
Abstract
Three different Na+/Ca2+ exchanger (NCX) isoforms, NCX1, NCX2, and NCX3, are expressed in brain where they play a relevant role in maintaining Na+ and Ca2+ homeostasis. Although the neuroprotective roles of NCX2 and NCX3 in stroke have been elucidated, the relevance of NCX1 is still unknown because of embryonic lethality of its knocking-out, heart dysfunctions when it is overexpressed, and the lack of selectivity in currently available drugs. To overcome these limitations we generated two conditional genetically modified mice that upon tamoxifen administration showed a selective decrease or increase of NCX1 in cortical and hippocampal neurons. Interestingly, in cortex and hippocampus NCX1 overexpression increased, where NCX1 knock-out reduced, both exchanger activity and Akt1 phosphorylation, a neuronal survival signaling. More important, mice overexpressing NCX1 showed a reduced ischemic volume and an amelioration of focal and general deficits when subjected to transient middle cerebral artery occlusion. Conversely, NCX1-knock-out mice displayed a worsening of brain damage, focal and neurological deficits with a decrease in Akt phosphorylation. These results support the idea that NCX1 overexpression/activation may represent a feasible therapeutic opportunity in stroke intervention.
Introduction
The brain is one of the few organs that express all three Na+/Ca2+ exchanger (NCX) proteins, NCX1, 1 NCX2, 2 and NCX3. 3 These exchangers are all present in neurons, astrocytes, oligodendrocytes, and microglia where they play a relevant role in maintaining Na+ and Ca2+ homeostasis under different neurophysiological4,5 and neuropathological conditions.6–11 Thanks to the generation of knock-out mice, the physiological and pathophysiological roles of the NCX2 and NCX3 isoforms in the brain have been elucidated.4,5,12 In contrast, studies investigating the role of NCX1 in stroke have been hampered by difficulties met in developing a global NCX1 knock-out mice due to its embryonic lethality. 13 Furthermore, a lack of highly selective compounds modulating NCX1 activity14–18 has generated an array of conflicting results on the role of this antiporter in stroke.6,19–21
We hypothesized that NCX1 might play a relevant role in neuroprotection during brain ischemia since: (1) an increase in NCX1 expression/activity raises Akt phosphorylation, 22 a well-known signaling of neuronal survival23–25 that increases neuronal resistance to brain ischemia; and (2) under anoxic conditions, NCX1 could work in the reverse mode, promoting Ca2+-influx and favoring Ca2+-refilling into the endoplasmic reticulum (ER), which is depleted under these conditions, thus allowing neurons to delay apoptosis. 21
To test this hypothesis and to overcome the genetic and pharmacological limitations, we used the well-known Cre/loxP system to obtain two genetically modified mice, that, upon tamoxifen administration show an increase or decrease in NCX1 expression in the hippocampus and cortex, two brain regions involved in stroke. In particular, after exposing these two new genetically modified mice to transient middle cerebral artery occlusion (tMCAO), we explored the effect of the overexpression or downregulation of NCX1 on Akt1 phosphorylation in neurons of adult mice and its putative neuroprotective role in stroke by measuring infarct volume and consequent neurological deficits.
Materials and methods
Generation of DNA constructs
The targeting vector for NCX1.4 overexpressing mice (Figure 1(a)) was obtained by subcloning three DNA cassettes in a ROSA26 plasmid ready for homolog recombination. The first DNA cassette was a promoterless neomycin resistance (neoR) gene flanked by two flippase recognition target (FRT) sequences; the second DNA cassette was a transcriptional stop sequence made of three bovine growth hormone polyadenylation signals flanked by two flox sequences; the third DNA region was the cDNA of the neuronal murine NCX1.4 splicing isoform. This isoform was obtained by reverse transcription of the mRNA extracted from the brain of C57BL/6 wild-type mice (Charles River) and amplified by high-fidelity Pfu (Roche). All constructs were verified by sequencing both DNA strands (Primm, Milan, Italy).
Gene targeting of NCX1.4 construct in ROSA26 locus and of exon 11 of the endogenous ncx1 gene. (a) Cartoon representing the structure of wild-type (wt) and targeted (tg) alleles for ncx1.4over mice in ROSA26 locus located on the E3 region of chromosome 6. The Xba I restriction site was the insertion site for flox-conditioned ncx1.4 DNA construct. Neomycin resistance cassette (neoR) (shaded box), flox sequences (black filled triangles), the probe used in Southern blot analysis (black bars), as well as the DNA fragments generated after digestion with EcoR I are represented. E, EcoR I; V, EcoR V. (b) Southern blot analysis of EcoR I digested genomic DNA isolated from ESCs clones with depicted DNA probe. Non-recombinant ESCs show a single band at 15 kb, whereas the ESCs heterozygous for NCX1.4 construct integration show a band at 15 kb for wild-type allele and a band at 6 kb for the targeted allele. (c) Cartoon representing the structure of exon 11 in wild-type (wt) and targeted (tg) ncx1 gene for ncx1neuko mouse strain on the E3 region of chromosome 17. Exon 11 (shaded box) with its neighboring genomic region (grey line), flox sequences (grey filled triangles), as well as the map of restriction sites are represented. H, Hind III; V, EcoR V; Ba, BamH I; Bg, Bgl II. (d) Topological model of NCX1 resulting after the excision of the amino acid region 722-813. The 10 cylinders represent transmembrane segments; the light grey represents the region that is not translated into protein after the recombination of exon 11 by CreERt recombinase. (e) Screening by PCR of ESCs by Flp enzyme for the recombination of neoR cassette. (f) Screening by PCR of mice carrying the NCX1.4 construct. (g) Screening by PCR of mice with a homozygous floxed exon 11.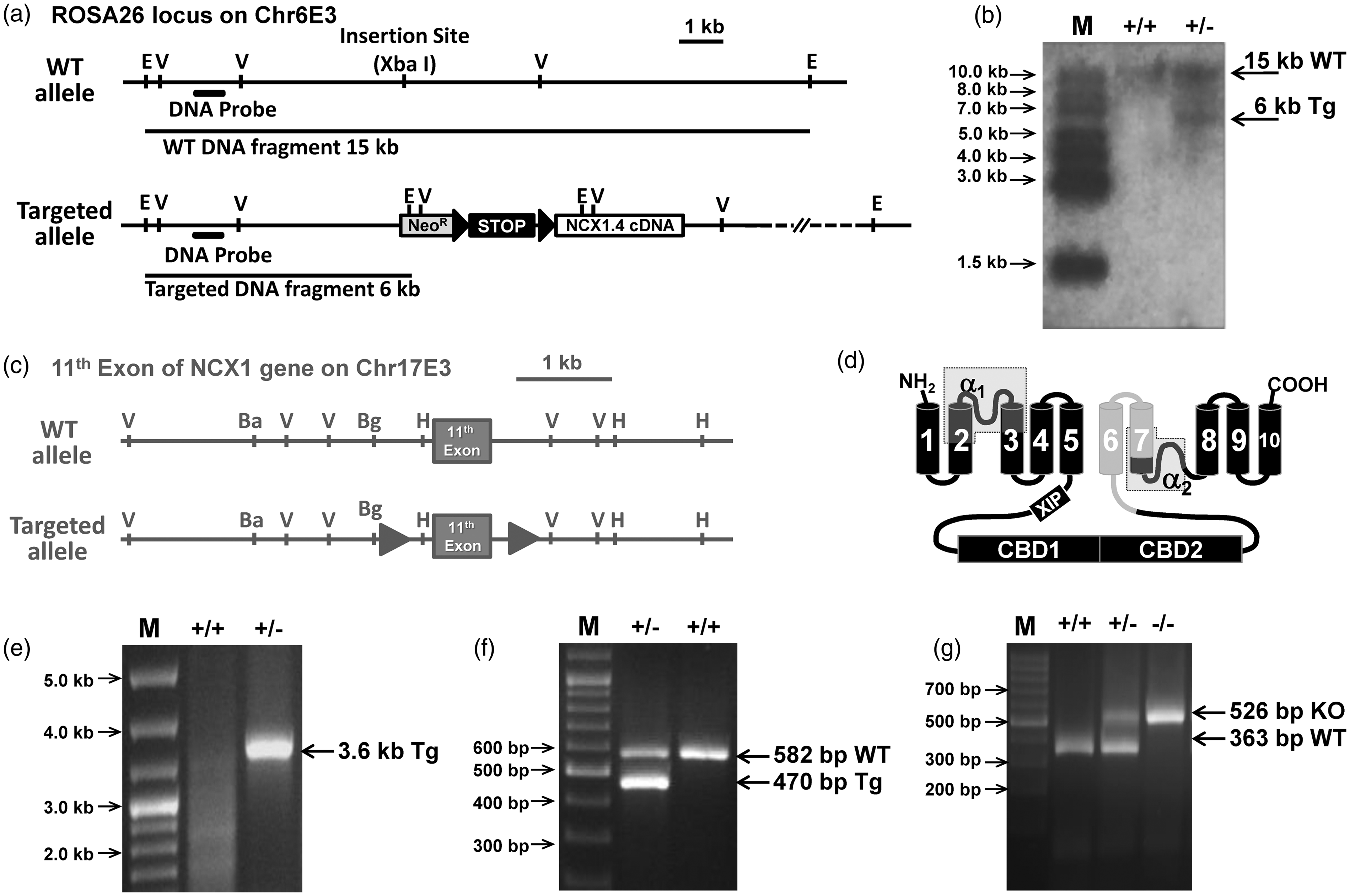
Genetically modified animals
Both genetically modified mouse lines, viz, neuronal specific NCX1.4 overexpressing mice (ncx1.4over) and NCX1 knock-out (ncx1neuko) mice, were generated by our research group. In particular, the ncx1.4over mouse line was obtained by mating transgenic mice carrying CreERt cDNA driven by a calmodulin kinase II alpha (CamKIIα) promoter 26 with our new genetically modified mouse strain carrying a flox-conditioned NCX1.4 construct in the ROSA26 locus on chromosome 6. This new mouse line is homozygous for the presence of the NCX1.4 construct gene and might have (ncx1.4over) or might not have (ncx1+/+) an additional CreERt gene. The other genetic modified mouse line, ncx1neuko, was obtained by mating transgenic mice carrying CreERt cDNA driven by a CamKIIα promoter 26 with NCX1 floxed mice. 27 This new mouse line is homozygous for the presence of the NCX1 floxed allele and might have (ncx1neuko) or might not have (ncx1+/+) an additional gene coding for the Cre recombinase fused with a mutated ligand-binding domain for the human estrogen receptor (CreERt). No evident abnormalities and no evident changes in cerebral vessels were observed in the brain of both ncx1.4over and ncx1neuko mice (n = 4) compared with the respective wild-type littermates (Supplementary Figure 1).
In this study, 165 male genetically modified mice, aged between 6 and 8 weeks and weighing 20–30 g, were used for experimental procedures. All the above mentioned animals were housed under diurnal lighting conditions. In particular, 11 animals for each experimental group, randomly selected from the four mouse genotypes, were considered to be sufficient to demonstrate the neuroprotective or neurodegenerative effect of NCX1 overexpression or downregulation. The sample size was chosen considering an expected average of 15% of mouse death during surgical procedure and of 15% excluded for cerebral blood flow reduction criteria. Six animals, randomly selected for each genotype, were considered to be sufficient per each experimental group to demonstrate any variation in CreERt-mediated DNA recombination, up- or down-regulation of analyzed proteins and mRNAs. Moreover, an up-regulation or down-regulation below 10% was not considered biologically relevant.
All protocols and procedures of the handling of animals were according to the international guiding principles for biomedical research proposed by the Council for International Organizations of Medical Sciences 28 and ARRIVE (Animal Research: Reporting In Vivo Experiments) guideline. 29 Experiments were approved by the Animal Care Committee of “Federico II” University of Naples, Italy.
Induction of CreERt recombinase with tamoxifen
Tamoxifen powder (Sigma Aldrich, Italy), stored at 4 ℃, was completely dissolved in a sunflower oil/ethanol (10:1) mixture at 10 mg/mL few hours before intraperitoneal (IP) injection. The solution was then stored at 4 ℃ in a light-proof closed tube and pre-warmed at 37 ℃ before IP injection. All experimental groups, i.e. ncx1.4over, ncx1neuko, and their respective ncx1+/+ congenic controls, received 50 mg/kg of tamoxifen every 12 h for 3, 5, or 10 consecutive days; the mice were used after at least three days of drug wash-out. The five-day treatment did not affect NCX1 mRNA or protein levels in congenic wild-type mice compared to vehicle-treated mice (Supplementary Figure 2).
Reverse transcription and real-time polymerase chain reaction
For the evaluation of gene expression, 4.0 µg of each RNA extract from the non-ischemic temporoparietal cortex, hippocampus, and striatum was digested with DNase and reverse-transcribed by SuperScript III (Life Technologies), according to the manufacture’s protocol. Data were normalized on the basis of the hypoxanthine-guanine phosphoribosyltransferase (HGPRT) signal. Melting curves of amplicons and gel electrophoresis were used to exclude the presence of nonspecific polymerase chain reaction (PCR) products.
The following sequences of primers were used for real-time PCR (7500fast, Applied Biosystems): NCX1-forward, 5-CCGTGACTGCCGTTGTGTT-3; NCX1-reverse, 5-GCCTATAGACGCATCTGCATACTG-3; NCX2-forward, 5-AGTGGATGATGAAGAGTATGAGAAGAAG-3; NCX2-reverse, 5-TTGGTTGAGTAGCAGAGCTGAGA-3; NCX3-forward, 5-CCTCTGTGCCAGATACATTTGC-3; NCX3-reverse, 5-CCAAACCAATACCCAGGAAGAC-3; HGPRT-forward, 5-TCCATTCCTATGACTGTAGATTTTATCAG-3; HGPRT-reverse, 5-AACTTTTATGTCCCCCGTTGACT-3.
Recombination measurement
For the evaluation of genetic recombination in ncx1.4over and ncx1neuko mice, genomic DNA was extracted from the temporoparietal cortex, hippocampus, and striatum of non-ischemic mouse. Recombination measurement was performed by real-time PCR (7500 fast, Applied Biosystems) on 100 ng of genomic DNA using standard curves on specific plasmids for each amplification and was expressed as the percentage of the absolute amount of recombinant floxed DNA versus the absolute amount of the genetic modified allele. In particular, in ncx1.4over mice, the recombination percentage was expressed as the absolute amount of recombinant stop cassette versus the absolute amount of the heterologous NCX1.4 cDNA on the genome. In ncx1neuko mice, the recombination percentage was expressed as the absolute amount of recombinant exon 11 versus the absolute amount of the exon 2 in the endogenous ncx1 floxed gene. Melting curves of amplicons and gel electrophoresis were used to exclude the presence of nonspecific PCR products.
Protein expression analysis
Whole-cell protein extracts from dissected areas of non-ischemic ncx1.4over and ncx1neuko mice were obtained and processed as previously described in Molinaro et al. 8 Briefly, nitrocellulose membranes were incubated with the anti-NCX1 antibody (rabbit polyclonal; Swant, Bellinzona, Switzerland; 1:1000 dilution), the anti-NCX2 antibody (rabbit polyclonal; Alpha Diagnostics International, San Antonio, TX; 1:1000 dilution), the anti-NCX3 antibody 30 (rabbit polyclonal; 1:2000), the anti-phospho-Akt antibody (monoclonal mouse antibody, Santa Cruz Biotechnology, Inc. CA, sc-81433), the anti-Akt antibody (polyclonal rabbit antibody, Santacruz Biotechnology), the monoclonal anti-β-actin antibody (1:1000; Sigma-Aldrich), and the anti-plasma membrane Ca2+-ATPase (PMCA) antibody (mouse monoclonal; Affinity BioReagents, Golden, CO; 1:1000 dilution). These nitrocellulose membranes were first washed with 0.1% Tween 20 and then incubated with the corresponding secondary antibodies for 1 h (GE Healthcare, Little Chalfont, UK). Immunoreactive bands were detected with the ECL (GE Healthcare). The optical density of the bands (normalized with β-actin) was determined by Chemi-Doc Imaging System (Bio-Rad, Segrate, Italy).
Immunohistochemistry
Immunostaining procedures were performed as previously described in Boscia et al. 31 Briefly, the mice were perfused transcardially with 4% w/vol paraformaldehyde in phosphate buffer. The brains were sectioned coronally at 60 µm on a vibratome. After blockade with Rodent Block M (Biocare Medical, CA, USA) for 40 min, sections were incubated with the following primary antisera: monoclonal anti-NCX1 (1:500; Swant, Bellinzona, Switzerland), monoclonal anti-Cre (1:2000, Abcam, Cambridge, UK). Subsequently, sections were incubated with Mouse-on-Mouse HRP-Polymer kit according to the manufacturer’s instructions (Biocare Medical, CA, USA). The reaction was visualized using 3,3-diaminobenzidine/4-HCl as a chromogen. Sections were mounted on chrome-alum gelatin-coated slides, dehydrated, and coverslipped. The brain sections from all animals examined (n = 3) were stained simultaneously in only one session with the same solutions and at the same incubation times. Images were observed using a Zeiss LSM510 META/laser scanning confocal microscope.
Purified synaptosomal preparation
For [Ca2+]i imaging, synaptosomes were prepared from cortex, striatum, and hippocampus of ncx1.4over, ncx1neuko mice, and their respective congenic wild-type (ncx1+/+) controls. Then, freshly dissociated nerve terminals were purified on discontinuous Percoll gradients, as previously described in Dunkley et al. 32 Briefly, each brain area was homogenized in a medium containing 0.32 M sucrose, 1 mM EDTA, and 0.25 mM DL-dithiothreitol (pH 7.4). The homogenate was centrifuged at 1000 g for 10 min at 4 ℃, and the supernatant was diluted at 14 mL/g with sucrose medium (pH 7.4). Two milliliters of the suspension were placed onto 8 mL Percoll discontinuous gradient containing 0.32 M sucrose and 3%, 10%, 15%, and 23% Percoll (pH 7.4). After centrifugation at 32,000 g for 15 min at 4 ℃, synaptosomes were recovered between the 15% and 23% Percoll bands, diluted five times with HEPES buffer medium containing (in mM): 125 NaCl, 2.5 KCl, 5 NaHCO3, 1.2 NaH2PO4, 1.2 MgSO4, 6 glucose, and 25 HEPES (pH 7.4), and centrifuged at 15,000 g for 15 min at 4 ℃. Finally, the pellet was resuspended in 1 mL of medium B (145 mM NaCl, 3 mM KCl, 1.2 mM MgCl2, 10 mM glucose, and 10 mM HEPES, pH 7.4) and stored on ice. Protein content was determined by the Bradford method. Percoll-purified synaptosomes were resuspended in medium B (1 mg/mL) and loaded with the ratiometric fluorescent Ca2+ indicator Fura-2AM (10 µM)33–35 in the presence of 16 µM bovine serum albumin for 45 min at 37 ℃. Dye-loaded synaptosomes were then washed by centrifugation, resuspended in medium B containing 1.2 mM CaCl2, and attached to poly-D-lysine-coated coverslips for 20 min at 37 ℃. Next, the coverslips were placed into a perfusion chamber (Medical System, Greenvale, NY, USA) mounted on the stage of an inverted Zeiss Axiovert 200 fluorescence microscope (Carl Zeiss) equipped with a 40-X oil objective lens. Experiments were carried out with a digital imaging system composed of MicroMax 512BFT cooled CCD camera (Princeton Instruments, Trenton, NJ, USA), LAMBDA 10-2 filter wheel (Sutter Instruments, Novato, CA, USA), and META-MORPH/METAFLUOR Imaging System software (Universal Imaging, West Chester, PA, USA). Synaptosomes were alternatively illuminated at wavelengths of 340 and 380 nm by a Xenon lamp (Osram, Berlin, Germany). The emitted light was passed through a 512-nm barrier filter. Fura-2AM fluorescence intensity was measured every 3 s. The emitted light was passed through a 512-nm barrier filter. Images were digitized and analyzed using METAFLUOR Imaging software. Assuming that the KD for FURA-2 was 224 nM, the equation of Grynkiewicz was used for calibration. 36 Ratiometric values were automatically converted by the software to [Ca2+]i. NCX activity was evaluated at 25 ℃ as Ca2+ uptake through the reverse mode by switching the normal Krebs medium to Na+-deficient NMDG+ medium named Na+-free (in mM): 5.5 KCl, 147 NMDG, 1.2 MgCl2, 1.5 CaCl2, 10 glucose, and 10 Hepes-Trizma (pH 7.4). Fura-2-detected NCX activity in transgenic mice was expressed as percentage of internal controls.
tMCAO model
Transient focal ischemia was induced as previously described in literature37,38 by transient suture occlusion of the middle cerebral artery for 1 h in male mice anesthetized with 1.5% sevoflurane, 70% N2O, and 28.5% O2. Achievement of ischemia was confirmed by monitoring regional cerebral blood flow (CBF) through laser Doppler flowmetry (PF5001; Perimed). Animals showing a CBF reduction lower than 70% were excluded from the study. All animals were sacrificed by decapitation 24 h after 60 min of tMCAO to quantify the infarct volume. Rectal temperature was maintained at 37 ± 0.5 ℃. Blood gas analysis was carried out in all animals, and no differences in pH, pCO2, and PO2 were detected in all the experimental groups.
Nine animals were excluded because they died during surgery: one in ncx1.4over and two in the congenic control ncx1+/+ experimental groups; four in ncx1neuko and two in the congenic control ncx1+/+ experimental groups. Five additional animals were also excluded because of low reduction in the CBF (<70%): 0 in ncx1.4over and one in the congenic control ncx1+/+ experimental groups; two in ncx1neuko and two in the congenic control ncx1+/+ experimental groups.
Evaluation of the infarct volume and neurological deficit scores
Mice were sacrificed by decapitation 24 h after tMCAO. Brains were quickly removed, sectioned coronally at 0.5 mm intervals, and stained by immersion in the vital dye (2%) 2,3,5-triphenyltetrazolium hydrochloride. The infarct volume was calculated by summing the infarction areas of all sections and by multiplying the total by slice thickness. To rule out expansion of infarct volume due to edema, the infarct volume was calculated by dividing the infarct volume by the total ipsilateral hemispheric volume and expressed as percentage of the infarct.39,40
Neurological scores were evaluated 24 h after reperfusion according to two scales: a general neurological scale and a focal neurological scale. In the general score, six general deficits were measured: (1) hair conditions, (2) position of ears, (3) eye conditions, (4) posture, (5) spontaneous activity, and (6) epileptic behavior. For each of the six general deficits measured, animals received a score ranging between 0 and 12 depending on the severity of the signs. The scores were then summed to provide a total general score. In the focal score, seven areas were assessed: (1) body symmetry, (2) gait, (3) climbing, (4) circling behavior, (5) front limb symmetry, (6) compulsory circling, and (7) whisker response. For each of these items, animals were rated between 0 and 4 depending on the severity of the signs. The scores were then summed to give a total focal score. All surgical procedures have been performed in a blinded manner by individuals who were not aware of the mouse genotype.
Statistical analysis
Values are expressed as means ± SEM, and statistical analysis was performed with unpaired t-test or two-way analysis of variance followed by Newman–Keuls test. Neurological deficits data were analyzed using the nonparametric Kruskal–Wallis test. Statistical significance was accepted at the 95% confidence level (p < 0.05).
Results
Generation of ncx1.4over knock-in mice
An engineered gene coding for the most abundant neuronal NCX1.4 splice variant was constructed as described in the Materials and Methods section and in Figure 1(a). Briefly, to increase the specificity of the cell selection, the targeting vector was provided with a promoter-free neomycin resistance (neoR) gene flanked by two FRT sequences. This was done to prevent the transfection of the targeting vector in embryonic stem cells (ESCs) from giving neomycin resistance by itself. However, after electroporation of the plasmid in ESCs, the homologous recombination of this DNA construct with the ROSA26 locus on chromosome 6 places the promoter-less neoR gene downstream to the ROSA26 promoter. This, in turn, leads to the expression of the neomycin resistance marker of selection only in the ESCs that have integrated the targeting vector by homologous recombination, thus increasing the efficiency of ESCs selection and identification. Positive mutant ESCs clones were confirmed by Southern blot (Figure 1(b)). Afterwards, mutant ESC clones were electroporated with a plasmid expressing the recombinase protein flippase, which recognizes and excises DNA regions flanked by FRT sequences. In this step, recombinant ESCs which had lost the neoR cassette from the genome were identified for the lack of neomycin resistance by PCR (Figure 1(e) and (f)). Selected ESC clones that had integrated the NCX1.4 cDNA allele were used to produce chimeric mice that, in turn, were bred with C57BL/6 females to obtain a non-chimeric knock-in mouse strain. Subsequently, this new knock-in mouse strain was mated with CreERt transgenic mice 26 to obtain a new mouse strain containing two genotypes: mice homozygous for the ncx1.4 construct allele with CreERt gene (ncx1.4over mice) and mice homozygous for the ncx1.4 construct allele without CreERt gene, which were used as the congenic wild-type control group (ncx1+/+).
Both ncx1.4over and ncx1+/+ mice were identified by PCR genotyping, subjected to multiple IP injections of tamoxifen, and used after at least three days of drug wash-out.
Generation of ncx1neuko knock-out mice
We used a genetically modified mouse strain carrying a loxP sequence into each of the two introns flanking exon 11 of the NCX1 gene 27 (Figure 1(c) and (d)). This mouse strain was mated with mice expressing CreERt recombinase driven by the CamKIIα promoter 26 to obtain a new mouse strain containing two genotypes: mice homozygous for NCX1 floxed allele with CreERt gene (ncx1neuko mice) and mice homozygous for NCX1 floxed allele without CreERt gene (ncx1+/+). The latter was used as the congenic control group.
Both ncx1neuko and ncx1+/+ mice were identified by PCR genotypization (Figure 1(g)), subjected to multiple IP injection of tamoxifen, and used at least after three days of drug wash-out.
ncx1.4over and ncx1neuko mice display a tamoxifen- and CreERt-mediated recombination in hippocampus and cortex
After 3, 5 and, 10 days of IP injections of tamoxifen, a recombination of floxed DNA regions was induced in cortical and hippocampal neurons of both ncx1.4over and ncx1neuko mice. In particular, three days of tamoxifen treatment induced a recombination of almost 25% of the stop cassette region in the cortical and hippocampal neurons of ncx1.4over mice. This recombination rose up to 50% in ncx1.4over mice treated with 5 or 10 days of tamoxifen (Figure 2(a) to (c)). In contrast, striatal neurons of ncx1.4over mice treated with tamoxifen showed no detectable recombination of STOP cassette by real-time PCR (Figure 2(b)). Similarly, three days of tamoxifen treatment induced a significant recombination of floxed exon 11 in the cortex and hippocampus of ncx1neuko mice, as measured by real-time PCR (Figure 2(d) to (f)). Moreover, five days of tamoxifen treatment in ncx1neuko mice showed a further increase in the percentage of CreERt-induced recombination in both cortical and hippocampal regions. In contrast, the striatum of ncx1neuko mice treated with tamoxifen did not show any PCR-detectable recombination of the floxed ncx1 gene (Figure 2(e)).
Effect of tamoxifen on genomic recombination in ncx1.4over and ncx1neuko mice. (a) Representation of the localization of the primers used for real-time PCR analysis in ncx1.4over mice. W, amplicon of non-recombined NCX1.4 DNA; C, amplicon of internal control for normalization; R, amplicon of recombined NCX1.4 DNA. (b) Percentage of recombined NCX1.4 locus in ncx1.4over mouse subjected to tamoxifen injection expressed as PCR signal of recombined NCX1.4 “R” normalized for internal control “C”. Values represent means ± SEM (n = 6 of two independent sessions). Statistical analysis was performed with two-way ANOVA, followed by Newman–Keuls test. *p < 0.05 versus vehicle treated control group, **p < 0.05 versus vehicle-treated mice and three days of tamoxifen treatment groups. (c) Representative gel electrophoresis of real-time PCR product obtained from genomic DNA of ncx1.4over mice. No nonspecific band was observed. (d) Representation of primers used for real-time PCR analysis in ncx1neuko mice. W, amplicon of non-recombinant exon 11 of endogenous ncx1 gene; C, amplicon of internal control for normalization on the exon 2 of endogenous ncx1 gene; R, amplicon of the recombinant exon 11. (e) Percentage of the recombinant exon 11 of endogenous ncx1 gene in ncx1neuko mouse subjected to tamoxifen injection expressed as the PCR signal of the recombinant exon 11 “R” normalized for internal control “C”. Values represent means ± SEM (n = 6 of two independent sessions). Statistical analysis was performed with two-way ANOVA, followed by Newman–Keuls test. *p < 0.05 versus vehicle treated control group, **p < 0.05 versus vehicle-treated mice and three days of tamoxifen treatment groups. (f) Representative gel electrophoresis of real-time PCR product obtained from genomic DNA of ncx1neuko mice. No nonspecific band was observed.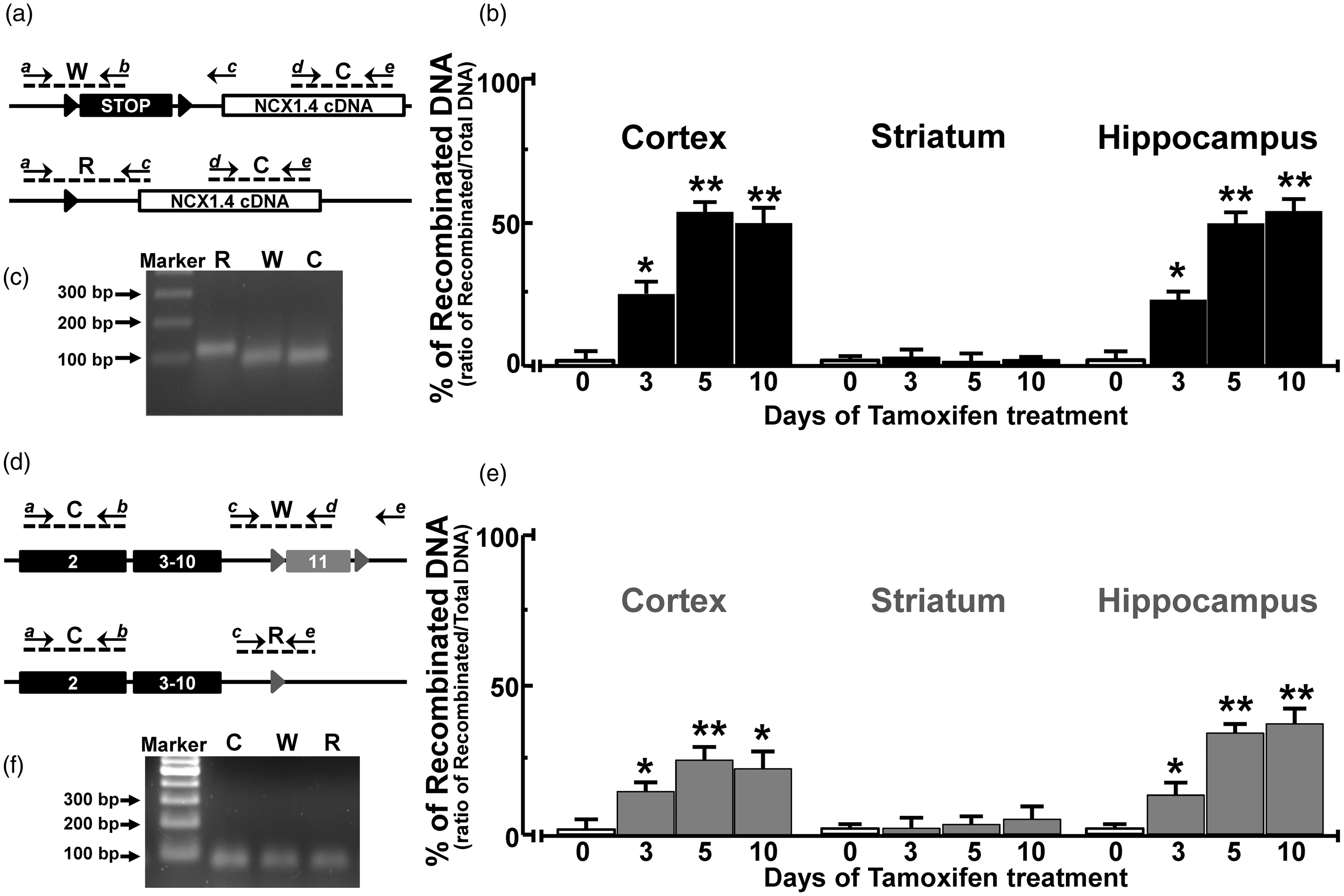
Both ncx1.4over and ncx1neuko mice do not show compensatory changes in the expression of NCX2, NCX3, and PMCA
Three days of tamoxifen treatment significantly increased the expression levels of NCX1 mRNA in the cortical and hippocampal regions of ncx1.4over mice (Figure 3(a)), whereas no PCR-detectable variation of NCX1 mRNA levels occurred in the striatum. A longer five-day treatment increased NCX1 mRNA and protein expression levels in the cortical and hippocampal regions (Figure 3(a) and (b)), whereas no PCR- or Western blot-detectable variation in NCX1 expression levels was observed in the striatum. Moreover, the five-day treatment had no effect on either NCX2 and NCX3 mRNA levels (Figure 5(a)) or NCX2, NCX3, and PMCA protein levels (Figure 5(c)), three plasma membrane partners of NCX1 in the regulation of [Ca2+]i. In accordance, NCX1 immunoreactivity was more intensely depicted within the isocortex and the hippocampal formation of ncx1.4over mice compared to congenic wild-type (ncx1+/+) mice after five days of tamoxifen injections (Figure 3(c)). In particular, in the somatosensory cortex of ncx1.4over mice, the anti-NCX1 antibody clearly revealed a strong NCX1 immunosignal in apical dendrites of neurons located in cortical layers III–V and all over the cortical neuropils (Figure 3(c), a–d). Furthermore, a more pronounced NCX1 signal was observed throughout the hippocampal layers, with the CA1 subregion being the most intensely immunostained (Figure 3C, (e) to (f)).
Expression of NCX1 transcript and protein in control or ncx1.4over mice following tamoxifen injection. (a) and (b) ncx1.4over mice treated for three or five days with tamoxifen show an increased expression levels of NCX1 mRNA (A) and protein (B) in cortex and hippocampus. Data were normalized on the basis of HGPRT for mRNA or of β-actin for protein signal and expressed as percentage of the vehicle control group taken as 100%. Values represent means ± SEM (n = 6 of three independent sessions). Statistical analysis was performed with two-way ANOVA, followed by Newman–Keuls test. *p < 0.05 versus control group, **p < 0.05 versus control and three days of tamoxifen treatment groups. (c) Distribution of NCX1 immunoreactivity in the somatosensory cortex of congenic ncx1+/+ (a, c) and ncx1.4over (b, d) mice treated for five days with tamoxifen. Distribution of NCX1 immunoreactivity in the hippocampal formation of congenic ncx1+/+ (e) and ncx1.4over (f) mice treated for five days with tamoxifen. Quantification of NCX2 and NCX3 mRNA, and NCX2, NCX3, and PMCA protein levels in ncx1.4over and ncx1neuko mice treated with tamoxifen. (a) and (b) quantification of NCX2 and NCX3 mRNA in cortex, striatum, and hippocampus of (A) ncx1.4over, in white and black on the left, and of (B) ncx1neuko, in white and grey on the right, treated with tamoxifen for five days. Data are normalized for HGPRT and expressed as percentage of congenic wild-type (ncx1+/+) treated for five days with tamoxifen taken as 100%. Values represent means ± SEM. (n = 4 of two independent sessions). Statistical analysis was performed with unpaired t-test. At the top, a schematic representation of the cerebral areas used for the evaluation of mRNA levels. (c) and (d) Quantification of NCX2, NCX3, and PMCA protein levels in (c) ncx1.4over mice, in white and black on the left, and in (d) ncx1neuko mice, in white and grey on the right, subjected to five days of tamoxifen treatment. Data are normalized for β-actin and expressed as percentage of congenic wild-type (ncx1+/+) control subjected to five days of tamoxifen treatment. Values represent means ± SEM. (n = 4 of two independent sessions). Statistical analysis was performed with unpaired t-test.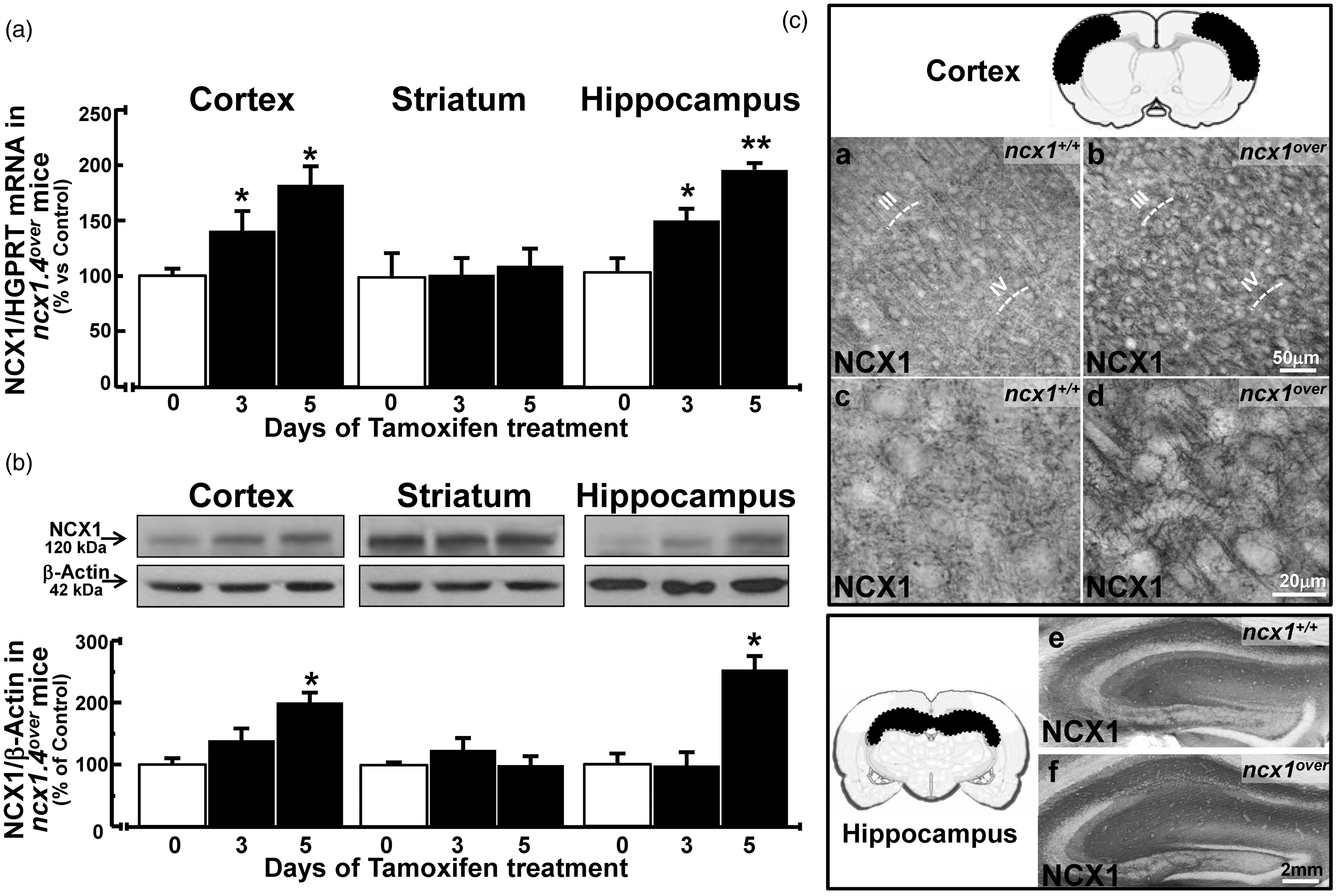
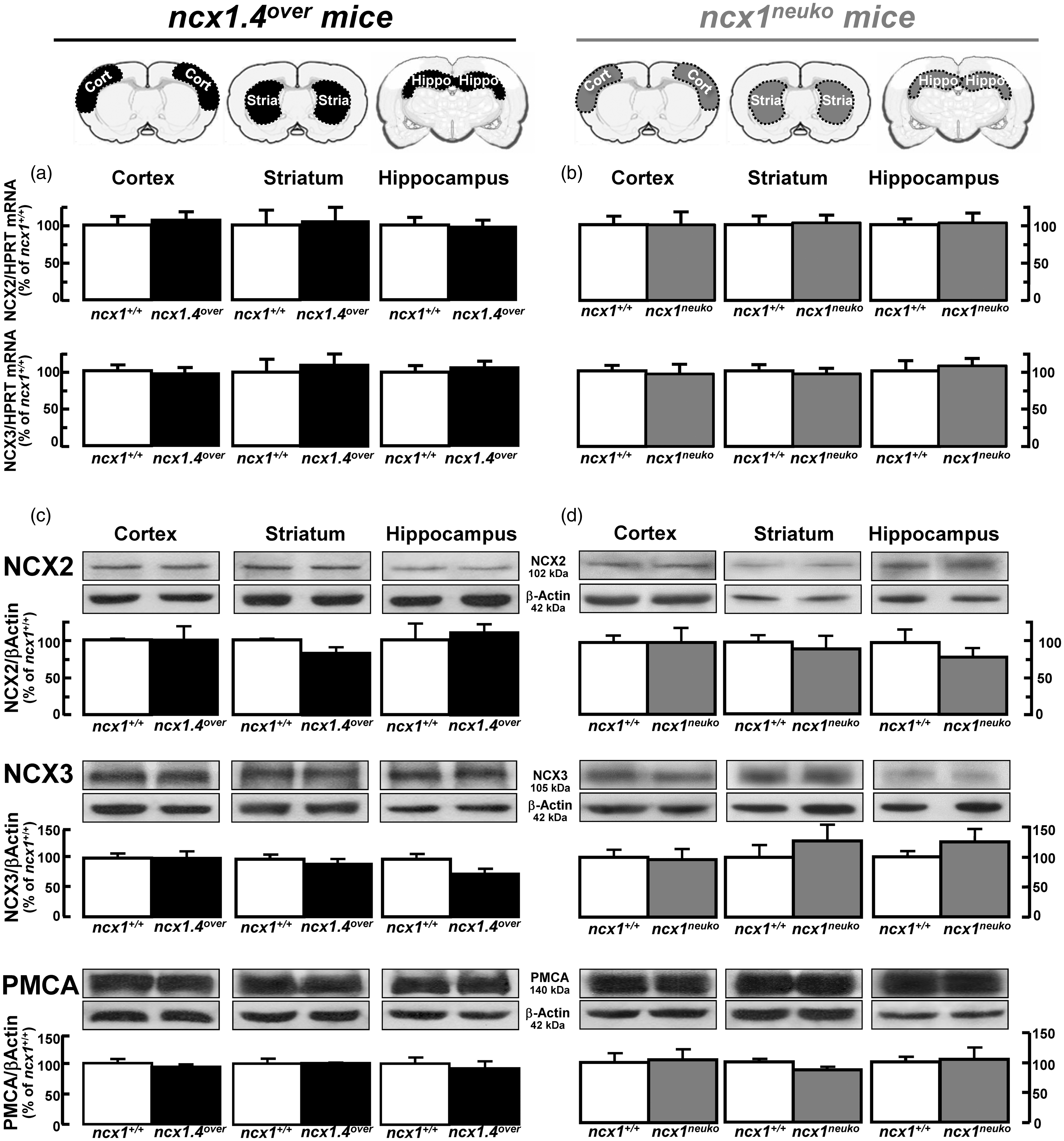
Regarding ncx1neuko mice, five days of tamoxifen treatment decreased the full-length of NCX1 mRNA and protein levels in the cortex and hippocampus (Figure 4(a) and (b)), whereas no PCR- and Western blot-detectable variations of NCX1 expression were observed in the striatum. Moreover, five days of tamoxifen injections affected neither NCX2 nor NCX3 mRNA levels (Figure 5(b)) nor NCX2, NCX3, and PMCA protein levels (Figure 5(d)). Accordingly, NCX1 immunoreactivity was less intense within the isocortex and the hippocampal formation of ncx1neuko mice compared to congenic ncx1+/+ mice after five days of tamoxifen injections (Figure 4(c)). In particular, in the somatosensory cortex of ncx1neuko mice, NCX1 immunostaining was clearly and extensively reduced both in apical dendrites and throughout the cortical neuropils (Figure 4C, (a) to (b)). Similarly, NCX1 signal was drastically reduced throughout the hippocampal layers (Figure 4C, (e) to (f)).
Expression of NCX1 transcripts and protein in ncx1neuko mice following tamoxifen injection. (a) and (b) levels of expression of full-length mRNA and protein of NCX1 in cortex, striatum, and hippocampus of ncx1.4neuko and congenic wild-type (ncx1+/+) mice after a five-day treatment with tamoxifen. Data were normalized on the basis of HGPRT for mRNA or of β-actin for protein signal and expressed as percentage of congenic wild-type (ncx1+/+) group taken as 100%. Values represent means ± SEM (n = 6 of two independent sessions). Statistical analysis was performed with unpaired t-test. *p < 0.05 versus congenic wild-type (ncx1+/+) group. (c) Distribution of NCX1 immunoreactivity in the somatosensory cortex of congenic wild-type (ncx1+/+) (a) and ncx1neuko (b) mice after a five-day treatment with tamoxifen. A schematic representation of the examined cortical area of the brain is shown in (c). Distribution of NCX1 immunoreactivity in the hippocampal formation of congenic wild-type (ncx1+/+) (e) and ncx1neuko (f) mice after a five-day treatment with tamoxifen. Distribution of CreERt immunoreactivity in the somatosensory cortex and hippocampal formation of ncx1neuko (d and h, respectively) mice. A schematic representation of the examined hippocampal area of the brain is shown in (g).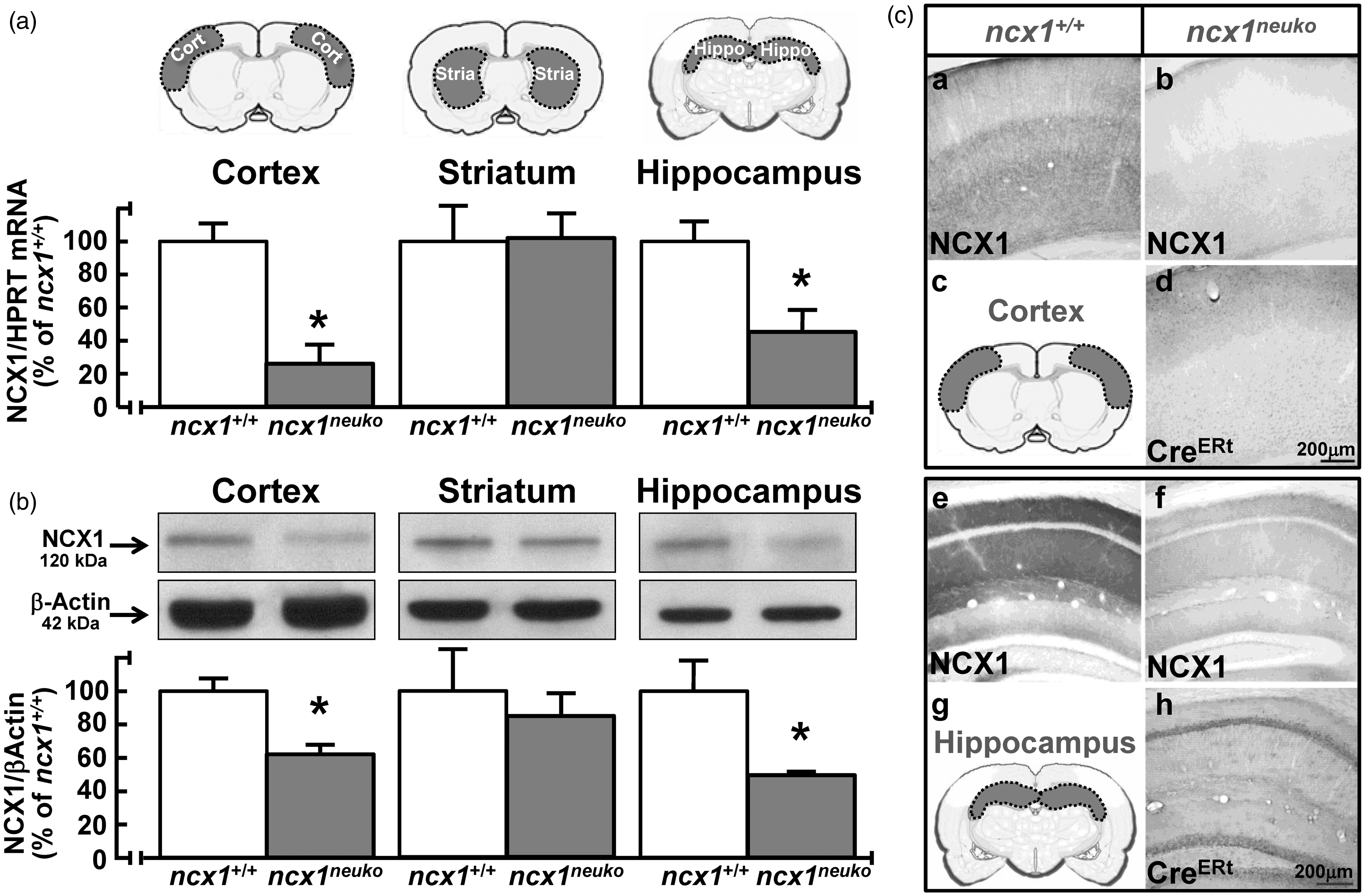
Ncx1.4over mice display an increase in both NCX activity and Akt1 phosphorylation, whereas ncx1neuko mice show a reduction
An increase of the total amount of Na+/Ca2+ exchange activity was observed in the cortex and hippocampus of adult ncx1.4over mice (Figure 6(a)), whereas a decrease in the exchanger activity was observed in the same brain areas of adult ncx1neuko mice (Figure 6(e) and Supplementary Figure 3), as measured by Na+-free induced [Ca2+]i increase in synaptosomes. On the other hand, Na+/Ca2+ exchange activity did not change in synaptosomes from the striatal region neither in ncx1.4over mice nor in ncx1neuko mice if compared with their respective congenic wild-type (ncx1+/+) controls. More relevantly, the phosphorylated form of Akt1 protein (pAkt1) increased in the brain cortex and hippocampus of ncx1.4over mice (Figure 6(b)) where it decreased in the same brain regions of ncx1neuko mice (Figure 6(f)). In contrast, pAkt1 did not change in the striatum of both genetic modified mouse strain, ncx1.4over and ncx1neuko, compared with respective congenic wild-type animals.
Effect of NCX1.4 overexpression and of NCX1 knock-out on NCX activity, Akt1 phosphorylation, ischemic volume, and neurological deficit scores. (a) and (e) quantification of NCX activity measured in the reverse mode of operation in synaptosomes from cortex, striatum, and hippocampus of adult (a) ncx1.4over and (e) ncx1neuko mice treated for five days with tamoxifen as measured by Na+-free induced [Ca2+]i increase. Each column represents means ± SEM of 30–60 synaptosomes prepared from four or five mice. Data are reported as percentage of NCX reverse mode in fresh preparation from respective congenic wild-type (ncx1+/+) mice. *p < 0.05 versus each respective wild-type (ncx1+/+) control. (b) and (f) Representative Western blots and relative quantification of Akt1 phosphorylation in the cortex, striatum, and hippocampus of (b) ncx1.4over and (f) ncx1neuko mice and their respective congenic wild-type (ncx1+/+) controls (n = 3 of two independent sessions). Statistical analysis was performed with unpaired t-test. Values represent means ± SEM. *p < 0.05 versus congenic wild-type (ncx1+/+) group. (c) and (g) Quantification of the infarct volume in (C) ncx1.4over and (G) ncx1neuko mice treated for five days with tamoxifen. The ischemic volume was normalized for the volume of the ipsilateral brain hemisphere and expressed as percentage of congenic control wild-type (ncx1+/+) mice treated for five days with tamoxifen taken as 100% (n = 7 of three independent sessions). Statistical analysis was performed with unpaired t-test. Values represent means ± SEM. *p < 0.05 versus congenic wild-type (ncx1+/+) group. (d) and (h) Quantification of the general (top) and focal neurological (bottom) deficits in (d) ncx1.4over and (h) ncx1neuko mice and their respective congenic wild-type (ncx1+/+) after tMCAO. Data were analyzed using the nonparametric Kruskal–Wallis test. *p < 0.05 versus congenic wild-type (ncx1+/+) group.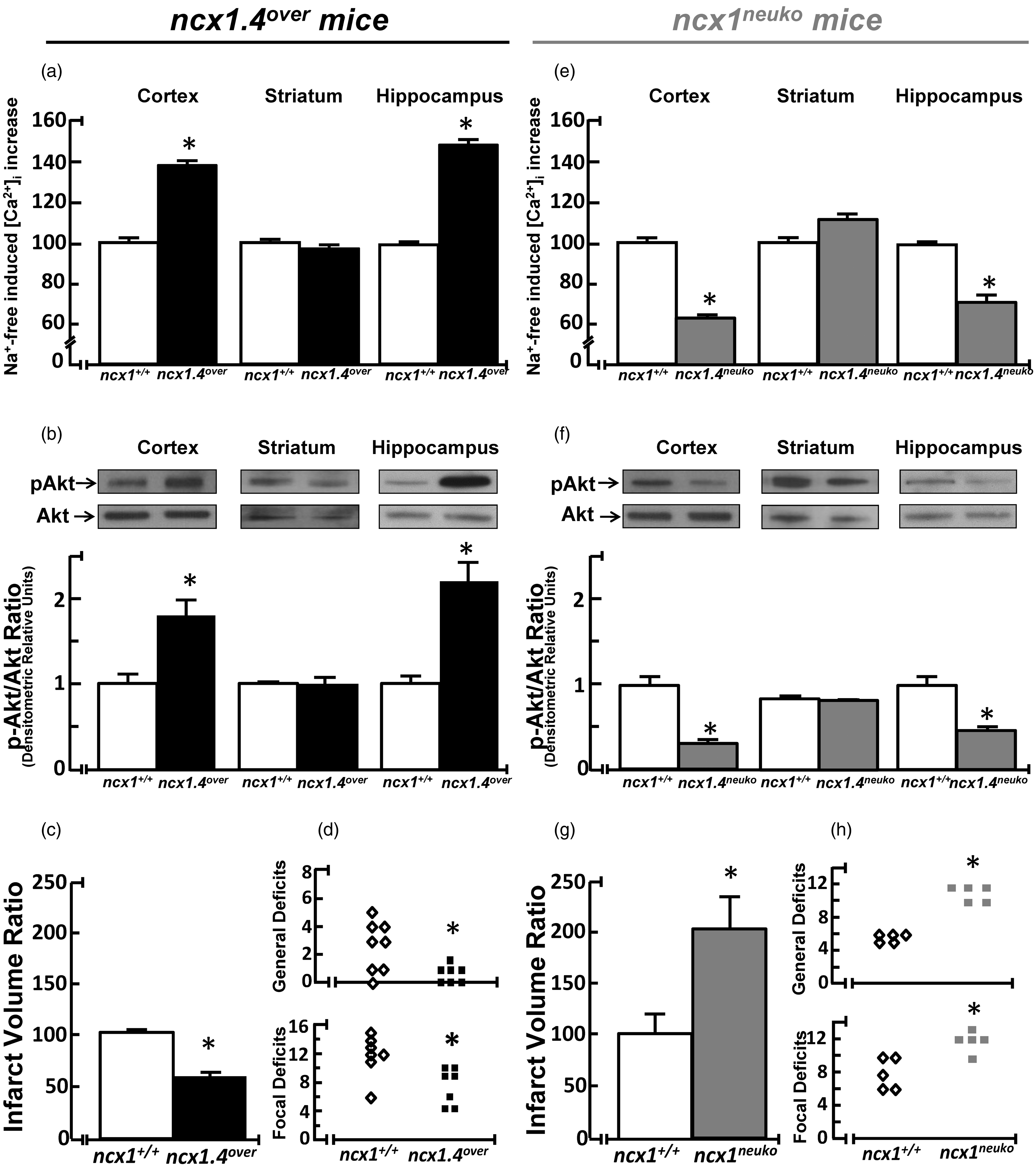
Overexpression of NCX1.4 reduces stroke injury, whereas NCX1 knock-out worsens ischemic damage
NCX1.4 overexpression, induced by CreERt-dependent recombination, significantly reduced the ischemic volume in ncx1.4over mice subjected to tMCAO, compared to congenic wild-type (ncx1+/+) control animals (Figure 6(c)). Consistently, NCX1.4 overexpression also reduced both general and focal neurological deficits in animals subjected to tMCAO (Figure 6(d)).
In contrast, NCX1 knock-out, induced by CreERt-dependent recombination, significantly increased ischemic volume in ncx1neuko mice subjected to tMCAO compared to congenic wild-type (ncx1+/+) control animals (Figure 6(g)). Consistently, general and focal neurological deficits were worsened in ischemic ncx1neuko mice (Figure 6(h)).
Discussion
The results of the present study demonstrate for the first time that the selective and conditional overexpression of NCX1.4 in cortical and hippocampal neurons increases the total amount of the phosphorylated form of Akt1 (pAkt1), thereby exerting a neuroprotective effect in mice subjected to tMCAO. In contrast, the selective and conditional knock-out of NCX1 in the same regions reduced the amount of pAkt1, thus worsening ischemic brain damage. Furthermore, in these two genetically modified mice, ncx1.4over and ncx1neuko, the neuroprotective and neurodetrimental effects on stroke injury were accompanied by an amelioration or a worsening, respectively, of both focal and general neurological deficits.
Notably, at variance with mice knock-out for NCX3 in which there is an adaptive increase in NCX1 and NCX2 mRNA and protein levels, 8 neither NCX1 overexpression nor NCX1 knock-out in the brains of ncx1.4over and ncx1neuko mice, respectively, was accompanied by a compensatory down-regulation or up-regulation of NCX2, NCX3, and PMCA, the other plasma membrane proteins that concertedly work with NCX1 to maintain [Na+]i and [Ca2+]i homeostasis. These results reinforce the selectivity and specificity of the data obtained on the role played by NCX1 in cortical and hippocampal neurons by means of our two genetically modified mice, ncx1.4over and ncx1neuko. Indeed, our results showed a selective and specific DNA recombination in the cortex and hippocampus of both ncx1neuko and ncx1.4over mouse brains, whereas the striatum did not display DNA CreERt-dependent recombination in both mouse strains, even in the experimental groups subjected to 10 days of tamoxifen treatment. The lack of loxP-mediated DNA recombination in this subcortical region was likely due to the insufficient expression levels of the heterologous recombinant CreERt enzyme to elicit DNA recombination between the two target loxP sites present in the genome, as it occurs in the parental CamK-CreERt mouse strain. 26
It should be mentioned that whereas the onset of CreERt-mediated DNA recombination and the changes in NCX1 mRNA levels coincide, variations in NCX1 protein levels require a longer time interval. The delay between CreERt-mediated DNA recombination and NCX1 protein expression may be due to the time needed for NCX1 protein synthesis, when it is up-regulated, and to NCX1 protein catabolism, when its gene is disrupted. Moreover, after five days of tamoxifen treatment, there is a correspondence between the up-regulation of NCX1 mRNA and protein expression in ncx1.4over mice, whereas a different amount of NCX1 mRNA and protein reduction in ncx1neuko mice occurs.
Consistent with NCX1 protein levels, a significant increase or decrease in total Na+/Ca2+ activity was observed in both cortical and hippocampal synaptosomes obtained from adult ncx1.4over or ncx1neuko mice, respectively. The most significant consequence of neuronal NCX1 up-regulation was the increase in the ratio of phosphorylated Akt1 on total Akt1 protein, which, in turn, increased neuronal resistance to stroke damage and improved the general and focal neurological deficits observed in ncx1.4over mice. Indeed, an increased phosphorylation of Akt1 stimulates the PI3K/Akt pathway and leads to a neuroprotective effect against in vitro41,42 and in vivo 43 stroke models. On the other hand, ncx1neuko mice displayed a decreased pAkt1/Akt1 ratio, a condition that exacerbates ischemic neuronal death.43,44 Consistent with these data, ncx1neuko mice showed an increased ischemic volume and a worsening of both general and focal neurological deficits. Interestingly, in the ischemic preconditioning, an endogenous approach causing neuroprotection that increases NCX1 expression/activity, there is also an increase in Akt1 phosphorylation under both in vivo 45 and in vitro 46 conditions.
All these results support the idea that NCX1 exerts a neuroprotective effect in stroke and that this neuroprotective effect might be partially due to an increase in Akt-mediated neuronal survival signaling. Furthermore, this potential neuroprotective role of NCX1 in stroke is corroborated by our previous in vitro data demonstrating that in primary cortical neurons subjected to anoxic conditions NCX1 exerts a neuroprotective effect by promoting Ca2+-influx and favoring Ca2+-refilling into the ER. 21 Indeed, NCX1-mediated prevention of ER-Ca2+ depletion delays neuronal apoptosis. Moreover, we have previously shown that the ncx1 gene participates in two prosurvival pathways42,47 and that two important transcriptional factors involved in stroke such as hypoxia-inducible factor-1, that increases NCX1 expression, 48 and RE1-silencing transcription factor, that represses NCX1 expression, 49 exert a neuroprotective or neurodegenerative effect on stroke damage, respectively.
Another point to emphasize is that ncx1neuko, ncx1.4over, and their respective congenic control mice do not show any difference in macroscopic brain morphology, cerebral vascular distribution, or blood physiological parameters, thus demonstrating that both the neuroprotective and neurodetrimental effects in ischemic mice can be ascribed to the selective overexpression or downregulation of NCX1 rather than to changes in brain morphology.
More relevant, since the amino acid sequence of NCX1 is highly conserved in mammals, 50 our present findings may provide valuable insights into the study of new druggable targets for stroke in human brain.
Altogether, these data suggest that the development of compounds capable of increasing NCX1 activity in brain might exert beneficial effects in stroke intervention. Indeed, the compound developed by our research group that enhances both NCX1 and NCX2 activity, neurounina-1, exerts a neuroprotective effect in stroke with a good potential time-window of therapeutic intervention. 17
Lastly, the generation of these two genetically modified mice, besides having firmly established a role for NCX1 in ischemic stroke, will allow to explore more deeply the role of NCX1 in other physiological and pathophysiological conditions of the central nervous system.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the following grants: Programma Operativo Nazionale (grant numbers PON_01602 and PON03PE_00146_1) from MIUR to L.A.; Futuro in Ricerca (grant number RBFR13M6FN) from MIUR to P.M.; Progetto Giovani Ricercatori (grant number GR-2010-2318138) from Ministero della Salute to A.S.; POR Campania FESR 2007-2013 FARMABIONET (B25C1300023007) to G.P.; POR Campania FESR 2007-2013 OCKEY (B25C13000280007) to G.D.R.; POR Campania FESR 2007-2013 MOVIE (B25C1300024007) to L.A. CaMKIIα mice were generous gift from Dr. Günther Schütz (German Cancer Research Center, Heidelberg, Germany).
Acknowledgements
The authors also wish to thank Dr. Paola Merolla for the editorial revision, and Vincenzo Grillo and Carmine Capitale for their technical assistance.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
PM designed the project and did most of the practical work and wrote the first draft of the paper together with the last author. RS, GP, TP, AS, FB, AV, OC did some experiments and helped in interpreting the data. MDF and RDL helped in the design and generation of the two genetically modified mice. KDP and GDR helped in the critical overview of the paper. LA supervised the project and wrote the paper.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.