
Abstract
The purpose of this study was to investigate the protective mechanism of leptin-mediated metabolic recovery against cerebral injury after ischemia and reperfusion. We determined the neurologic deficit score, extent of brain edema, and infarct volume after reperfusion. The histopathologic alterations and changes in glucose uptake in the brain were also observed. Moreover, the levels of lactate dehydrogenase (LDH), lactic acid, pyruvate, and ATP in brain tissue were detected. Leptin levels in serum were also detected. To further define leptin-induced neuroprotective signaling pathways, we examined the levels of phosphorylated Akt (p-Akt) in the brain and in cultured cells. After transient ischemia, leptin treatment markedly reduced the neurologic deficits, cerebral infarct volume, and brain edema. After leptin injection, ATP, leptin, and p-Akt levels were significantly increased, LDH levels and lactic acid/pyruvate ratio were noticeably reduced, and histopathologic injuries were alleviated, which were all reversed by the PI3K inhibitor LY294002. These data show that leptin ameliorates cerebral ischemia/reperfusion injury by enhancing p-Akt, which in turn improves the supply of energy. The PI3K/Akt pathway was found to be the critical pathway for the mediation of leptin-induced neuroprotection, a finding that may prove to be useful in the treatment of ischemic stroke.
INTRODUCTION
Acute ischemic stroke injuries to brain tissue are among the leading causes of death and long-term disability in humans and are instigated by a number of physiologic factors. These factors include the cessation of the glucose supply to challenged brain tissues that have high energy demands and the impediment of the delivery of oxygen, which is vital for cellular respiration.
Leptin, a centrally acting hormone secreted by adipocytes, has significant biologic activity in the central nervous system and is known to inhibit food intake and stimulate energy expenditure. 1 Moreover, leptin is generally considered to be a strong and rapid stress mediator after injuries and has been found to exert neuroprotective effects in a mouse model of transient focal cerebral ischemia. 2 Leptin receptors are expressed in a number of specific brain regions, 3 and the binding of leptin leads to the regulation of a range of biologic functions and processes, including energy intake and expenditure and the glucose balance.4,5 The activation of the leptin receptor recruits several intracellular signaling pathways, including the PI3K/Akt pathway. Akt (PKB), a PH domain-containing serine/threonine kinase, regulates growth factor signaling 6 and thereby stimulates glucose uptake, 7 glycogen synthesis, and protein synthesis. 8 This led us to ask whether and how leptin protects the brain against cerebral ischemia/reperfusion via the PI3K/Akt signaling pathway. Furthermore, the downstream molecules that are involved in the metabolism of brain ischemia/reperfusion injury after the inhibition of PI3K/Akt by LY294002 remain to be identified. Here, we clarified the major role of the PI3K/Akt pathway in the leptin-mediated protection against metabolic alterations induced by cerebral ischemia/reperfusion injury.
MATERIALS AND METHODS
Animal Model and Leptin Infusion
Male Kunming mice (age, 42 ± 1.5 days old; weight, 25 ± 0.3 g) were supplied by the Experimental Animal Center of our hospital and treated in accordance with the Guide for Care and Use of Experimental Animals approved by the Animal Care Committee of the 301 Hospital. In brief, blood pressure, blood gases, and blood glucose concentrations were monitored through the catheterization of the left femoral artery and remained within the normal range throughout the experiments. Rectal temperature was continually monitored and maintained at 37 to 37.5°C using a heating pad and a heating lamp. Brain temperature was monitored using a 29-Gathermocouple implanted in the left caudate-putamen and maintained at 35.8 ± 0.2°C during ischemia.
Mice were deprived of food for 12 hours before the start of the experiments, while drinking water was available ad libitum. Focal cerebral ischemia/reperfusion injury was induced by the unilateral middle cerebral artery occlusion (MCAO) model as described previously. 9 Briefly, mice were anesthetized with pentobarbital sodium (60 mg/kg, intraperitoneally), and their skin was sterilized. The common carotid artery, the external carotid artery, and the internal carotid artery on the right side were exposed through a ventral midline neck incision. A 6-0 nylon monofilament coated with silicon resin was introduced into the right common carotid artery and advanced until faint resistance was felt. After a 2-hour ischemia period, 24 hours of reperfusion were achieved by withdrawing the monofilament, thus restoring the blood supply to the MCA territory.
All animals were randomly divided into four groups as follows: (1) the sham-operated group (Sham), in which the common carotid artery, the external carotid artery, and the internal carotid artery on the right side were exposed without occlusion; (2) the cerebral ischemia/reperfusion group (I/R), which were given 200 μL normal saline (via tail vein injection) immediately after occlusion; (3) the leptin injection group (Leptin), which were given leptin (1 mg/kg in 200 μL normal saline via tail vein injection) immediately after occlusion; and (4) the LY294002 + leptin group (LY), which were pretreated with LY294002 (15 nmol/L via tail vein injection) 30 minutes before the operation and given a tail vein injection of 1 mg/kg of leptin dissolved in 0.2 mL of physiologic saline immediately after occlusion. The incision was closed in layers, and resuscitation with isotonic saline (30 mL/kg, intraperitoneally) was performed. The suture sites were then sterilized with iodophor, and the mice were released back to their cages, where drinking water was available ad libitum.
Indicators of General Wellbeing
Mice were weighed as an indicator of their general wellbeing at 24 hours post surgery for the short-term study. The survival rates of sham, I/R, leptin-treated, and LY-pretreated animals are presented as a percentage of the number of animals undergoing surgery. The body weights are presented as a percent change compared with the values recorded immediately before undergoing MCAO.
Evaluation of Neurologic Deficits
Twenty-four hours after MCAO, Longa's ‘5 Law’ 10 was used to determine the global neurologic function according to the following scoring system: 0. no deficit; 1, forelimb flexion; 2, decreased resistance to lateral push; 3, unidirectional circling; 4, longitudinal spinning; 5, no movement. The ratings were performed in a blinded manner. Neurologic function was scored twice: initially after full recovery from anesthesia during the reperfusion period, and subsequently after the completion of 24 hours of reperfusion.
The exclusion criteria were as follows:
Death within 24 hours after MCAO.
Subarachnoid hemorrhage (as assessed macroscopically during brain sampling).
Bederson score = 0 (24 hours after MCAO).
PET/CT Studies and Image Analysis
The PET/CT images of mice were obtained using a small-animal PET/CT scanner (Explore VISTA Micro-PET/CT; GE Healthcare, Amersham, UK); the procedure has been described elsewhere. 11 Briefly, the mice were anesthetized using 1% chloral hydrate (0.45 mg/g of body weight) and positioned prone on the scanning table. 18 F-FDG was injected via the tail vein at a dose of 20 ± 1.84 MBq in 0.2 mL of saline. Image data were acquired for 10 minutes using list mode, starting 45 minutes after injection. For image reconstruction, list mode data were sorted into 3-dimensional sinograms followed by Fourier rebinning and 2-dimensional ordered-subset expectation maximization reconstruction with 2 iterations and 50 subsets. The image pixel size was 0.385 × 0385 × 0.335 mm. Image J software was used on reconstructed images for the quantification of brain uptake. The right/left hemisphere (background) uptake ratio was also calculated.
Measurement of Local Infarct Volume
Mice were killed 24 hours after reperfusion, and their brains were isolated to estimate the infarct volume (n = 10/group) as previously described. 12 Briefly, the mice were killed under deep anesthesia, their brains were removed and immediately frozen in isopentane cooled with dry ice, and serial sections from anterior to posterior were prepared as 2 mm thick slices. Samples were placed in 2% 2,3,5-triphenyl tetrazolium chloride (TTC; Sigma, St Louis, MO, USA) stain for 20 minutes at 37°C and fixed in 10% buffered formaldehyde for 24 hours. The stained brain sections were then digitally photographed. Cerebral infarct volumes were measured using microscope image analysis software (Image-Pro Plus, San Rafael, CA, USA). The data were expressed as the percentage of infarct volume/ipsilateral hemisphere volume in the coronal slices, as recently described. 12
Investigation of Histopathology
For histologic analysis, the animals were grouped randomly, with eight mice in each group. Mice were perfusion fixed with 4% paraformaldehyde in 0.1 mol/L pH 7.4 phosphate-buffered saline (PBS) under anesthesia after 24 hours of reperfusion. The brains were immediately removed, additionally fixed in 10% paraformaldehyde overnight at 4°C and embedded in paraffin. Serial coronal sections (5 μm thick), obtained 1.5 mm posterior to bregma, were stained with hematoxylin and eosin. The normal morphology and the presence and nature of ischemic damage in the fronto-parietal cortex were verified by two neuropathologists who were blind to the experimental design or the results of TTC staining.
Detection of Brain Edema
After 24 hours of reperfusion, brains were immediately removed, and the two hemispheres were separated and weighed individually. The data on the extent of brain edema were expressed as follows: brain edema = the damaged ipsilateral hemisphere weight – contralateral hemisphere weight.
Preparation of Serum Samples
Blood was collected by extracting eyeballs at 2, 6, and 24 hours post reperfusion. The whole blood samples were centrifuged by 3,000 g at 4°C for 10 minutes to separate mouse serum samples. The supernatants were collected and stored at −20°C. Serum levels of leptin were measured with the ELISA kit (Murine Leptin ELISA Development Kit; PEPROTECH, Rocky Hill, NJ, USA).
Detection of Tissue Lactic Acid, Pyruvate, and Lactate Dehydrogenase
After the 24-hour reperfusion period, samples of the ischemic cerebrum hemisphere were harvested, frozen immediately, and homogenized by a high-speed homogenizer five times at 559 g for 10 seconds on an ice bath in 10 volumes of physiologic saline. The homogenates were centrifuged at 6,000 g for 10 minutes at 4°C. The lactic acid, pyruvate, and lactate dehydrogenase (LDH) levels were detected using their respective assay kits according to manufacturers' recommendations (Jiancheng Bioengineering Institute, Nanjing, JS, China).
Determination of Tissue ATP Levels
Changes in ATP levels were measured using an ATP assay kit according to manufacturer's recommendations. Briefly, brain tissue was dissected to separate the stroke-affected tissue for the measurement of ATP concentrations. The tissue was immediately homogenized in a manual Dounce-type glass/glass homogenizer on ice in distilled water according to a weight/volume ratio of 1:9 and boiled for 10 minutes. The homogenates were centrifuged at 6,000 g for 10 minutes at 4°C. The ATP levels in the supernatant were detected using the respective assay kits according to manufacturers' recommendations (Jiancheng Bioengineering Institute, Nanjing, JS, China).
Tissue Collection and Western Blot Analysis
Mice were killed at 24 and 48 hours post reperfusion. The ischemic cerebrum hemisphere was separated, homogenized in cell lysis buffer, and sonicated. The protein level was normalized to total protein on the membrane using 0.05% Coomassie brilliant blue. A total of 60 μg protein was separated on 10% or 4% to 15% SDS-polyacrylamide gels (Mini-PROTEAN; Bio-Rad, Hercules, CA, USA) and transferred onto nitrocellulose filter membranes. After blocking in 5% milk/TBS/0.05% Tween-20, the membranes were incubated with an antibody overnight at 4°C followed by an HRP-coupled secondary antibody (Pierce, Rockford, IL, USA) and enhanced chemiluminescence onto light-sensitive film. Blots were quantified using Image J (NIH).
Cell Culture and Hypoxia/Reoxygenation
SH-SY5Y cells were plated onto poly-D-lysine (10 μg/mL) precoated 6-well plates for the analysis of cellular p-Akt expression. For all immunostaining experiments, cells grown on coverslips were randomly divided into four groups: Control, H/R injury, Leptin treatment, and LY (cells were treated with LY for 30 minutes, and leptin was then added to the medium to reach a final concentration of 500 μg/L). To model ischemic-like conditions in vitro, primary cultures were exposed to oxygen-glucose deprivation.12,13
In brief, the culture medium was replaced with serum- and glucose-free media. The cultures were then put into an air-tight box, and 95% N2 and 5% CO2 mixed gas was continually aerated into the box for 5 minutes to completely replace the air. The box was placed in a water-jacketed incubator at 37°C for 1 hour and then returned to 95% air, 50% CO2, and glucose-containing medium for 3 hours (hypoxia for 1 hour and reoxygenation for 3 hours, H1 h/R3 h).
Immunocytochemical Analyses
Immunofluorescence was performed as described previously. 14 In brief, SH-SY5Y cells were grown on glass coverslips coated with poly-L-lysine. After hypoxia/reoxygenation injury, coverslips were washed twice in PBS and fixed for 15 minutes at room temperature in 4% (wt/vol) paraformaldehyde in PBS, permeabilized in 0.1% (vol/vol) Triton X-100 in PBS for 15 minutes, blocked with 1% (vol/vol) BSA-PBS for 30 minutes and probed with p-Akt (Thr308) (1:100, #40565, USA) antibody. The antibody was diluted in blocking solution at 37°C for 1 hour. After a wash in PBS (3 times for 15 minutes each), coverslips were placed in a humidity chamber for 1 hour and covered with FITC-conjugated anti-rabbit secondary antibody (Sigma, 1:200 dilution in PBS). The coverslips were inverted and mounted onto glass slides with Vectashield containing 4′,6-diamidino-2-phenylindole (DAPI, Vector Laboratories, Burlingame, CA, USA) and viewed using an immunofluorescence microscope.
Statistical Analysis
All data are expressed as the mean ± SD Stata 7.0 software (Stata Corp, College Station, TX, USA) was used to process the data. For all data except the grading of neurologic deficits, a one-way analysis of variance and Student's t-tests were applied. For the grading of neurologic deficits, a Wilcoxon signed-rank test was used. A P-value of <0.05 was chosen as the threshold for statistical significance.
RESULTS
Leptin Did Not Significantly Affect the Survival Rate or Body Weight
In Figure 1A, the survival data are presented in a histogram, which revealed no significant differences in the survival rate between experimental groups regardless of whether the animals were treated with leptin or LY294002. All experimental groups lost weight over the 24-hour period after cerebral ischemia/reperfusion injury (Figure 1B).
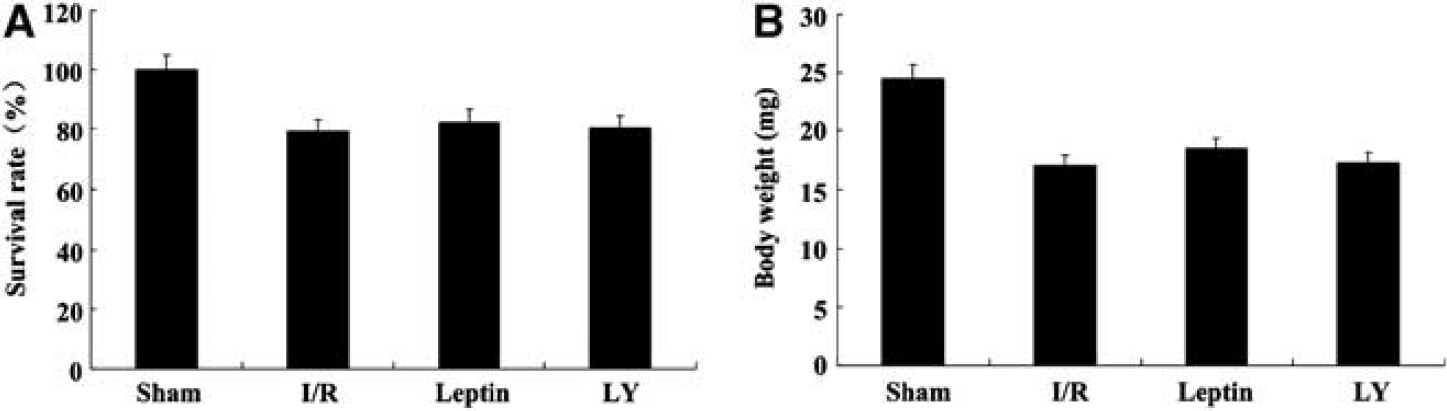
Leptin did not significantly affect the survival rate (
Leptin Reduced the Brain Infarct Volume, Histologic Alterations, and Brain Edema and Improved the Neurologic Deficits in Mice
After TTC staining, the infarcted brain was visualized as an area of unstained (white) tissue in a surrounding background of viable (brick red) tissue. The TTC staining showed that all of the sham brain slices were stained red, and the right frontal cortical and striatal infarct volumes in the I/R group were clearly white (Figure 2A). The infarct volume of mice in the leptin group was significantly smaller than in the I/R mice (P < 0.001); LY294002 markedly reversed this effect (P < 0.01), as shown in Figure 2B. The normal morphology and the presence and nature of ischemic damage in the fronto-parietal cortex were verified by two neuropathologists who were blind to the experimental design and the results of TTC staining.
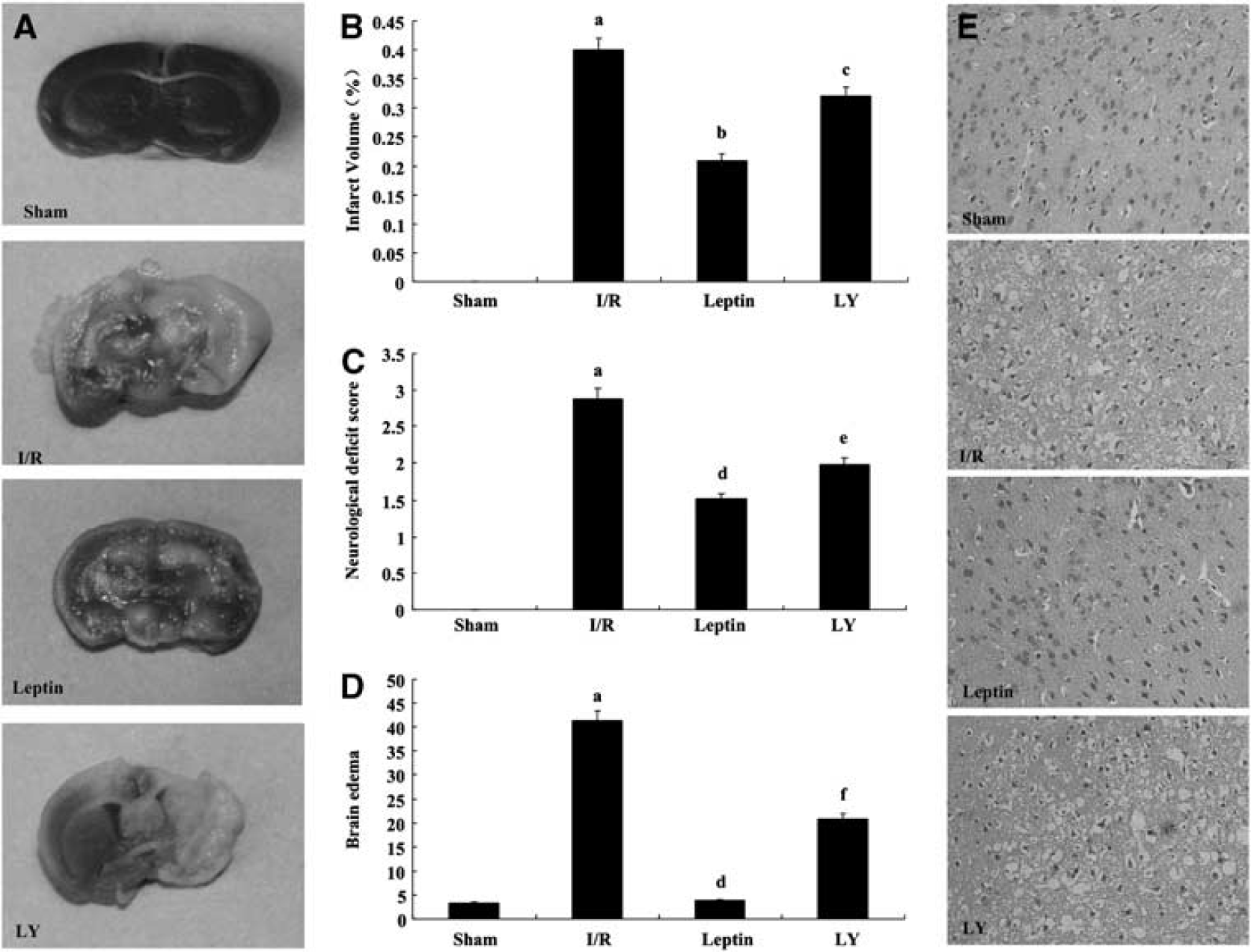
Leptin reduced brain infarct volume (
Two hours after reperfusion, the mice presented varying degrees of neurologic deficits when regaining consciousness. The neurologic deficit scores in the leptin-treated group were significantly lower than in the I/R group (P < 0.001). The neurologic deficit scores in the LY group were significantly higher than those in the leptin group (P < 0.001), indicating that the effect of leptin was reversed by LY294002 (Figure 2C).
After injury, the brain tissues in each group showed differing degrees of edema. The edema was greatest in the I/R group. The edema in the leptin group was significantly reduced compared with the I/R group (P < 0.001). The edema in the LY group was significantly increased compared with the leptin group (P < 0.05), indicating that the effect because of leptin was reversed by LY294002 (Figure 2D).
Histopathologic changes in the cerebral cortex after injury were detected by H&E staining. Normal morphology or the presence and nature of ischemic damage in the fronto-parietal cortex were verified by two neuropathologists who were blind to the experimental design and the results of the TTC staining. Compared with the histologic alterations in the ischemic fronto-parietal cortex in sham mice, the neurocytes in I/R mice showed a disorderly arrangement with loosened cytoplasms and karyopyknosis. There was also evidence of diffuse vacuolization and edema in the interstitial spaces. Similar but nonstatistically significant alterations were observed in the leptin group, and LY294002 reversed the effect of leptin (Figure 2E).
Leptin Increased Glucose Uptake in the Brain
To investigate brain glucose metabolism, we studied the changes in glucose uptake as detected by Micro-PET/CT scanning of living mice from the sham, I/R, leptin injection, and LY pretreatment groups (Figure 3). Micro-PET/CT scanning results of 18 F-FDG uptake in the brain showed that glucose uptake was equivalent between the left and the right hemispheres in the sham group. The mice in the I/R group exhibited large decreases in glucose uptake and energy metabolism in the cerebral cortex. After a 24-hour reperfusion, we observed a large area in the right hemisphere devoid of glucose uptake, indicating that glucose metabolism in the right hemisphere was decreased significantly compared with the left. The observed decrease was 50.5% compared with the contralateral hemisphere (P < 0.01). Compared with the I/R group, the ratio of glucose uptake in the right/left hemisphere in the leptin group was increased by 66.7% (P < 0.001). LY294002 reversed this effect.
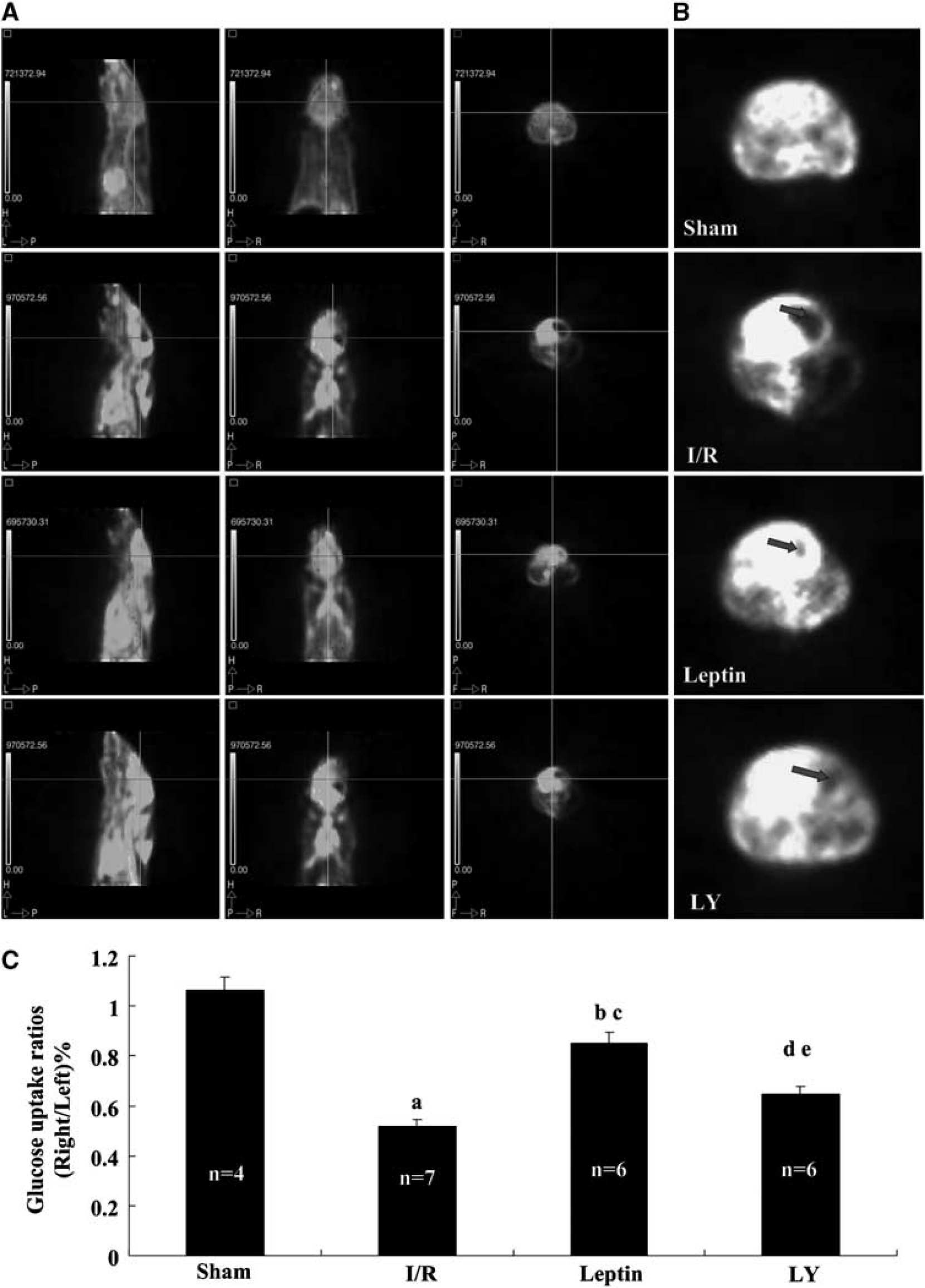
Leptin treatment increased glucose uptake in the brain. After 24 hours reperfusion, mice were reanesthetized.
18
F-FDG was injected via the tail vein at a dose of 20 ± 1.84 MBq in 0.2 mL of saline. Image data were acquired for 10 minutes beginning 45 minutes after injection by list mode. Mice were subjected to the PET/CT scanning to obtain typical images as shown in (
Exogenous Leptin Increased the Leptin Levels in Blood at the Earlier Period
To investigate changes of leptin levels in blood after reperfusion, we measured the serum leptin levels at 2, 6, and 24 hours post reperfusion with the ELISA kit. As shown in Table 1, after 2 hours reperfusion, serum leptin levels in Leptin and LY groups were significantly higher than Sham (P < 0.001) and I/R (P < 0.001) groups. Serum leptin level at I/R6h in Leptin group was much higher than Sham (P < 0.05) and I/R (P < 0.05) groups, but markedly lower than Leptin group at I/R2h (P < 0.001). Serum leptin level at I/R6h in LY group was also higher than I/R (P < 0.05) group. Serum leptin levels at I/R24h in each group have no significant difference. Serum leptin levels in I/R, Leptin, and LY groups were reduced with the reperfusion time increase.
Leptin levels in blood after reperfusion at different time points
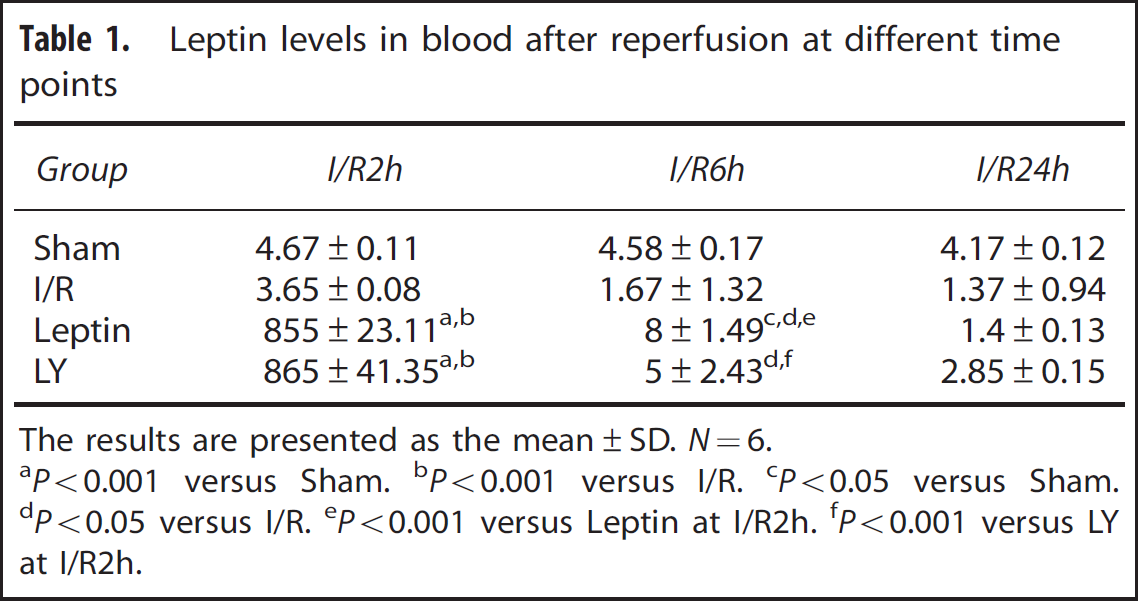
The results are presented as the mean±SD. N=6.
P<0.001 versus Sham.
P<0.001 versus I/R.
P<0.05 versus Sham.
P<0.05 versus I/R.
P<0.001 versus Leptin at I/R2h.
P<0.001 versus LY at I/R2h.
Leptin Reduced Brain Tissue Lactate Dehydrogenase and the Lactic Acid/Pyruvate Ratio, and Increased ATP Levels
Tissue LDH in the ischemic cerebrum of I/R mice was significantly greater compared with the sham group (P < 0.05), and leptin markedly reduced the tissue LDH levels after injury (P < 0.05). This effect was reversed by LY294002 (P < 0.001) (Figure 4A).
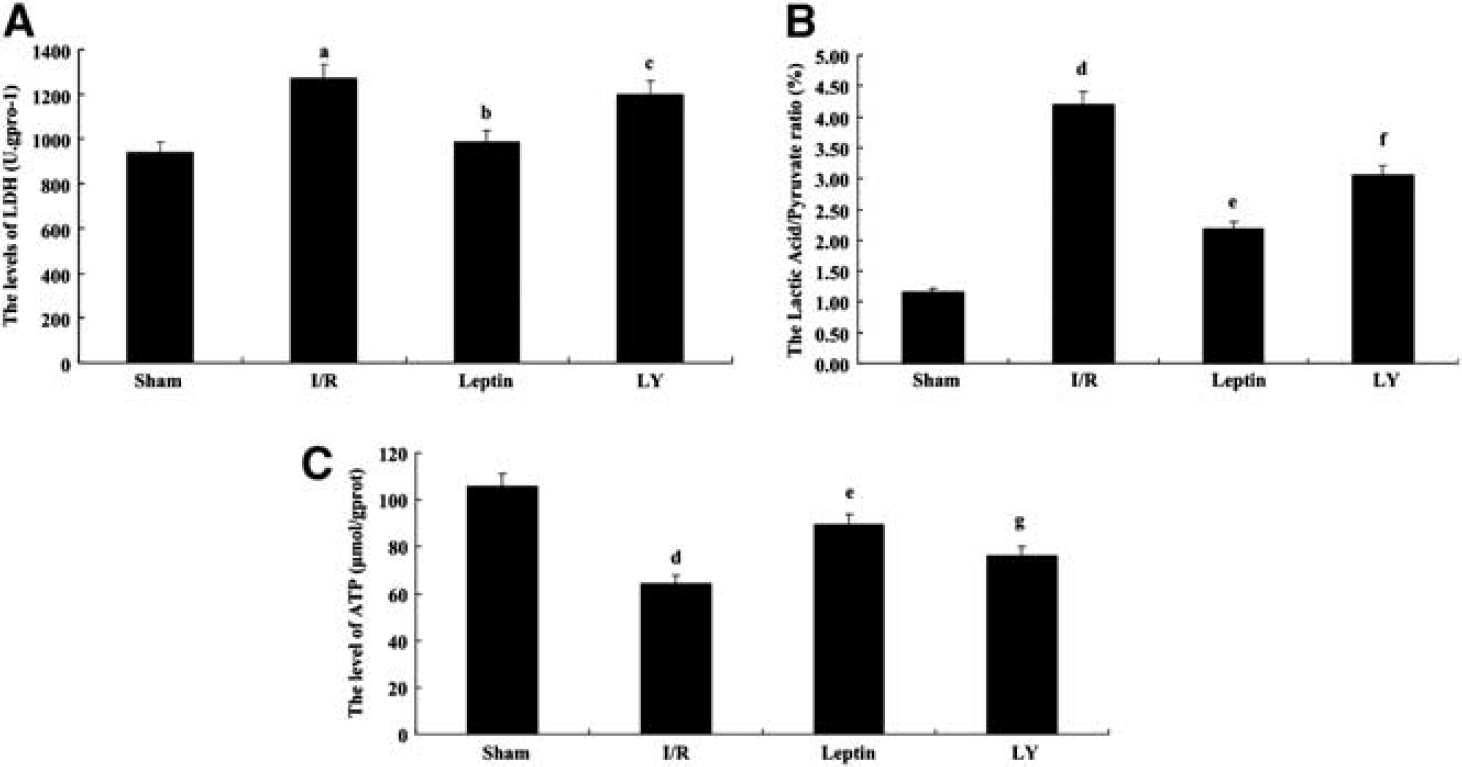
Leptin reduced brain tissue lactate dehydrogenase (LDH) (
The tissue lactic acid/pyruvate ratio was significantly increased in the I/R group, and leptin markedly reduced the tissue lactic acid/pyruvate ratio after injury. This effect was reversed by LY294002. In contrast, leptin conferred protection against cerebral ischemia/reperfusion injury by reducing the lactic acid/pyruvate ratio. As shown in Figure 4B, these results indicated that leptin can mitigate acidosis by inhibiting the PI3K/Akt pathway in brain tissue after injury.
After ischemia/reperfusion injury, the levels of ATP were significantly reduced, and leptin could partially recover the levels of ATP (P < 0.001). LY294002 reversed this effect, and the levels of ATP were reduced compared with the leptin group (P < 0.01) (Figure 4C).
Leptin Enhanced AKT Phosphorylation in the Brain
To evaluate the role of the PI3K/Akt signaling pathway in the observed protective mechanisms on cerebral ischemia/reperfusion because of leptin, we detected the levels of Akt phosphorylation in the cerebral cortex after I/R injury. p-Akt was increased in the I/R group (P < 0.01); this effect was further enhanced by leptin at 24 and 48 hours after reperfusion (P < 0.01 and P < 0.05, respectively), indicating that the PI3K/Akt pathway may have a critical role in the leptin-mediated neuroprotection against injuries resulting from energy failure induced by transient focal cerebral ischemia in mice (Figure 5A).
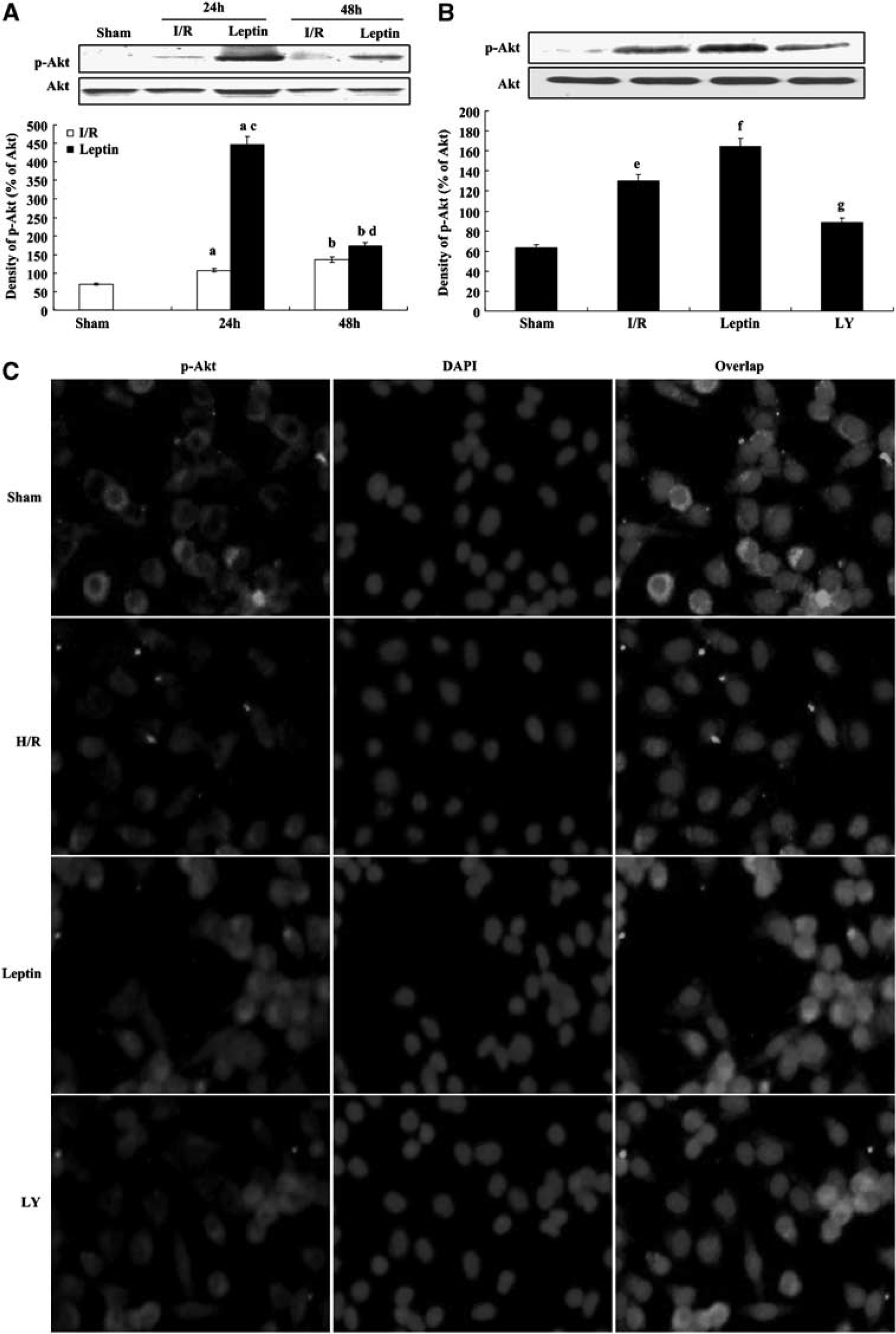
Leptin promoted Akt phosphorylation in vivo and in vitro. (
To further evaluate the role of the Akt signaling pathway in the protective mechanisms of leptin during cerebral ischemia/reperfusion, we investigated the effects of the PI3K inhibitor LY294002, which inhibits the production of PIP3 and blocks the activation of Akt. After 24 hours of reperfusion, p-Akt expression was detected (Figure 5B). Phosphorylated Akt was increased in the I/R group (P < 0.01); this effect was further enhanced by leptin (P < 0.01), indicating that the PI3K/Akt may have a critical role in the leptin-mediated neuroprotection against injuries resulting from energy failure induced by transient focal cerebral ischemia in mice. However, compared with the leptin group, this effect was attenuated by LY294002 (P < 0.01), which strongly indicated that the PI3K/Akt signaling pathway is a critical pathway in the leptin-mediated protection against cerebral ischemia/reperfusion injury.
Furthermore, an immunofluorescence assay was used to detect p-Akt expression in SH-SY5Y cells after H/R injury (Figure 5C). In SH-SY5Y cells, p-Akt expression was promoted by leptin after H/R injury, and this effect was partly reversed by LY294002, indicating that the PI3K/Akt pathway may be a critical mediator of leptin-induced protection from H/R injury (Figure 6).
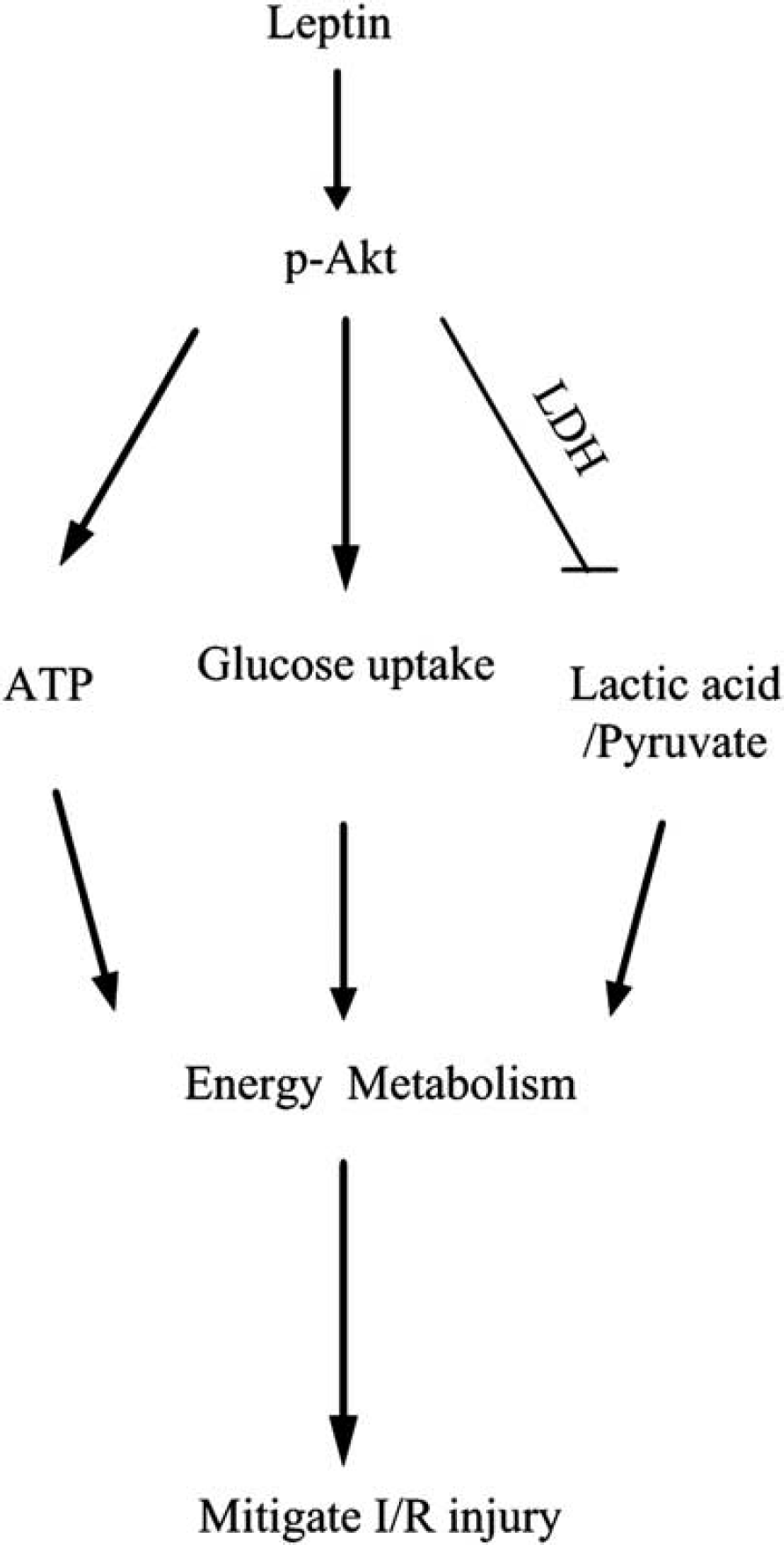
Molecular pathways involved in the action of leptin on cerebral ischemia/reperfusion (I/R) injury. LDH, lactate dehydrogenase.
DISCUSSION
Stroke is the third-leading cause of death and the leading cause of long-term disability in the United States and Europe. The pathophysiologic mechanisms of cerebral ischemia/reperfusion injury are primarily related to the energy deficiency of neurons, cell excitatory responses, inflammation, and the start of the apoptosis cascade. The disruption of the energy metabolism of the brain is the hallmark of this pathology because of the restricted delivery of oxygen and glucose, which slows or stops the synthesis of the ATP molecules that are required to maintain ionic gradients. 15 The ischemic brain responds to the rapid decline in ATP supplies by increasing the rate of anaerobic glycolysis, which leads to lactate accumulation and consequently to acidosis. 16
Previous studies have shown that endogenous leptin may increase hippocampal neurogenesis via the induction of Akt and STAT signaling pathways without changes in newborn cell differentiation. 17 The effect of leptin on the increase of angiogenesis and neurogenesis after brain ischemia may be mediated at least in part via increases in the leptin receptor, activated AMPK, and TRPV1 expression. 18 In our present study, we found that after ischemia injury the levels of leptin were markedly decreased with the reperfusion time increase and exogenous leptin could complement the deficiency of endogenous leptin and increase the levels of leptin in brain. Furthermore, ischemia-induced behavioral changes, brain edema, and infarct volume are significantly reversed by leptin. In a previous study, we also showed that leptin had protective effects after cerebral ischemia/reperfusion,12,19 which was consistent with a previous report.2,20 To verify the neuroprotective ability of leptin against cerebral ischemia/reperfusion injury via the PI3K/Akt pathway, we performed a PI3K inhibition experiment by injecting LY294002 into the tail vein 30 minutes before ischemia. Leptin minimizes the brain infarct volume and histologic alterations and improved neurologic deficits in mice, results that are reversed by LY294002.
After hypoxia, two major changes occur in the brain: the decrease in aerobic sugar decomposition and the increase in anaerobic glycolysis. As a highly active metabolic organ, the brain has very low reserves of energy and oxygen. The energy required for brain function is entirely dependent on aerobic metabolism, which is dependent upon the oxygen and glucose provided by the blood. Therefore, the brain is highly sensitive to ischemic or hypoxic injury. Without a fresh oxygen source, the brain produces energy only through the consumption of reserved high-energy phosphate compounds and the conversion of stored glucose and glycogen to lactic acid. After focal cerebral ischemia, the decrease in oxygen and glucose supplies leads to an oxidative phosphorylation disorder, eventually resulting in energy reduction and even energy exhaustion, which causes an increase in anaerobic glycolysis. Subsequently, the increase in anaerobic glycolysis leads to a large accumulation of lactic acid, which can cause tissue acidosis and thus tissue damage and death through multiple pathways. When there is an insufficient oxygen supply, pyruvate generated from the glycolytic pathway is reduced to lactic acid. The lactic acid/pyruvate ratio is an important parameter for the health of brain tissue. The greater the hypoxia, the higher the lactic acid/pyruvate ratio. Lactate dehydrogenase is an important enzyme that participates in energy metabolism and can catalyze lactic acid to pyruvate by dehydrogenation. In our study, leptin decreases tissue LDH levels and thereby decreases the lactic acid/pyruvate ratio, resulting in a mitigation of acidosis because of anaerobic metabolism within the brain. This effect is reversed by LY294002, indicating that the PI3K/Akt signaling pathway has a critical role in leptin-mediated neuroprotection.
In our experiment, we further applied micro-PET/CT to investigate the leptin-mediated glucose uptake. After leptin treatment, 18 F-FDG uptake is significantly increased, showing that leptin can regulate glucose uptake after cerebral ischemia. However, this effect is reversed by LY294002, indicating that the PI3K/Akt signaling pathway has a critical role in leptin-mediated neuroprotection.
As the most important energy molecule in the body, ATP has various important physiologic and pathologic roles. Therefore, changes in ATP levels will affect many cellular functions. According to a previous report, brain ATP levels decreased by one-third within the first 15 minutes of cerebral ischemia. 21 Leptin has a role in regulating energy metabolism. 22 In this study, we find that after cerebral ischemia/reperfusion injury, leptin can promote ATP synthesis and thus attenuate injury in the brain, a result that was reversed by LY294002. Furthermore, the phosphorylation of Akt is found to be significantly enhanced by leptin treatment, and this effect was reversed by LY294002. In an in vitro experiment, after hypoxia/reoxygenation injury, we also detected the activation of Akt in human neurons (SH-SY5Y) (Figure 5). All of the above results strongly show that PI3K/Akt signaling is an important pathway in leptin-induced neuroprotection.
In summary, after cerebral ischemia/reperfusion injury, leptin treatment significantly increases the expression of activated Akt in the ischemic brain, which successively reduces the levels of LDH and the lactic acid/pyruvate ratio and increases brain glucose uptake and ATP levels. This phenomenon results in the repair of the brain energy deficit; reduces the infarct volume, histologic alterations and brain edema; and improves neurologic deficits (Figure 6). Collectively, leptin is effective in reducing experimental ischemic injury induced by MCAO in mice, suggesting that leptin may be useful in treating ischemic incidents in humans.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.