
Abstract
Cytokines are recognized to play an important role in acute stroke. Tumor necrosis factor-α (TNF) is one of the pro-inflammatory cytokines and is expressed in ischemic brain. We hypothesized that TNF might play a role in the regulation of tolerance to ischemia when administered prior to the ischemic episode. We studied the effects of pretreatment of TNF administered intravenously, intraperitoneally, or intracisternally in mice that were subjected to middle cerebral artery occlusion (MCAO) 48 h later. MCAO was performed in BALB/C mice by direct cauterization of distal MCA, which resulted in pure cortical infarction. A significant reduction in infarct size was noted in mice pretreated by TNF at the dose of 0.5 μg/mouse (p < 0.01) intracisternally. At the doses used in this study, administration of TNF by intravenous or intraperitoneal routes was not effective. Immunohistochemical analysis of brains subjected to 24 h of MCAO revealed a significant decrease in CD11b immunoreactivity after TNF pretreatment compared with control MCAO. Preconditioning with TNF affects infarct size in a time- and dose-dependent manner. TNF induces significant protection against ischemic brain injury and is likely to be involved in the signaling pathways that regulate ischemic tolerance.
Animal models of ischemic tolerance induced by “preconditioning” offer the possibility to examine the mechanisms selected by nature to limit ischemic damage. Many kinds of pretreatment, such as sublethal ischemia (Kitagawa et al., 1990; Kato et al., 1991; Kirino et al., 1991; Aoki et al., 1993), hyperthermia, and oxidative stress (Ohtsuki et al., 1993), are able to induce tolerance against delayed neuronal death after transient ischemia in rodents. The mechanisms of induced tolerance are not known precisely; however, several possible explanations exist, such as induction of heat shock proteins in neurons (Aoki et al., 1993; Nowak and Jacewicz, 1994) and increased expression of bcl-2 oncoprotein (Shimazaki et al., 1994).
In focal cerebral ischemia, such as permanent middle cerebral artery occlusion (MCAO), the ischemic core cannot be salvaged by any presently known means of treatment without reestablishment of cerebral blood flow. The border zone, or penumbra, can be salvaged by several kinds of treatment. One of the factors affecting the fate of the penumbra is thought to be a gradually progressing disturbance in microcirculation. Excitotoxicity, energy imbalance due to ischemic depolarization, free radical injury including synthesis of nitric oxide, and inflammatory reactions that include leukocyte infiltration may affect outcome of the penumbra (Dalkara and Moskowitz, 1994; Hossmann, 1994; Garcia et al., 1995; Siesjö et al., 1995). We have observed that systemic administration of lipopolysaccharide (LPS) can induce ischemic tolerance in spontaneously hypertensive rats subjected to permanent MCAO. Co-administration of TNF binding protein (Amgen, Boulder, CO, U.S.A.) with LPS blocked acquisition of ischemic tolerance in that model (Tasaki et al., 1997).
TNF is one of the pro-inflammatory cytokines and is expressed in ischemic brain (Liu et al., 1994). TNF stimulates acute-phase protein production in the liver (Guy et al., 1991; Marino et al., 1991), enhances permeability of the blood-brain barrier, and induces expression of endothelial adhesion molecules and release of inflammatory mediators from macrophages, endothelial cells, and glial cells (Briscoe et al., 1992; Feuerstein et al., 1994). TNF also activates or induces synthesis or gene expression of manganese superoxide dismutase in mitochondria (Suzuki et al., 1993; Mokuno et al., 1994), up-regulates nitric oxide synthetase in endothelial cells (Tureen, 1995), and induces growth factors from astrocytes contributing to neuronal survival after insults (Donato et al., 1989; Feuerstein et al., 1994).
Induction of tolerance by TNF and interleukin-1 against a subsequent ischemic insult has been reported in the heart and lung (Colletti et al., 1994) but not in the brain. In this study we investigated a role of TNF in acquisition of tolerance to ischemic brain damage.
MATERIALS AND METHODS
Experimental groups
Mature male BALB/C mice, weighing 22–26 g, were used. Animals were obtained from Charles River Labs (Wilmington, MA, U.S.A.) and were given free access to food and water prior to surgery. Animals were housed and cared for in accordance with Guide for the Care and Use of Laboratory Animals [DHEW (DHHS) publication no. (NIH) 85-23, rev. 1985, Office of Science and Health Reports, DRR/NIH, Bethesda, MD 20205, U.S.A.]. Procedures using animals were approved by the NIH Animal Care and Use Committee.
Sixty-one mice were divided into 10 experimental groups. MCAO was performed without any pretreatment in the “control” group (n = 8). In the “vehicle i.p.” group (n = 6), mice were pretreated by intraperitoneal injection of vehicle [phosphate-buffered saline (PBS) with 50 μg/ml of LPS-free bovine serum albumin (BSA)]. In the “TNF 40 μg/kg i.p.” (n = 6) and the “TNF 200 μg/kg i.p.” (n = 5) groups, mice were pretreated by an intraperitoneal injection of an appropriate dose of TNF. In the “vehicle i.v.” group (n = 7), mice were pretreated by an intravenous injection of vehicle (PBS with 50 μg/ml of LPS-free BSA). In the “TNF 40 μg/kg i.v.” group (n = 7) and the “TNF 200 μg/kg i.v.” group (n = 2), mice were pretreated by an intravenous injection of an appropriate dose of TNF. In the “vehicle cisternal” group, mice (n = 7) were pretreated by an intracisternal injection of vehicle (PBS with 50 μg/ml of LPS-free BSA). In the “TNF 0.05 μg cisternal” (n = 7) and the “TNF 0.5 μg cisternal” group (n = 6), mice were pretreated by an intracisternal injection of the appropriate dose of TNF.
Preconditioning by mouse recombinant TNF
In the first experiments, the effect of peripheral administration of recombinant mouse TNF (Sigma Chemical Co.) was studied. Preconditioning was performed by a single intraperitoneal or intravenous injection of recombinant mouse TNF at a dose of 40 or 200 μg/kg body wt 48 h before MCAO. Body weight and temperature were measured before and after pretreatment with TNF.
Second, the effect of central administration of recombinant mouse TNF was studied. Intrathecal injection of recombinant mouse TNF (0.05 μg or 0.5 μg/mouse) was performed through the cisterna magna under direct microsurgical exposure of the occipitoatlantic region. To avoid an abrupt increase in intracranial pressure, 10 μl of cerebrospinal fluid was initially aspirated and then 5 μl of TNF (0.05 μg or 0.5 μg/mouse) was injected slowly. For the prevention of infection, especially meningitis, all surgical procedures were done in a completely aseptic manner. A control group of animals (“vehicle cisternal”) received the same volume of vehicle (PBS with 50 μg/ml of LPS-free BSA). Forty-eight hours later, left MCAO was performed in animals as described herein.
Permanent MCAO in mice
Animals were anesthetized with 3% halothane for induction and 1.5% halothane for maintenance in 30% O2/70% N2O via a face mask. The rectal temperature was measured and maintained at 37 ± 0.2°C with a heating blanket. The exposure of the MCA was based on the description by Welsh et al. (1987). Briefly, the left temporoparietal region of the head was shaved and a 5-mm incision was made between the orbit and ear. Under an operating microscope, an incision was made by dividing the temporal muscle, and the left lateral aspect of the skull was exposed by reflecting the temporal muscle and surrounding soft tissue. A small burr hole (1 mm) was made using a high-speed microdrill, through the outer surface of the semi-translucent skull just over the visibly identified MCA at the level of the inferior cerebral vein. Saline was applied to the area throughout the procedure to prevent heat injury. The inner layer of the skull was removed with fine forceps, and left MCAO was performed by electrocoagulation, using a small vessel cauterizer directly through the dura without damaging the brain surface. If the brain surface was visibly damaged or if the MCA was noted to bleed due to incomplete artery occlusion/coagulation, the animal was killed and eliminated from the study. The muscle and soft tissue were replaced and the skin was closed using 5-0 nylon suture. The duration of anesthesia in all animals was <30 min. During anesthesia, the left femoral artery was catheterized using a P-10 tube for arterial blood sampling and continuous monitoring of arterial blood pressure.
Measurement of size of cortical infarcts using 2,3,5-triphenyltetrazolium chloride
Twenty-four hours after MCAO, the animals were injected with a lethal dose of thiopental (150 mg/kg body mass i.p.); the brains were removed, cut into 1-mm-thick coronal sections, and immersed in 2% 2,3,5-triphenyltetrazolium chloride saline solution at 37°C for 30 min. The infarcted areas of the brain slice were measured using a computer-assisted image analyzing system (NIH image 1.57). The infarct volume was calculated by taking the sum of the infarct areas of different brain slices times the thickness of the slices. The contribution of the edema volume to the infarct volume was corrected by subtracting the ipsilateral uninvolved hemisphere volume from the total contralateral hemisphere volume and dividing by the total contralateral hemisphere volume (Swanson et al., 1990; Lin et al., 1993). Infarct volumes in each group were assessed by analysis of variance followed by Bonferroni/Dunn test to ascertain significance between groups. Statistical significance was set at p < 0.05.
Histopathological and immunohistochemical evaluation
Twenty-four hours after MCAO with or without pretreatment by cisternal 0.5 μg of TNF, the animals were injected with a lethal dose of thiopental (150 mg/kg body mass i.p.) and fixed transcardially by perfusion with 4% paraformaldehyde in 150 mM NaCl and 50 mM sodium phosphate (pH 7.4). The brain was removed, stored in fixative for several hours, and then immersed in 20% sucrose solution in PBS for 1 day. The brains were then frozen in 2-methylbutane with dry ice. Coronal sections at the level of the dorsal hippocampus that represented the midportion of the infarcts were cut on a cryostat at 10 μm of thickness at −25°C. The sections were mounted onto silanized slides and were processed for histology and immunohistochemistry. The sections were stained with hematoxylin and eosin to confirm neuronal damage and leukocyte infiltration after ischemic insults. The extent of the infarction was measured using a computer-assisted image analyzing system (NIH image 1.57). The infarcted areas in each group were assessed by analysis of variance followed by Bonferroni/Dunn test to ascertain significance between groups. Statistical significance was set at p < 0.05.
The sections were immunohistochemically stained with a polyclonal rabbit anti-cow glial fibrillary acidic protein (GFAP) antibody, a rat anti-mouse CD11b antibody, a monoclonal rat anti-mouse mature macrophage, or a polyclonal rabbit anti-mouse TNF. After blocking of endogenous peroxidase activity with 3% hydrogen peroxide for 10 min at room temperature and blocking of nonspecific antibody binding by incubation with normal BSA for 20 min at room temperature, the sections were incubated for 1 h with a polyclonal antibody to GFAP raised in rabbits (Dako A/S, Glostrup, Denmark) diluted 1:100 in PBS, a rat anti-mouse CD1 lb antibody diluted 1:50 in PBS with 2% BSA (Serotec, Washington, D.C., U.S.A.), a monoclonal rat anti-mouse mature macrophage (F4/80) (Biosource International, Camarillo, MA, U.S.A.) diluted 1:10 in PBS, or a polyclonal rabbit anti-mouse TNF (Genzyme, Cambridge, MA, U.S.A.) diluted 1:100 in PBS. The sections were washed with PBS, treated with biotinylated anti-rabbit immunoglobulin [F(ab′)2] for GFAP and TNF or biotinylated F(ab′)2 anti-rat IgG for CD11b and F4/80 for 10 min, washed with PBS again, incubated with peroxidase-labeled streptavidin for 10 min, and incubated with 3,3′-diaminobenzidine solution. Finally, nuclear counterstaining was performed lightly with Mayer's hematoxylin. To compare the immunoreactivity of each brain slice semiquantitatively, the amount and concentration of antibodies and reagents and the duration of incubation were all identical. The mean optical densities of GFAP, CD11b, F4/80, and TNF and immunoreactivity in the infarcted area, periinfarct area, and contralateral cortex were measured using a computer-assisted image analyzing system (NIH image 1.57). Optical densities in each group were assessed by analysis of variance followed by Bonferroni/Dunn test to ascertain significance between groups. Statistical significance was set at p < 0.05. Immunohistochemical controls, completed on adjacent sections from brains of both groups of animals, consisted of the same reaction procedures described before in the absence of primary antibodies.
RESULTS
Preischemic physiological variables did not differ significantly among the various groups of animals (Table 1). There was also no significant elevation of body temperature or reduction of body weight following pretreatment with TNF at any of the doses used in this study.
Physiological variables in each experimental group
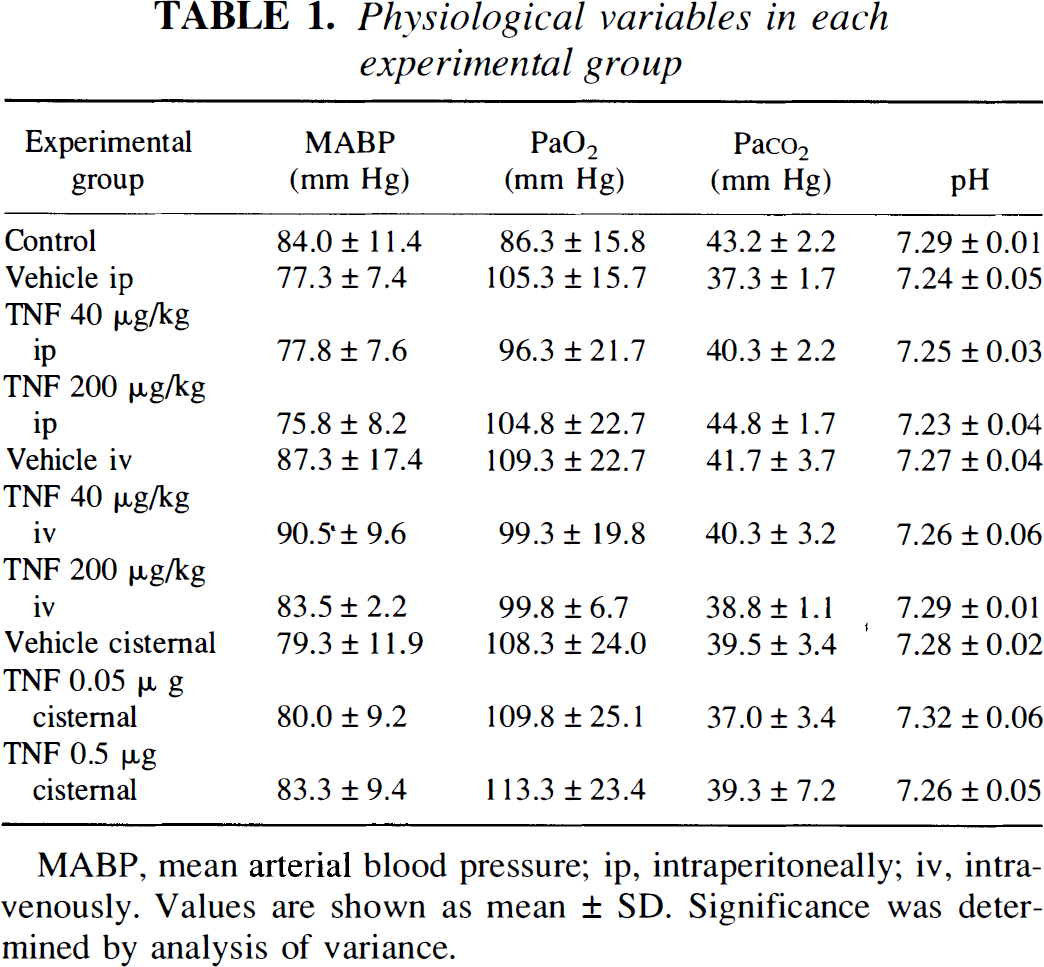
MABP, mean arterial blood pressure; ip, intraperitoneally; iv, intravenously. Values are shown as mean ± SD. Significance was determined by analysis of variance.
Volumes of brain damage (adjusted for the contribution of edema) were 29.5 ± 3.5 mm3 in control animals (n = 8), 27.2 ± 3.7 mm3 in vehicle i.p. (n = 6), 27.3 ± 3.0 mm3 in TNF 40 μg/kg i.p. (n = 6), 28.5 ± 2.7 mm3 in TNF 200 μg/kg i.p. (n = 5), 28.0 ± 4.7 mm3 in vehicle i.v. (n = 7), 29.2 ± 4.8 mm3 in TNF 40 μg/kg i.v. (n = 7), 31.2 ± 0.8 mm3 in TNF 200 μg/kg i.v. (n = 2), 30.3 ± 4.0 mm3 in vehicle cisternal (n = 7), 23.0 ± 9.9 mm3 in TNF 0.05 μg cisternal (n = 7), and 22.5 ± 3.6 mm3 in TNF 0.5 μg cisternal (n = 6) animals (values expressed as means ± SD) (Fig. 1). Pretreatment by cisternal injection of TNF produced a significant reduction in infarct volume as compared with the values of vehicle cisternal animals. The reduction of the infarct size was 26% of control values in TNF 0.5 μg cisternal animals and 24% in TNF 0.05 μg cisternal animals. No dose-dependent effect of TNF pretreatment was noted. There was no difference in the infarct size between TNF 40 μg/kg i.p., TNF 200 μg/kg i.p., and vehicle i.p. animals. There was also no difference between TNF 40 μg/kg i.v., TNF 200 μg/kg i.v., and vehicle i.v. animals. Representative photographs of brain slices in control vehicle cisternal animals and TNF 0.5 μg cisternal animals are shown in Fig. 2. In the pretreated animals that received intracisternal TNF, border zones between anterior and middle cerebral arteries appeared to become more viable as compared with control animals. In perfusion-fixed brain slices, the infarcted area (mm2) was also significantly (p < 0.01 by analysis of variance followed by Bonferroni/Dunn test) smaller in the pretreatment group animals compared with the control MCAO (Fig. 3). The reduction of the infarcted area was 27% of control values in the pretreated group animals.
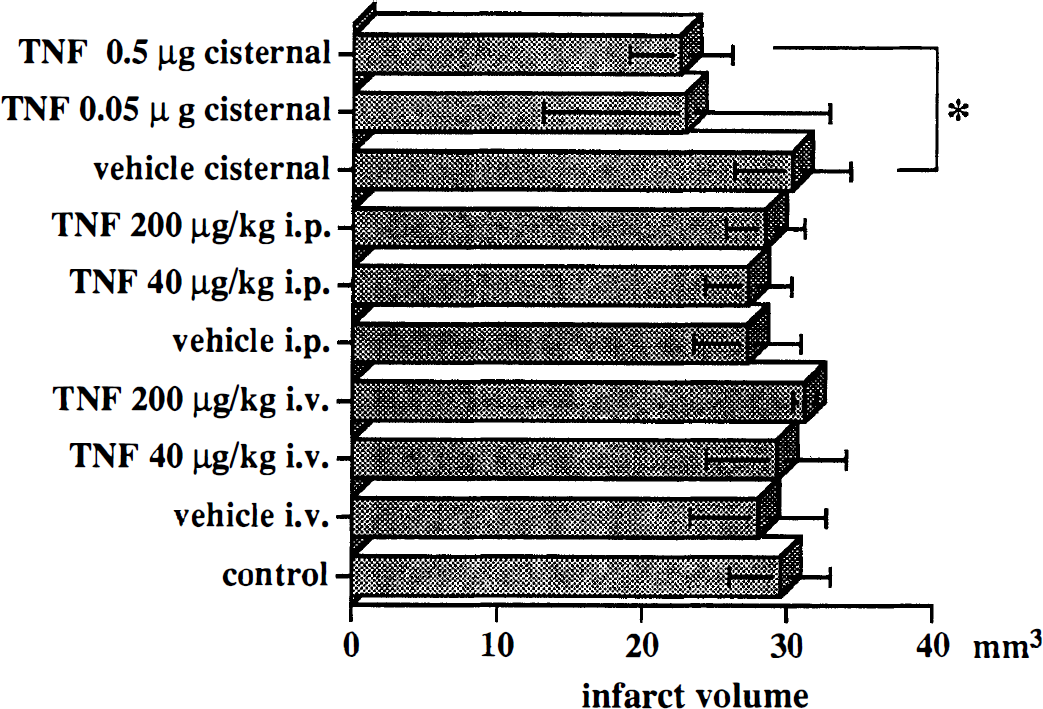
Infarct size in each experimental group. Average ± SD infarct volumes in each group are shown. Significance was determined by analysis of variance. *p < 0.01 when compared with the values of vehicle cisternal group. i.p., intraperitoneally; i.v. intravenously.
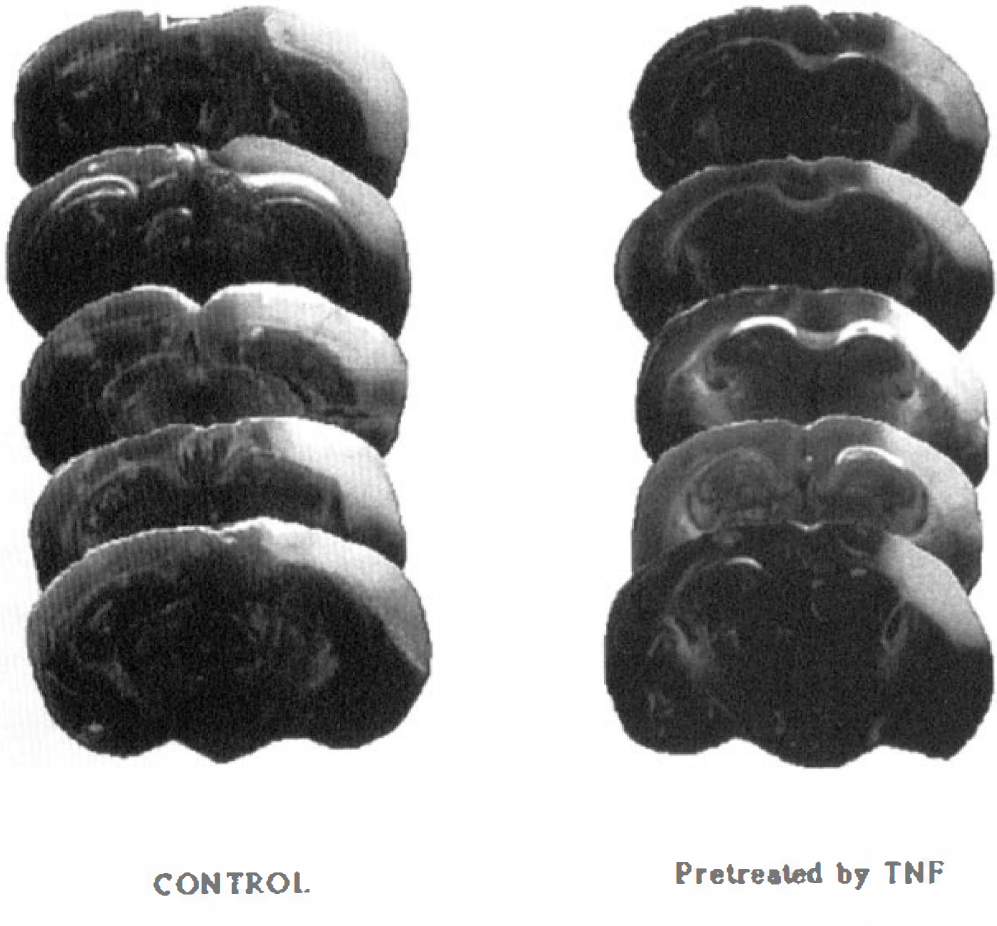
Coronal brain sections obtained 24 h after middle cerebral artery occlusion from each of five different animals in control vehicle cisternal and tumor necrosis factor-α 0.5 μg cisternal groups. Sections are from the level of dorsal hippocampus 5.0 mm from the frontal pole and are stained by 2,3,5-triphenyltetrazolium chloride.
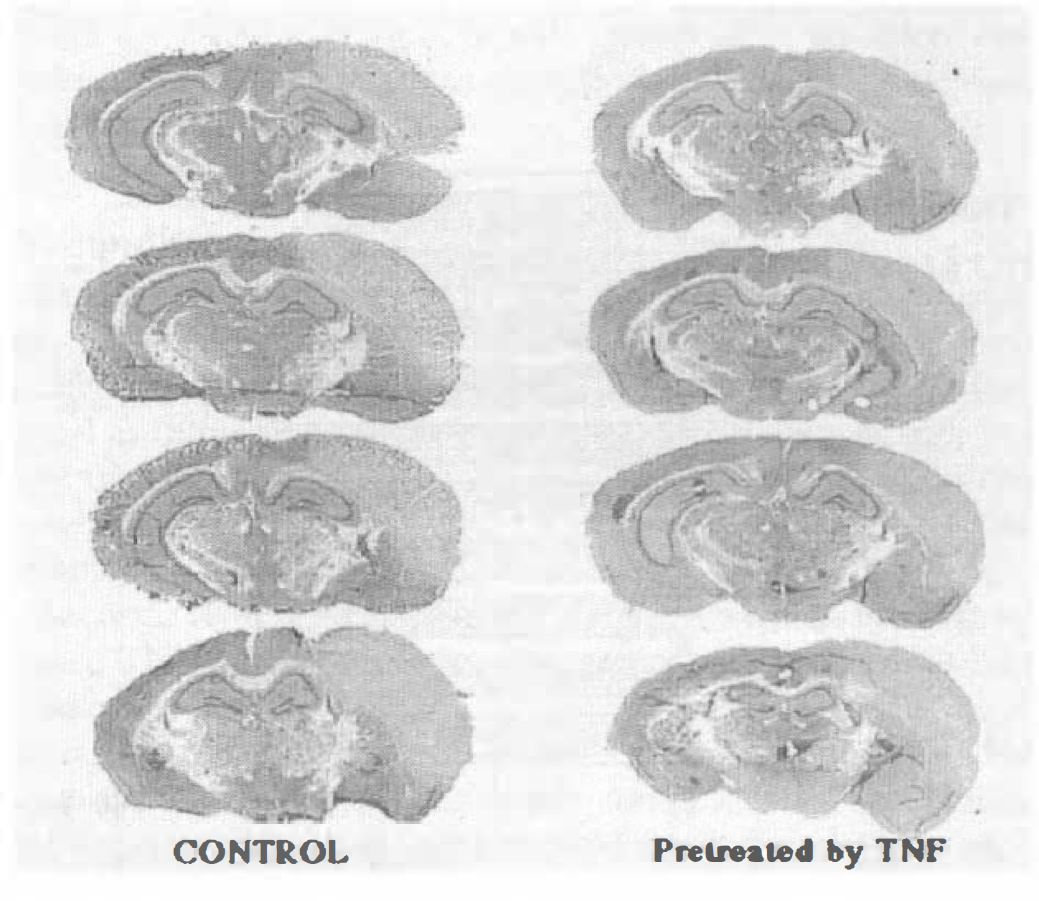
Coronal brain sections from each of four different animals in control vehicle cisternal and tumor necrosis factor-α 0.5 μg cisternal groups at the level of the dorsal hippocampus. The sections were stained with hematoxylin and eosin to confirm neuronal damage and leukocyte infiltration after ischemic insults.
Immunohistochemical analysis of brains subjected to 24 h of MCAO revealed a significant decrease in CD11b immunoreactivity after TNF pretreatment (n = 4) compared with control MCAO (n = 4) (Fig. 4). The representative immunohistochemical stainings for CD11b and GFAP of brain slices in control vehicle cisternal animals and TNF 0.5 μg cisternal animals are shown in Fig. 5. CD11b immunoreactivity was prominent in the ipsilateral corpus callosum of all control MCAO animals, but only one of four animals in the pretreatment group showed CD11b immunoreactivity within the corpus callosum. The CD11b immunostained cells were predominantly neutrophils and macrophages in the core of infarct and activated microglia in the periinfarct area as identified morphologically. The representative photomicrographs of CD11b immunostaining in the infarcted area in both TNF-pretreated and control groups are shown in Fig. 6. GFAP immunoreactivity increased significantly in the infarcted area in both TNF-pretreated and control groups with no significant difference between these two groups (Figs. 4 and 5). F4/80 immunoreactivity decreased significantly in both the core and the periinfarct areas in the TNF-pretreated group (n = 4) as compared with vehicle control group (n = 4) (Fig. 7). The F4/80 stains exclusively ramified microglial cells in the periinfarct area and ameboid macrophages/microglia in the core of infarct. The representative photomicrographs of F4/80 immunostaining in the periinfarct area in both TNF-pretreated and control groups are shown in Fig. 8. TNF immunoreactivity increased significantly after MCAO in the infarcted area in both TNF-pretreated and control groups with no significant difference between these two groups (Figs. 4 and 9). The type of TNF immunostained cell was exclusively macrophage and ameboid microglia by morphologic criteria. Some neurons were also positively immunostained for TNF. No discernible immunoreactivity was noted when the primary antibody was omitted from any of the staining procedures.
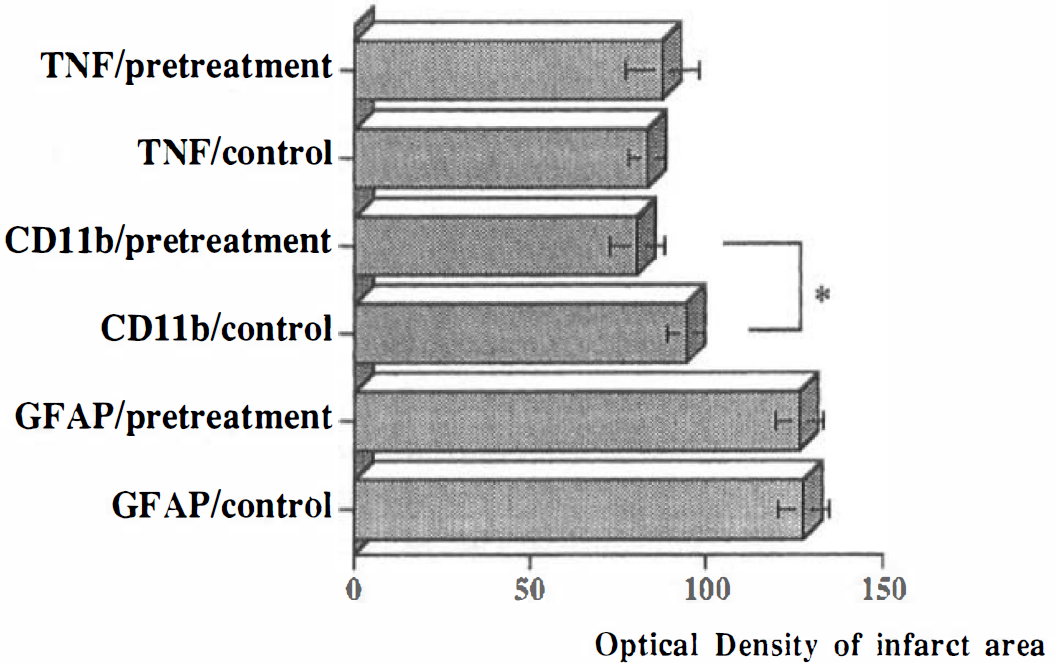
Tumor necrosis factor-α (TNF), CD11b, and glial fibrillary acidic protein immunoreactivity in the infarcted area of both TNF-pretreated (n = 4) and control (n = 4) animals. Optical densities in each group were determined and compared by analysis of variance followed by Bonferroni/Dunn test to ascertain significance between groups. Statistical significance was set at p < 0.05. *p < 0.05.
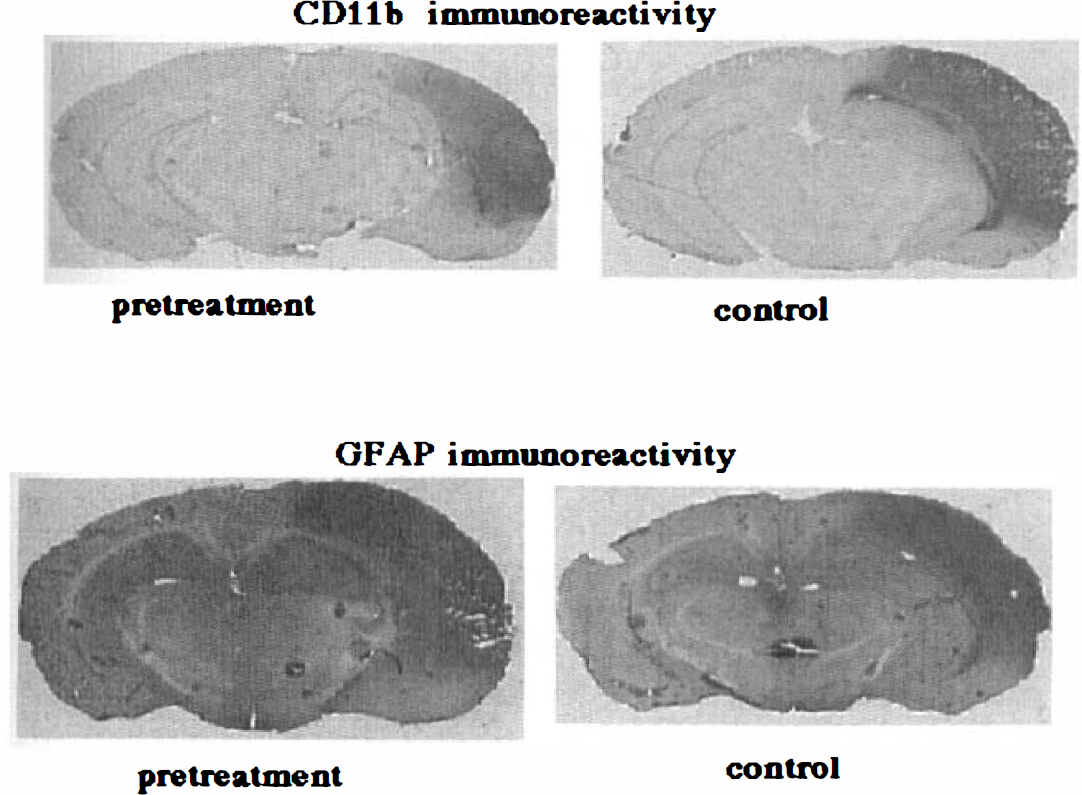
Representative immunohistochemical staining for CD11b and glial fibrillary acidic protein (GFAP) in brain slices from control vehicle cisternal animals and tumor necrosis factor-α (TNF) 0.5 μg cisternal animals. CD11b immunoreactivity was prominent in the ipsilateral corpus callosum of all control middle cerebral artery occlusion animals, but only one of four TNF-pretreated animals showed CD11b immunoreactivity within the corpus callosum. GFAP immunoreactivity increased significantly in the infarcted area in both TNF-pretreated and control groups with no significant difference between the two.
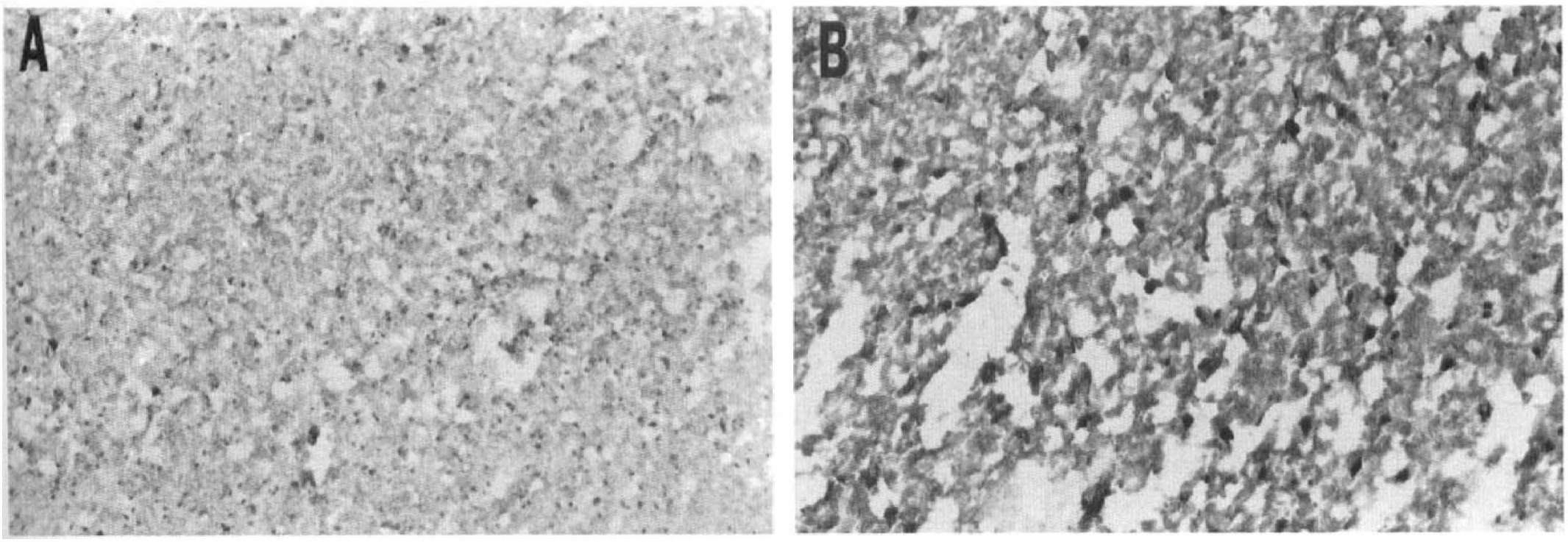
Photomicrographs of CD11b immunostaining in the core of the infarct of tumor necrosis factor-α (TNF)–pretreated (
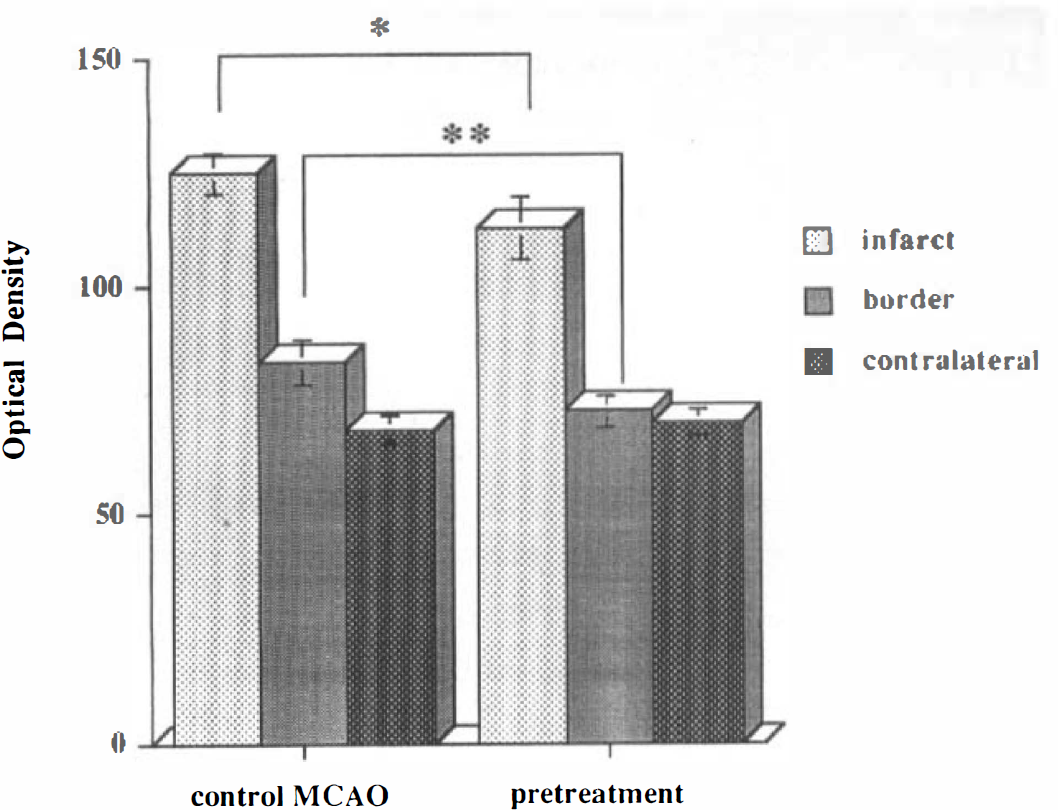
F4/80 immunoreactivity in the infarcted, periinfarct, and contralateral frontal cortex of both tumor necrosis factor-α (TNF)–pretreated (n = 4) and control (n = 4) animals. Optical densities in each group were compared by analysis of variance followed by the Bonferroni/Dunn test to ascertain significance between groups. Statistical significance was set at p < 0.05. A significant decrease in F4/80 immunoreactivity in pretreated animals was noted in the core and periinfarct area. *p < 0.05; **p < 0.01.
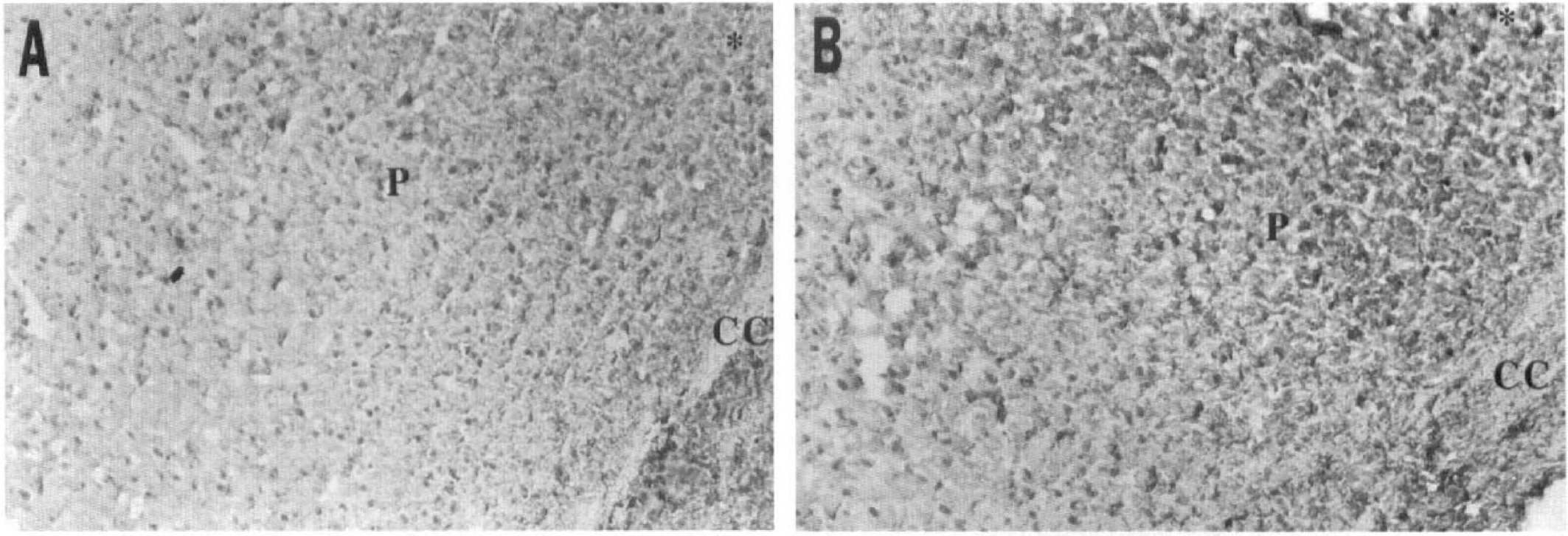
Photomicrographs of F4/80 immunostaining in the periinfarct area (P) of tumor necrosis factor-α (TNF)–pretreated (A) and control (B) animals. The F4/80 immunostained cells are activated macrophage/microglial cells. CC, corpus callosum; *, infarcted area. The sections are counterstained lightly with Mayer's hematoxylin. Original magnification, ×250.
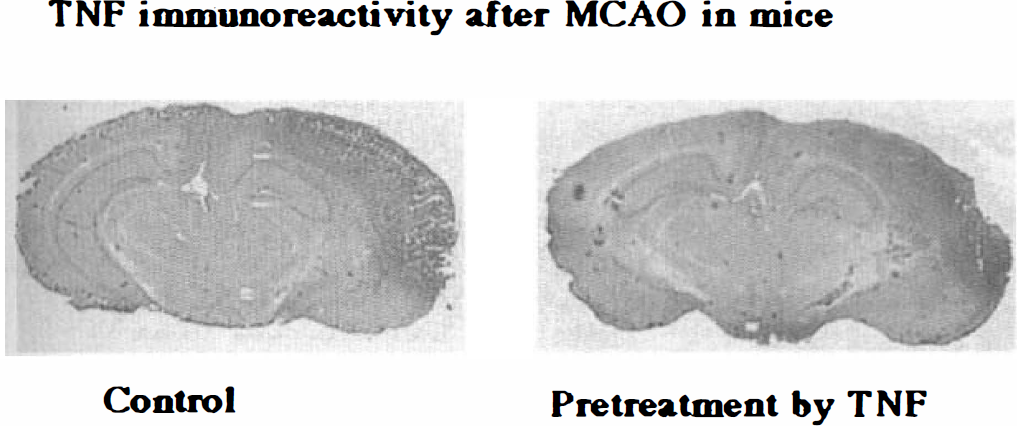
Representative immunohistochemical staining for tumor necrosis factor-α (TNF) in brain slices from control vehicle cisternal animals and TNF 0.5 μg cisternal animals. TNF immunoreactivity increased significantly after middle cerebral artery occlusion in the infarcted area in both TNF-pretreated and control groups with no significant difference between these two groups.
DISCUSSION
In this study, we investigated the effects of TNF pretreatment on the outcome of a subsequent MCAO in mice. We chose this model because distal MCAO can produce a relatively large cortical infarct with a small variation in BALB/C mice (Barone et al., 1993). Recombinant mouse TNF is a 17-kDa protein that penetrates the intact blood-brain barrier at low levels. It has been reported that intravenous or intraperitoneal injection of LPS can elicit a rapid increase of TNF in serum but no prominent increase in cerebrospinal fluid (Hallenbeck et al., 1991; Sirén et al., 1993). However, peripheral administration of TNF, which can stimulate blood cells and endothelium, had no significant protective effects after permanent MCAO in the doses used in this study. To evaluate the central effect of TNF, we administered TNF intrathecally. Cisternal injection of TNF induced significant protection against focal cerebral ischemia. We found no dose-dependent effect of TNF pretreatment. This may be because the doses chosen were maximal and a 26% reduction in infarct volume is all that TNF pretreatment can do in this model. The degree of neuroprotection, ∼25% reduction of control value, was smaller than that reported previously using the noncompetitive N-methyl-
Neurons, astrocytes, and microglial and endothelial cells have both types of TNF receptors, p55 and p75, and all could participate in TNF-induced tolerance (Armitage, 1994; Beutler and Van Huffel, 1994). Immunohistochemical analysis of TNF expression demonstrated that TNF immunoreactivity increased significantly after MCAO in the infarcted area. However, TNF immunoreactivity was not different between the control and tolerant groups. Thus, down-regulation of TNF expression in pretreated animals appears not to be responsible for the induced tolerance in this model. Secondary effects of TNF might play a role, however. One important inflammatory effect of TNF is up-regulation of adhesion molecules on endothelial cells (Briscoe et al., 1992; Feuerstein et al., 1994). This facilitates migration of activated leukocytes into the ischemic brain and might result in secondary damage following brain ischemia (Kochanek and Hallenbeck, 1992; Hallenbeck, 1996). In fact, TNF administered simultaneously with MCAO or 24 h before MCAO exacerbated focal ischemic injury (our unpublished data; Barone et al., 1996).
To elucidate the role of leukocytes in TNF-induced tolerance, we examined expression of a leukocyte integrin, Mac-1 (CD11b/CD18), in the infarcted hemisphere. The immunohistochemical analysis of CD11b showed a significant decrease in its immunoreactivity after pretreatment by cisternal TNF when compared with control MCAO. The rat anti-mouse CD11b antibody recognizes the murine α-integrin chain, which exists as a heterodimer with the β2-integrin (CD18) on neutrophils and monocytes (Springer, 1990). CD11b immunostaining suggests that leukocyte infiltration is reduced. In fact, a related finding is that leukocyte infiltration was lessened 48 h after cisternal injection of TNF (Fig. 6). This seems to be a possible contributing mechanism for the acquisition of tolerance after intracisternal TNF. Since CD11b/CD18 integrin is also expressed by macrophages and microglial cells, a role for these cells is also possible. Indeed the degree of immunoreactivity for F4/80 in microglial cells was lessened in the periinfarct area of tolerant animals (Figs. 7 and 8). Excessive activation of microglial cells might exacerbate outcome during cerebral ischemia. The role of activated astrocytes in acquisition of tolerance induced by TNF pretreatment may be small because the immunoreactivity for GFAP was very similar in the groups that received intracisternal TNF or vehicle prior to MCAO.
Our data, demonstrating a decrease of CD11b immunoreactivity and low leukocyte counts in the infarcted area of tolerant animals, are consistent with the recent observation that pretreatment of cultured endothelial cells with TNF for 72 h inhibited TNF-mediated activation of nuclear factor-κB (NF-κB) (Ferran et al., 1995). NF-κB is known to initiate transcription of the genes encoding ICAM-1 on endothelial cells, a counterreceptor for leukocyte integrins. NF-κB activation is associated with protein phosphorylation that results from TNF binding to its receptor (Laegreid et al., 1995). There are reports of TNF-mediated activation of serine kinases, microtubule-associated protein-2 kinases, cap-binding protein kinases, protein kinase C, and tyrosine kinases (Guy et al., 1991; Marino et al., 1991). These intracellular processes may participate in acquisition of tolerance against ischemia.
In summary, our results indicate a role for TNF in the induction of tolerance to ischemia. Elucidation of mechanisms that regulate the acquisition of such tolerance could guide efforts to develop effective measures to reduce ischemic injury.