spreading ischemia, 62 due to impaired neurovascular coupling and reduced perfusion pressure.54,64,141–144 As in hyperemia, the perfusion decrease follows the depolarization, and does not precede it. 64 This is also a graded phenomenon, with the perfusion waveform changing shape as SD traverses the penumbra. The blood flow response is a transient decrease followed by increase in the outer penumbra, a transient decrease only in a middle zone, and a persistent decrease in the inner penumbra (Figure 2(b)).
As a fourth piece of direct evidence of SDs adverse effect, the persistent decreases in perfusion have a cumulative effect with each repetitive SD, progressively increasing the degree and spatial extent of ischemia in a stepwise manner.54,65,124,141 This effect of SD to exacerbate ischemia results in prolongation of depolarization by reducing energy supply and perhaps vascular clearance of [K+]e.7,145 In the most severe cases, it enables terminal depolarization (Figure 2(b)).51,64,143 In other words, SD sows the conditions for its own destructive effect. The reader is referred to previous reviews on the vicious cycle of SD-induced [K+]e elevation leading to vasoconstriction and hence prolonged depolarization in nitric oxide-depleted tissue.8,146
Comparison of initial terminal and secondary spreading depolarizations
As discussed, reasonable doubt of the causal role of secondary SDs in time-dependent growth of focal infarcts has been obviated by use of advanced real-time monitoring, including MRI, NADH fluorescence, two-photon and laser speckle imaging, in combination with electrophysiology (Table 2). The direct effect of SD as executioner of vulnerable tissue is observed, as spreading waves induce further terminal depolarization and expand the infarct core. Here, the effect of terminal depolarization to induce persistent loss of ion homeostasis and structural damage is very similar to that of the initial terminal depolarization in the ischemic core.71,147 There are, however, critical differences between initial and secondary SDs apart from the triggering mechanisms described above. The first is that the initial terminal depolarization in the ischemic core follows the critical reduction in blood flow from arterial occlusion, whereas delayed secondary SDs induce the perfusion decreases that prevent or delay recovery in the penumbra (Figure 2). Second, in the case of secondary SDs, neuronal damage can result not only from a single persistent depolarization, but also from the cumulative effects of multiple transient SDs, particularly if they are prolonged or recur in high numbers.40,68,148,149 Dendrites, for instance, can undergo repeated rounds of beading and recovery with each transient SD until eventually beading becomes terminal.147,150 This progressive course toward permanent injury depends in part on the proximity of cells to capillaries and may develop in specific microdomains of more severe hypoxia/ischemia.61,147 During repetitive SDs, the failure of subsets of neurons to repolarize may underlie the shallow negative ultraslow potential (NUP) that is sometimes observed.10,151 Third, in comparison to the initial depolarization of ischemia, neuronal injury from secondary SDs may be more dependent on excess accumulation of extracellular glutamate and NMDA receptor-mediated Ca2+ loading. 152 In metabolically compromised but viable neurons, a single SD causes cell death by inducing an irrecoverable Ca2+ increase in apical dendrites that then spreads to the soma. 137 Excess glutamate release and NMDA receptor activation are required, since persistent Ca2+ loading and cell death are prevented in the same conditions by NMDA receptor antagonism. Moreover, bursts of glutamate release that occur in vivo during delayed penumbral lesion growth are observed only in synchrony with SDs, and not independently. 69 Thus, it appears that glutamate excitotoxicity occurs only in connection with SD and that excess glutamate signaling and SD are merely different facets of the same secondary injury process. The different role of NMDA receptors in secondary SDs as opposed to the initial persistent depolarization in the ischemic core has important treatment implications.6,8
Other models of cortical lesion development
Ischemic stroke is a model disease for the study of acute brain injury since the cause of cortical dysfunction is singular (vascular occlusion), the onset of pathology is extreme and abrupt, and animal models are well established. Furthermore, ischemia is an important component of other types of stroke as well as TBI, which are more multifactorial, and the concepts of secondary injury and neuroprotection were conceived largely from the study of ischemic stroke. Accordingly, characterization of the SD continuum in ischemic stroke, as described above, has provided a conceptual framework for understanding how SD may be involved in acute neuronal damage from other causes.
Subarachnoid hemorrhage
Aneurysmal SAH is a stroke subtype in which cortical ischemic lesions are observed in two primary phases, early and delayed. Early lesions develop in laminar or band-like patterns of contiguous cortex or symmetrically in midline regions (Figure 6(d)).153,154 Delayed lesions develop around 6–7 days after aneurysm rupture, often in conjunction with neurologic decline in a condition known as delayed cerebral ischemia (DCI). Cortical lesions are often found in regions covered with blood, such as a sulcus or fissure with a thick subarachnoid clot.155–158 Furthermore, the likelihood of DCI correlates with the amount of subarachnoid blood 159 and the time-course of DCI development corresponds with that of hemolysis. 160
These observations led to the idea that delayed lesions develop from the release of hemolysis products into the subarachnoid space. To model this, the rat cortex was superfused with artificial cerebrospinal fluid (ACSF) containing high concentrations of K+ and free hemoglobin. 62 These conditions were sufficient to trigger SDs that recurred repeatedly over several hours. Importantly, the electrophysiologic changes developed independently, without prior decrease in blood flow. Rather, it was found that SDs induced a profound constriction in the microvasculature, termed “cortical spreading ischemia.” Repeated SDs thus induced and sustained ischemic conditions reaching ∼10% basal cerebral blood flow for up to 2 h. Despite return of baseline blood flow and tissue repolarization after washout with normal ACSF, band-like cortical infarcts were observed at sacrifice 24 h later. 161 Therefore, cortical infarcts developed in this model through a pathophysiologic cascade similar to that in ischemic penumbra: SD induces ischemic conditions that preclude rapid recovery from depolarization. Also similar is that the commitment point for cell death was reached, despite subsequent recovery of perfusion. Importantly, spreading ischemia is an essential mechanism in this sequence, since lesion development could be blocked by application of a nitric oxide donor or the calcium channel antagonist, nimodipine, both of which at least partially reverted the spreading ischemia to spreading hyperemia.162,163
Endothelin model
Another model of cortical neuronal necrosis investigates the effects of mild but prolonged focal cerebral ischemia. Endothelin is a peptide that promotes vasoconstriction and is implicated in a number of disease states. When 1 µM endothelin-1 (ET-1) is topically applied to the rat cortex, CBF remains above the ischemic threshold (∼75 ± 15% of baseline), but several spontaneous SDs are observed in 50% of animals. 164 Interestingly, only the animals with SDs develop regions of selective neuronal death after 24 h. 148 To determine whether SDs were a cause or consequence of neuronal injury, SDs were elicited experimentally in remote cortex and propagated into the ET-1 window in animals that did not develop SD spontaneously. In each case, necrosis developed similar to animals with spontaneous SDs, and neuronal death only occurred in regions exposed to ET-1. Similar to studies of severe focal ischemia,134–136 these results support the causal role of SDs as a determinant of neuronal cell death even in mild ischemia.
Traumatic brain injury
Is the SD continuum observed in acute brain lesions developing from causes other than cerebrovascular disease? TBI is the only other condition in which this question has been addressed experimentally, albeit to a much lesser extent. The injury cascade following cerebral contusion is very similar to that after ischemia, including an early rise in [K+]e, elevation of extracellular glutamate, and neuronal injury resulting from excitotoxic Ca2+ overload.165–167 While an initial SD at the time of injury has been shown in multiple studies,168–171 to date only two studies have specifically examined early ionic changes in relation to histologically confirmed lesions. Nilsson et al. 172 found an increase in [K+]e (3.0 to 40 mM) and decrease in [Ca2+]e (1.1–0.1 mM) immediately after contusion that corresponded with a negative DC shift. These changes persisted for 5–15 min in the region where a lesion developed, but for only a few minutes in adjacent tissue that survived. Similarly, Takahashi et al. 165 found a permanent increase in [K+]e (4.0–50 mM) in tissue with a histologic contusion after a high impact injury, but only a transient increase in [K+]e (4.0–30 mM) in animals that did not develop a contusion after low impact injury. These results demonstrate that SD occurs at the moment of traumatic impact, and that the SD is prolonged when a region of neuronal injury develops.
After the initial impact, spontaneous secondary SDs have been observed for hours to days in some TBI models, but not others.171,173–178 Whether they occur and their impact on lesion development seem to depend on injury severity,169,178 the extent of a metabolic penumbra, and the presence of secondary insults (e.g., elevated intracranial pressure and hypotension). In the controlled cortical impact model, von Baumgarten et al. found no effect of remotely elicited SDs on contusion volume, despite a 6-fold increase in the number of SDs in experimental versus control animals. 168 Sword et al. 150 by contrast, showed that remotely elicited SDs caused reversible but then terminal dendritic beading in the same model. In fluid percussion injury, increases in intracranial pressure to levels observed in clinical severe TBI (30 mm Hg) were associated with continuous repetitive SDs, even in the contralateral hemisphere. 169
The anatomic pathology of clinical TBI consists not only of contusions, however, but may also include intracerebral hemorrhage, diffuse axonal injury, subdural or epidural hematoma, or any mix of these. Subdural hematoma in particular is one of the most common TBI lesions and is associated with high morbidity and mortality. To study its pathophysiology, Miller et al.
179
developed a rat model in which 400 µl of blood is injected into the subdural space. This results in a large cortical lesion, comprising 15% of hemispheric volume, within 4 h. The lesion is ischemic in nature, as indicated by H&E histopathology and measurement of blood flow, and is associated with cytotoxic edema and a large glutamate surge.180,181 Considering the similarities to focal ischemia, including neuroprotection by hypothermia and NMDA receptor antagonists,182,183 electrophysiology was performed to investigate the SD continuum (Figure 7). Preliminary results suggest that the infusion of subdural blood acutely induces terminal depolarization in cortex that evolves to infarction. In the infarct periphery, the initial SD is prolonged, lasting up to 15 min, and spontaneous SDs of shorter duration are subsequently observed throughout 4 h of monitoring (JMH and JAH, unpublished). SDs are also observed in a swine model of intracerebral hemorrhage,
184
further suggesting that hemorrhagic lesions might explain the high incidence of SD in patients with severe TBI13,185 as compared with contusional rodent models. In general, the mixed pathology and heterogeneous nature of clinical TBI present serious challenges for animal modeling, especially considering that the degree and volume of such injuries that are survivable in humans are orders of magnitude greater than those in rodents.
The continuum of spreading depolarizations in a rat model of acute subdural hematoma. Electrophysiologic recordings of spontaneous electrocorticographic activity (top traces; 0.5–50 Hz) and DC potential (bottom traces) from two micropipette electrodes in cerebral cortex. Slow infusion of 0.4 ml of arterial blood into the subdural space (large arrow, left) causes an immediate depolarization that begins at electrode 2 and spreads to electrode 1 at 4 mm/min. At electrode 2, the depolarization is persistent through 4 h of monitoring and TTC-staining confirms tissue infarction at this location. The initial depolarization at electrode 1 is prolonged with recovery after 15 min, and spontaneous short-lasting depolarizations are subsequently observed. When the animal is killed by asphyxiation after 4 h (large arrow, right), terminal depolarization is observed in viable cortex (electrode 1) but not in the infarct core. Scale bars are 1 and 20 mV for top and bottom traces, respectively, and apply to both electrodes.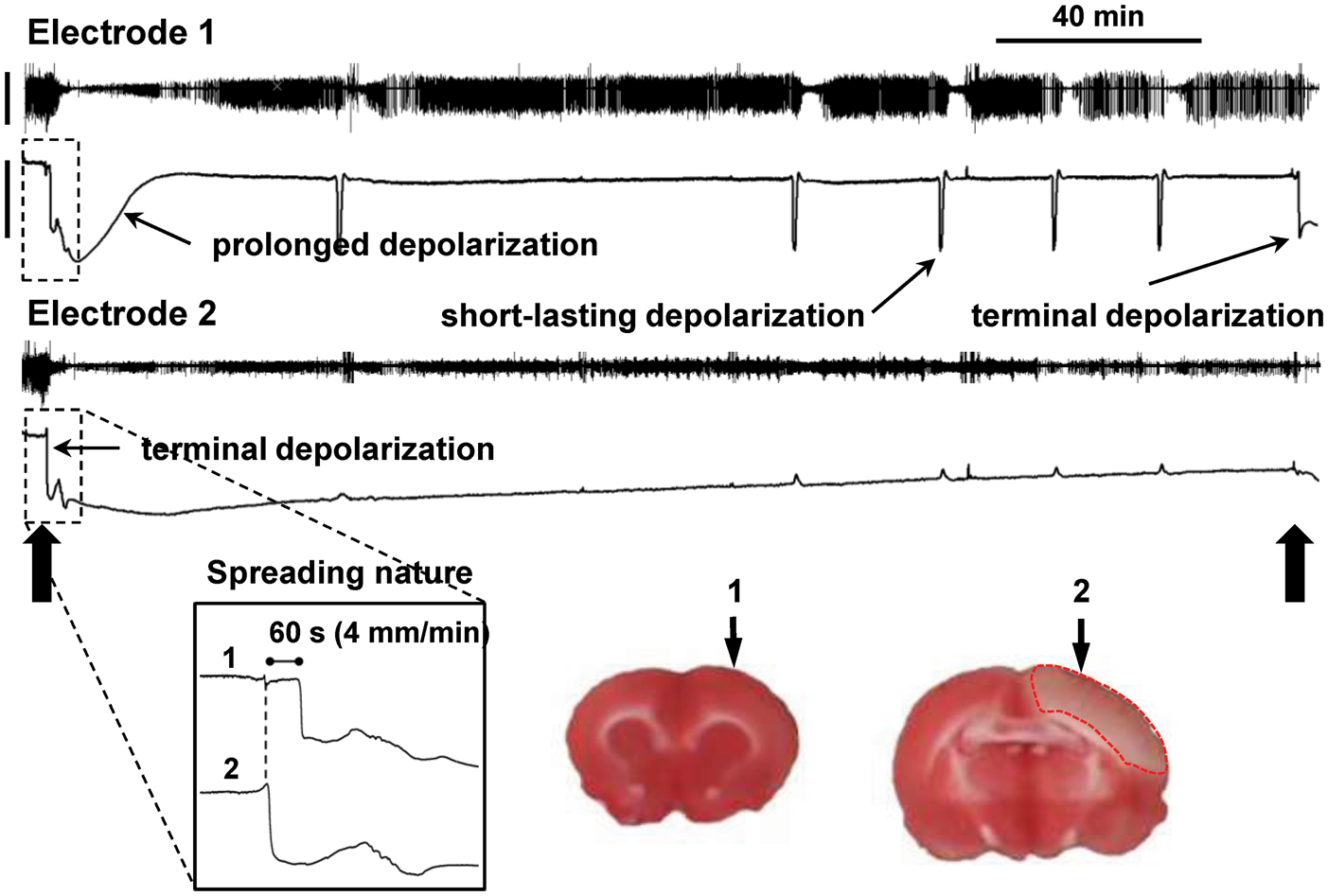
Pre-conditioning: Is spreading depolarization protective?
A substantial literature has accumulated showing that pre-conditioning the cortex with SDs has a neuroprotective effect against subsequent ischemia.186–190 In such experiments, SD is typically provoked with KCl application to the cortex for 2–3 h at an interval of 1–7 days prior to induction of global or focal ischemia. Compared to control hemispheres or animals, pre-conditioning results in significant reduction of the final cell death count or infarct size. The mechanism of neuroprotection likely involves upregulation of cytokines and growth factors,190–192 down-regulation of metabolism, 193 and altered neurotransmission. 194
The protective effects of SD pre-conditioning offer potential insight into mechanisms of ischemic tolerance, but have also been advanced as a reason for skepticism concerning the adverse effects of SDs that occur after brain injury. We note, however, that the pre-conditioning and post-injury concepts are entirely congruent. First, it is a general principle of physiology that sub-lethal challenges can build resistance and protection against subsequent threats. Thus, neuroprotective pre-conditioning is a phenomenon also observed with ischemia, hypoxia, and glutamate. Second, pre-conditioning is not applicable to an acutely developing brain injury since SDs are initiated by the injury and not before. An exception occurs in patients who are at risk for a “second hit” within the time window (days) for pre-conditioning, as in the case of DCI after SAH. However, clinical data demonstrate the deleterious nature of both early and delayed SDs even in this condition.63,195,196 Third, the effects of SD can be spatially disparate depending on local conditions of cortex, as explained above. A wave could confer a net benefit in normally perfused regions remote from an injury focus, but the same wave may trigger vasoconstriction and persistent depolarization in the injury penumbra. Aside from pre-conditioning, remote beneficial effects of SD may include induction of neurogenesis or facilitation of plasticity.197–199 Whether such effects influence functional recovery or counterbalance the harm of SD deserves further study.
Conclusions
In view of evidence from these diverse animal models, it appears SD is a necessary and ubiquitous mechanism for the development of pannecrotic and selective neuronal lesions of the cerebral cortex. In every model studied to date, including global ischemia, focal ischemia, products of hemolysis, endothelin, contusion, and hematoma, tissue fate is determined by the occurrence of prolonged or persistent depolarization. The role of depolarizations is causal, as shown by real-time monitoring/imaging of multiple physiologic variables during spontaneous SDs, and also by experimental induction of exogenous SDs. Furthermore, observations from these studies unite the historically separated phenomena of spreading depression and anoxic depolarization into a singular class, as presaged by Leão and described as the SD continuum. The continuum is most notably evidenced by the continuity of singular waves that transform from persistent to progressively shorter SDs as they propagate peripherally from an initial ischemic event, and the same full continuum of singular waves that arise spontaneously as a secondary injury process.
Importantly, the SD continuum is not a semantic formulation, but rather is a pathophysiologic concept that is essential for understanding how lesions develop in cerebral cortex and some other gray matter structures. After ischemic onset, the occurrence of persistent depolarization is the causal switch between mild ionic changes and the near-complete breakdown of transmembrane gradients, coupled with the structural damage of cytotoxic edema. The study of persistent depolarization in the ischemic core further informs the mechanism of secondary ischemic lesion growth: spontaneous SDs propagate to the innermost penumbra where they expand the core region of persistent depolarization that is fated for infarction. Finally, when it is considered that the commitment point for cell death is reached after a finite duration, the lessons of persistent/terminal depolarization also apply to transient depolarizations occurring in high numbers or with prolonged durations. As illustrated in the hemolysis and endothelin models, such repetitive SDs are a necessary and determining factor for neuronal lesion development. In both the hemolysis model and in secondary ischemic lesion growth, SDs cause not only the ionic and structural changes, but also the ischemic conditions (through spreading ischemia) necessary for their lasting detrimental effects.
Results of ongoing translational research suggest that these pathophysiologic concepts are also essential to understanding human disease. Continuous electrocorticographic (ECoG) monitoring of more than 500 patients by the COSBID group has shown that SDs occur in a very high proportion (55–90%) of patients with acute brain injury, and that they often occur in high numbers for days to weeks after injury.13,14,15,195,200 As in animals, SDs in patients are associated with excitotoxic injury,201–203 metabolic crisis,202,204–207 decreased blood flow,63,208,209 new lesion development,63,195 and poor outcomes.13,16 The framework of the SD continuum is necessary to understand these events, since they often have characteristics that defy categorization in the historic dichotomy of “spreading depression” and “anoxic depolarization.” 10 Furthermore, the continuum provides a working hypothesis and informs interpretation of clinical neuromonitoring data. For instance, the evidence from experimental stroke that each SD has detrimental effects when it reaches the most metabolically compromised tissue, inducing decreased perfusion and terminal depolarization, suggests that detection of any SD may be interpreted by the neurointensivist as a warning sign that secondary injury is occurring somewhere in the brain, even if the SD has a short duration with rapid recovery of spontaneous activity at the recording site. This is analogous to detection of a tsunami by an ocean buoy, providing warning of expected effects at distant shores. Experimental studies further demonstrate that SDs are an essential link in the causal chain between triggering insult and neuronal cell death. Thus, it is a fallacy to conclude that SDs in patients are an epiphenomenon based on their upstream triggers, whether systemic insults such as hypoxia, hypotension, hypoglycemia, or pyrexia,65,127,210,211 or more subtle changes in tissue microenvironment.212,213 Rather, identifying causes of secondary SDs may lead to refinements in neurointensive care—whether medications, improved perfusion and temperature management, or minimizing patient stimulation—that minimize the probability of SDs and therefore limit damage and improve outcomes.
Footnotes
Funding
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: This work was supported by the Mayfield Education and Research Foundation (JAH), Deutsche Forschungsgemeinschaft (DFG DR 323/6-1 to JPD; DFG DR 323/5-1 to JPD, JW, OS, RG), the Bundesministerium für Bildung und Forschung (Center for Stroke Research Berlin, 01 EO 0801; BCCN 01GQ1001C B2 to JPD), Era-Net Neuron 01EW1212 (JPD), the Hungarian Scientific Research Fund (Grant No. K111923 to EF), the Bolyai János Research Scholarship of the Hungarian Academy of Sciences (BO/00327/14/5 to EF), U.S. Department of Defense (CDMRP PR130373 to KCB), the National Institutes of Health (NS085413 to KCB; NS083858 to SAK; NS055104, NS061505 to CA), the Wellcome Trust (HICF 0510-080 to MGB), and the Fondation Leducq, the Heitman Foundation, and the Ellison Foundation (CA).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
JAH wrote the initial draft, conceived and prepared the figures and tables, and edited and approved the manuscript. CWS, SAK, CA, JMH, and BF contributed to the initial draft, prepared figures, and edited and approved the manuscript. RDA, MGB, KCB, APC, MAD, CD, CD, MF, EF, DF, RG, RH, ML, SM, AIO-F, FR, ESR, OWS, RS-P, ES, MS, AJS, AU, MBW, MKLW, OWW, and JW edited and approved the manuscript. JPD contributed to conceptual design and initial draft of the article and edited and approved the manuscript.