
Abstract
The inhaled route is still a relatively novel route for delivering biologics and poses additional challenges to those encountered with inhaled small molecules, further complicating the design and interpretation of toxicology studies. A working group formed to summarize the current knowledge of inhaled biologics across industry and to analyze data collated from an anonymized cross-industry survey comprising 12 inhaled biologic case studies (18 individual inhalation toxicity studies on monoclonal antibodies, fragment antibodies, domain antibodies, oligonucleotides, and proteins/peptides). The output of this working group provides valuable insights into the issues faced when conducting toxicology studies with inhaled biologics, including common technical considerations on aerosol generation, use of young and sexually mature nonhuman primates, pharmacokinetic/pharmacodynamic modeling, exposure and immunogenicity assessment, maximum dose setting, and no observed adverse effect levels determination. Although the current data set is too small to allow firm conclusions, testing of novel biologics remains an active area and is likely to remain so for molecules where delivery via the inhaled route is beneficial. In the future, it is hoped others will continue to share their experiences and build on the conclusions of this review to further improve our understanding of these complex issues and, ultimately, facilitate the safe introduction of inhaled biologics into clinical use.
Keywords
Introduction
Following discussion of the challenges associated with developing inhaled biologics at a number of recent conferences held by learned societies, including the Association of Inhalation Toxicologists (AIT) and the British Society of Toxicological Pathology (BSTP) in 2017 to name just 2, it was recognized that the inhaled route is still a relatively novel route for delivering biologics. Surprisingly, there are only a few examples of approved inhaled biologics drugs, despite the fact that the first protein-based inhaled drugs were developed several decades ago (reviewed by Wolff 1 ). During discussions across different society conferences, it became apparent that many groups trying to develop inhaled biologics were experiencing similar problems in designing inhalation toxicology studies and interpreting the resultant findings. Furthermore, the lack of consensus on interpretation of findings meant it could be difficult to reach an agreement with Regulatory Agencies on a particular molecule's risk–benefit profile, which in turn could have prevented safe molecules from progressing into First-In-Human (FIH) clinical trials and beyond. It was recognized that there was a paucity of published data describing recent experience and a lack of consensus around what constitutes an expected and/or nonadverse response to inhaling proteinaceous materials or oligonucleotides regardless of therapeutic target, whether any responses would differ depending on modality, and/or what responses could be attributed to immunogenicity (and thus would not translate to the clinic) and, perhaps most importantly, how these can be differentiated from a true toxic, adverse response. This is further compounded by the lack of guidelines or guidance relevant to inhaled biologics specifically and thus relevant information has to be extrapolated from the International Committee on Harmonisation (ICH) guidance on biologics, ICHS6(R1) 2011, 2 or from established methods used for inhalation toxicology studies with low-molecular-weight chemicals. 3 –5 In November 2017 at the Annual Scientific Meeting of the BSTP entitled “The Pathology and Toxicology of Biologics and Biotherapeutics,” a proposal was put forward to produce a review to collate the current state of knowledge across the pharmaceutical and contract research organization (CRO) industries on inhaled biologics, and this proposal gained wide approval from several other professional societies.
Purpose
Subsequently, an anonymized cross-industry survey was conducted to gather case studies which could form the basis of this review. This survey is described in more detail in Hall et al (2021).
6
In total, data on 12 molecules were received (referred to as case studies and covering 18 individual inhalation toxicity studies), 10 of which had respiratory system targets, collected from 8 pharmaceutical companies, and these represent the current thinking and working practices of many of the experts involved in developing large molecules (biologics) as inhaled therapies worldwide. The 12 molecules comprised 5 peptide/proteins, 3 domain antibodies (Dabs), 2 fragment antibodies (Fabs), 1 monoclonal antibody (mAb), and 1 oligonucleotide, representing a reasonable spectrum of the types of biologics typically found in current drug development (see Table 1). A working group was formed of representatives from each of the companies supplying either case studies and/or subject matter expertise. The resulting article focuses on the technical, toxicological, pharmacokinetic/pharmacodynamic (PKPD), and immunogenicity aspects of the preclinical studies required to support clinical testing of inhaled biologics. A sister review focusing on the pathology aspects is published alongside this toxicology review.
6
The working group collaborated, using the case study data set where appropriate, to gather opinion and increase understanding by asking the following questions: What technical difficulties are associated with inhaled biologics toxicology studies? What study design aspects are specific to inhaled biologics studies? What are the characteristic responses to inhaled biologics and how can exposure be demonstrated at the site of administration? How can we differentiate between a physiological response to foreign biological material, an immunogenic response to proteinaceous material, and true, human translatable toxicity? What particular PKPD aspects are important for and/or peculiar to inhaled biologics? How are no observed adverse effect levels (NOAELs) defined for inhaled biologics (vs small molecules).
Summary of Study Design Parameters From the Inhalation Toxicity Case Studies.a
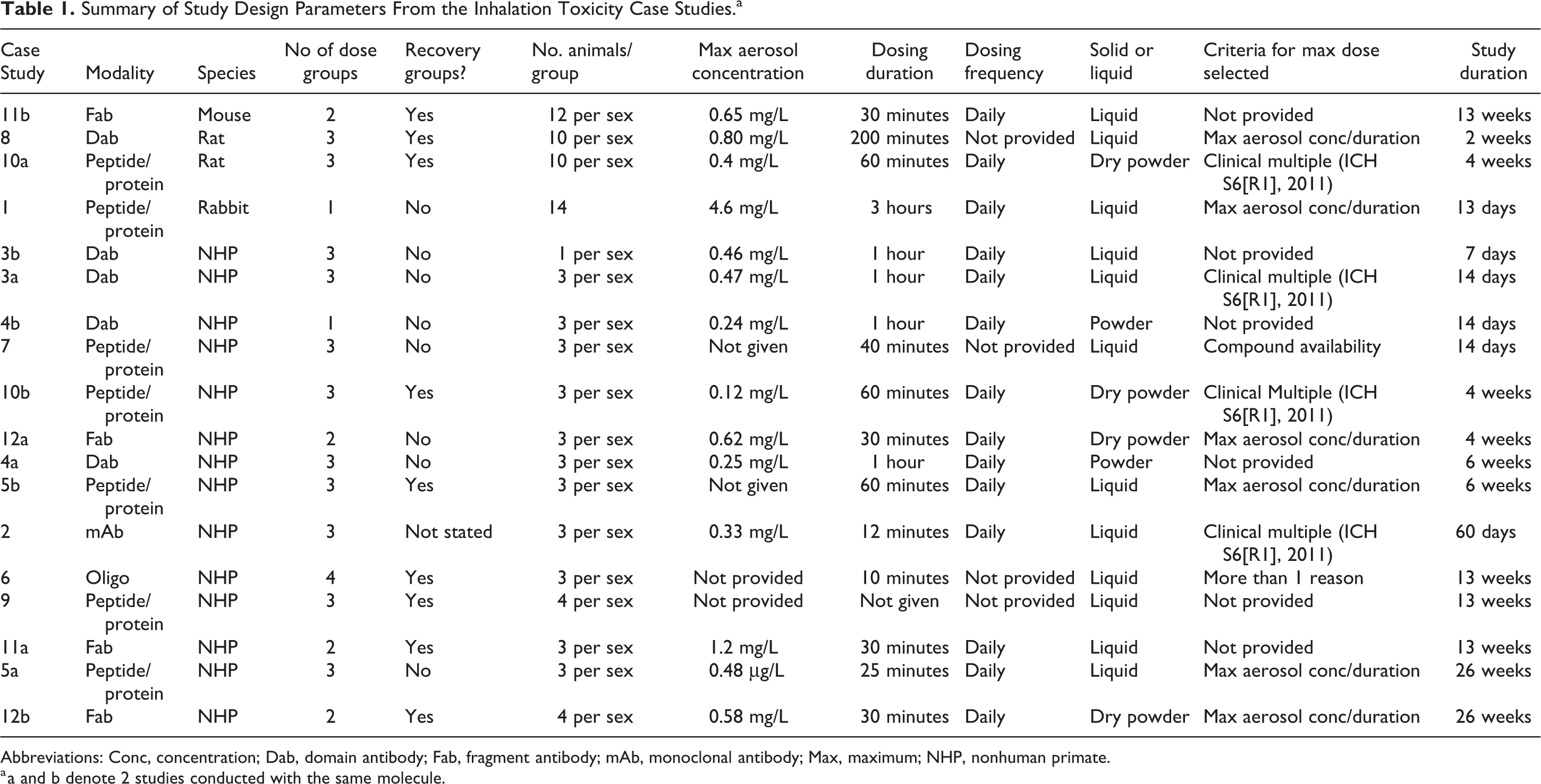
Abbreviations: Conc, concentration; Dab, domain antibody; Fab, fragment antibody; mAb, monoclonal antibody; Max, maximum; NHP, nonhuman primate.
a a and b denote 2 studies conducted with the same molecule.
The goals of this toxicology-focused article (and its associated pathology-focused article) are to serve as sources of collective information, knowledge, and experience to aid those continuing to work in this field. This includes companies and individuals designing inhalation toxicity studies/programs to support safe clinical use and regulatory agencies worldwide who are responsible for reviewing the study data to ensure patient safety. More general considerations on the conduct of inhalation toxicity studies are available elsewhere 3 –5 and will not be discussed further. To our knowledge, the industry survey is the largest collation of technical data from unpublished inhalation toxicology studies conducted on biologics to date and so provides a relatively rich data source. It is acknowledged that the data set comprises 12 case studies (Table 1) and thus it is hoped that others will continue to share their experiences to expand this data set. Ultimately, such data will improve our understanding of these complex toxicological issues and enable inhaled biologics to safely enter clinical use and provide novel pharmaceuticals to patients in need where the inhaled route is the optimal or only route of administration.
Firstly, it is important to define what is meant by a biologic in the context of this publication. Biologics (also referred to as biotechnology-derived pharmaceuticals, biotherapeutics, or biopharmaceuticals) are therapeutic drug products manufactured in, extracted from, or semisynthesized from biological sources or expression systems, such as bacterial, yeast, insect, plant, or mammalian cells. In the broadest sense, biopharmaceuticals include vaccines, blood, blood components, cytokines, recombinant plasma factors, growth factors, fusion proteins, enzymes, receptors, hormones, oligonucleotides, mAbs, and antibody-derived molecules (antibody fragments, single-chain variable fragments, single Dabs). For the purposes of this publication, the biologics assessed were mAbs or antibody-derived fragments, peptides/proteins, and oligonucleotides. 2,7 –9 Subsequently, any future use of the term biologics herein will refer to this subset of biopharmaceuticals only. The only approved inhaled biologics are Pulmozyme (DNAse), Exubera (insulin, withdrawn from the market), and Afrezza (insulin), and all would belong to the class of peptides/proteins in this article.
As biologics are subject to proteolytic degradation, particularly in the gastrointestinal tract, delivery of these therapies has historically been limited to parenteral routes of administration, primarily intravenous or subcutaneous injection. However, inhaled delivery of biologics can be advantageous for a number of reasons. For systemic therapeutic targets, the inhaled route can be advantageous for any molecule type due to the large absorptive area of the lung, rapid absorption potential (depending on the modality), potential for a rapid onset of activity if the target is local, ease of administration, and potential self-administration by the patient. Moreover, there are a wide variety of delivery options, for example, nebulizers and dry powder inhalers, which allow for optimized formulation development. 7 For therapeutic targets in the respiratory tract, inhalation maximizes delivery of the drug to the target organ and reduces the systemic exposure, thus minimizing the potential for systemic toxicity, as the dose administered directly to the lung is typically much lower than an equivalent lung dose administered systemically. However, in contrast to the benefits, inhalation of a foreign biologic has a higher potential to produce antidrug antibodies (ADAs) than administration via a parental route, owing to the presence of mucosal tissues. 10
Nonclinical assessments of drug candidates of any modality are conducted to determine pharmacology, pharmacokinetics, and the safety profile of the drug product (active ingredient and/or vehicle or formulation) in animals, typically such assessments comprise a battery of in vitro, ex vivo, and in vivo tests, and this is unchanged for biologics administered via the inhalation route. Of particular importance for biologics, a good understanding of the manufacturing method, purity, sequence, structure, immunoglobulin (Ig) class/isotype, anticipated pharmacological effects (including target biology), and the intended patient population and clinical dosing plan is also required. 11 The data generated are then integrated to inform the safe starting dose for FIH studies, dose escalation and maximum dose or exposure and safety, and pharmacodynamic markers for clinical monitoring. If there are 2 pharmacologically relevant species for a biologic (1 rodent and 1 nonrodent), then both species should be used for short-term studies, but this could be reduced to a single species for longer-term studies if the findings from both species are similar. The use of 1 species for all toxicity studies for mAbs, their derivatives, and Fabs is justified when the clinical candidate is pharmacologically active in only 1 species. Generation of a homologous product to allow testing in 2 species is not recommended for a variety of reasons. 2 As biologics are highly specific for their target, any true toxicity observed is most often related to exaggerated or prolonged pharmacology, to effects secondary to induction of ADAs, or to target expression on multiple cells or tissues, other than the tissue of interest. Such high specificity should be confirmed for each individual molecule during development. For clinical testing of longer durations and for marketing authorization, toxicity studies assessing chronic dosing (26 weeks), reproductive end points, and developmental toxicity evaluations are subsequently performed. As many biopharmaceuticals are only pharmacologically active in NHPs, in vivo carcinogenicity assessments are often not feasible and written assessments evaluating the carcinogenicity risk of the candidate are submitted for health authority review and endorsement. 2 The focus of this working group was solely on repeat-dose toxicity studies conducted with inhaled biologics regardless of the duration of dosing, and the other study types are not discussed further.
The data generated from the 18 anonymized toxicology studies were evaluated with the following 5 questions in mind. Additional expert opinion was provided by the subject matter experts in the working group. The key questions are discussed in turn.
Question 1: What Technical Difficulties Are Associated With Inhaled Biologics Toxicology Studies?
For many of the technical aspects of conducting an inhalation toxicity study with biologics, the considerations are the same as for a novel small molecule. However, for each of these aspects, there are specific issues associated with the use of proteinaceous material that may require specific modifications to the dosing equipment and/or procedure. These need to be considered on a case-by-case basis.
Definition of “dose”
To understand the importance of maximum aerosol concentration, it is important to define “dose” in the context of an inhalation toxicology study. Unlike most other modes of administration, the dose in an inhalation toxicology study is not the amount/weight of the drug in solution administered to an animal or the concentration of drug in the air for inhalation—this is referred to as the nominal dose. Rather the delivered dose or pulmonary deposited dose is preferentially referred to and these are estimated doses. The delivered dose is generally calculated using the equation presented by the AIT
3
and represents the inhaled dose at the breathing zone of the animal: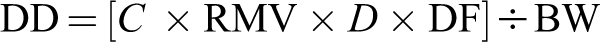
where DD = delivered dose (mg/kg), C = concentration of substance in air (mg/L), RMV = respiratory minute volume or the volume of air inhaled in 1 minute (L/min), D = duration of exposure (minute), DF = deposition factor and BW = body weight.
The RMV is normally calculated using the body weight of the animal and cannot be changed outside the normal variability for the species in question. This requires special consideration for sexually mature primates, which are significantly heavier than standard aged monkeys and for which the standard RMV allometric scaling calculations are flawed (see Special considerations for use of sexually mature primates below). For the determination of delivered dose, the deposition factor is set to 1.0. For the determination of the pulmonary deposited dose, the same equation is used but the deposition factor is changed based on data indicating that 10% of any aerosol concentration will reach the lung of a rodent and 25% will reach the lung of a nonrodent. 5,12 Thus, the deposition factor would be 0.1 and 0.25 for rodent and nonrodent, respectively. For humans, this is 1.0. As RMV and DF are inflexible factors in the equation, dosing duration and aerosol concentration become the 2 key determinants of the delivered dose administered in an inhalation toxicology study and so are intrinsically linked when establishing the delivered dose or pulmonary deposited dose. It is clear from the equation that, for any given delivered dose, the higher the aerosol concentration achieved, the shorter the dose duration needs to be and, conversely, the lower the aerosol concentration, the longer the dose duration needs to be, provided the inhalable fraction of the aerosol concentration remains in the acceptable range and similar to any previous studies conducted.
Aerosol generation of a biologic
The aerosol generated for inhalation of a novel biologic will be governed by 3 aspects—the physical state of the material, that is, solid (dry powder blend or neat material) or liquid; the concentration of the biologic in its formulation; and the ability to generate a stable respirable particle size. Unlike small molecules, novel biologic drug substances usually require complex formulations to ensure molecular stability and retention of the correct physicochemical properties. These are generally either solutions of the molecule in an appropriate buffer (with or without additional excipients) or (more typically) a spray dried powder blend, but there are also other novel particle engineering techniques currently under development. In many cases, the biologic is already formulated at the maximum practical concentration in the buffer and higher concentrations would compromise the protein, either structurally, that is, change its folding, encourage aggregation or precipitation; or alter its binding affinity. As a result, the formulation can have considerable influence on the achievable aerosol concentration, as it may not be technically possible to generate the required high aerosol concentration to provide a sufficiently large dose multiple of the intended clinical dose. In addition, it may be difficult to generate stable, low aerosol concentrations or, if a liquid, the available formulation may be too viscous to allow reproducible and stable aerosol concentrations to be generated repeatedly over the required dosing duration. These issues will require extensive discussion with manufacturing and formulation representatives for the molecule to evaluate whether any alternative options are available. Preferentially, the toxicology studies should be conducted using the same formulation as will be used in the clinic. Some dry powder formulations of biologics may be particularly insoluble, in which case it may be possible to suspend in a simple buffer and dose using a nebulizer. However, this is not recommended and should only be used if there are no viable alternatives.
There are no specific guidance or guidelines on conducting an inhalation toxicology study with a novel biologic. At the time these inhaled studies were performed, it was generally accepted that the majority of the particles in the aerosol for any modality should be between 1 and 5 µm in diameter (mass median aerosol diameter, MMAD) with a geometric standard deviation (GSD) less than 3, in line with the Organization for Economic Co-operation and Development (OECD) test guidelines (TG) for inhalation toxicology testing of novel chemicals (TG412 and TG413, first issued in 1981). These OECD TGs are aimed at exposure of rodents to inhaled chemicals, although the TGs do allow a different MMAD to be used for nonrodents, provided sufficient exposure of the lower respiratory tract and the size range chosen can be justified. Both TGs were reissued in 2018 13,14 and now request the MMAD to be ≤2 µm for rodents to accommodate nanoparticles. Although the OECD TGs do not apply to pharmaceuticals, it is possible they could influence the application of MMAD requirements in all inhalation studies, including pharmaceutical inhalation studies, despite the low feasibility of routinely achieving such a small particle size. However, in 2018, Food and Drug Administration (FDA) stated that these OECD TGs are unsuitable for the testing of novel inhaled pharmaceuticals, which would indicate this is less likely to be an issue in reality. 15 General requirements on particle size are discussed in more detail in the publication by Tepper et al 4 and so will not be discussed further here, as the principles are the same for inhaled small molecules and inhaled biologics regardless of the required MMAD.
In the 18 case studies collated for this industry survey, 16 reported the maximum aerosol concentration used (Table 1). Regardless of the physical state of the molecule, maximum aerosol concentrations ranged from 0.12 to 1.2 mg/L and are in line with other publications on maximum aerosol concentrations for inhalation toxicity studies.
4,16
There was a single use of a higher concentration of almost 5 mg/L (Table 1), however this was unusual and was only used on a short-term study (13 days). For all the biologics collated in the case studies, the MMAD and GSD were measured and confirmed to be in the respirable range as defined at the time the studies were conducted (MMAD ≤ 5 µm and GSD < 3). A. Solids
As for any solid material, the primary limitation will be the ability to generate a respirable particle size at the desired aerosol concentration. For repeat dose studies in any species, 2 mg/L is normally considered the appropriate maximum concentration. With the exception of 1 case study of very short duration (discussed above), none of the case studies used aerosol concentrations of the biologic in excess of 2 mg/L, with aerosol concentrations ranging from 0.12 to 1.2 mg/L. However, for at least 2 of the case studies, the biologic was dosed as part of a dry powder formulation including excipients, meaning the aerosol concentration of the formulation was higher than that reported for the biologic. Care should be taken that the particle sizes don’t differ between the active and the excipients when dosing a formulation, as this could alter the relative dose of each the animal receives. High powder aerosol concentrations could also cause upper respiratory deposition, with the animal’s nostrils showing evidence of powder residue which could cause physical irritation and distress. In addition, the animal may lick their nostrils leading to unintentional and usually unwanted oral exposure, particularly of any excipients as biologics are typically degraded in the gastrointestinal tract before absorption can occur. Such effects are not desirable and are likely to reduce the pulmonary dose reaching the animal or result in localized findings which are solely attributable to powder load or irritation rather than a true treatment- or molecule-related effect.
Many biologic materials are not stable at room temperature for long periods and should be stored at low temperatures. This may require the preparation of smaller aliquots of material, in order to avoid repeated freeze–thaw cycles, which places additional emphasis on ensuring batch-to-batch comparability for the characterization, integrity, and potency of the biologic during the study. In rare cases, certain parts of the exposure system may need to be stored under refrigerated conditions between dosing days. Additionally, biologics also tend to require low humidity storage conditions and should be stored under desiccant or nitrogen to avoid water condensate. B. Liquids
Nebulization is usually the simplest method of liquid aerosol generation. There are many nebulizers available using 3 main methods to deploy the droplets: (1) jet nebulizers, which rely on compressed air to generate shear force to separate the liquid into fine droplets; (2) vibrating mesh nebulizers, which provide a gentler way of creating droplets using a vibrating mesh to literally sieve out droplets, but which may become blocked when using suspensions and require frequent cleaning, sometimes during use; and (3) ultrasonic nebulizers, which use ultrasonic sound waves to break liquid into tiny particles, which may increase the temperature of the formulation and denature delicate molecules. If it is not possible to nebulize the material, for example, if it is highly viscous, it may be necessary to use a jet atomizer, which has other issues related to low generation efficiency, very high material use, and production of a high proportion of large droplets, which may need to be elutriated pre-exposure as they are not respirable. In all cases, thorough investigation of the selected nebulization method is required to ensure that the biologic material is not being degraded by the nebulization process and that a similar activity profile is maintained.
A final option, for both solids and liquids, is use of the inhaler device intended for clinical use, but this inherently limits the flexibility to administer larger doses to the nonclinical species as the dose emitted is already pre-established. Such a device is also likely to be associated with various other problems, such as clogging of the device after multiple uses and selection of a suitable solvent. It should be clear, however, that it is rarely necessary to use the clinical device, as any method that delivers the proper concentration/dose and is respirable and stable should be sufficient.
For any liquid administration, it is difficult to control the droplet size of the aerosol and, while droplets can be elutriated to remove large, nonrespirable particles, this can lead to significant loss of material during dosing, necessitating manufacture of an even larger quantity of material to meet the objectives of the study.
Dosing frequency
For biologics with a larger molecular weight, such as full-length mAbs, once weekly dosing may be sufficient if the target is systemic, as these molecules are known to have a long serum half-life. It is not known whether the same would be true for a lung half-life, but interestingly, the only mAb assessed in the case studies used a daily dosing regimen. For smaller molecular weight biotherapeutics such as Fabs, peptides, or oligonucleotides, the half-life in serum, and it is assumed the lung, is much shorter and daily dosing is likely to be required. Typically for any novel pharmaceutical, the dosing frequency used in the toxicology studies should mimic or exceed that intended for use in the clinic but may be altered based on the need to achieve a certain dose to provide the required 10-fold and 5- to 6-fold pulmonary deposited dose multiples for rodents and nonrodents, respectively, requested by FDA. 4 Other countries may accept a safety margin less than 10-fold between the nonclinical and clinical delivered doses, but as most molecules are developed for international use, the FDA requirement is often the more conservative default. If the dosing frequency is increased, particularly to more than once per day, it is important to establish the tolerability of both the animal, in terms of stress and clinical observations, and the equipment, in terms of the ability to generate a stable, reproducible, and respirable aerosol particle, for the total dosing duration on each dosing day. The practical feasibility is also important, in terms of the ability to complete dosing within a day while also allowing time for the other required animal study activities, such as multiple blood samplings for hematology, clinical chemistry, pharmacodynamics, biomarkers, and toxicokinetics, as well as any functional/pharmacology parameters that need to be assessed. Generally, it is recommended that dosing is not conducted more than once daily, unless necessary to generate a suitable safety margin or to support the dosing regimen intended for clinical use.
In the case studies, the frequency of dosing was provided for 14 of the 18 studies and all of these used once daily dosing, confirming that dosing more than once per day is uncommon.
Criteria for setting the maximum dose assessed
One of the key considerations for a well-designed good laboratory practice (GLP) inhalation toxicology study to support First-in-Human (FIH) exposure is selecting the maximum dose level and the lower dose range, assessed in the pivotal toxicology studies. For small molecules, it is common to assign the maximum dose based on toxicities observed in previous dose-range finding studies. However, biologics are often nontoxic at therapeutic and significantly higher doses, and any toxicity observed is frequently associated with exaggerated pharmacology. As a result, rather than being based on toxicity, the maximum dose is selected based on scientific judgment following evaluation of a multitude of factors, including the ability to routinely dose the required aerosol over the duration of the study without inducing undue stress or irrelevant toxicities, for example, particle overload leading to localized irritation. Attempting to set a maximum tolerated dose for a biologic, as described in OECD TG412 or TG413, 13,14 is not appropriate in the majority of cases, as these guidelines are intended to guide the hazard identification of novel small molecular weight chemicals. Similarly, use of the guidance for maximal dose of a novel small molecular weight pharmaceutical as discussed in ICH M3(R2) 2009 is 2 not appropriate for inhaled biologics.
The following factors should be evaluated when assigning the maximum dose of a novel biologic: A. Regulatory guidelines
There are no guidelines or guidance specific to administration of biologics via inhalation. ICH S6(R1) 2011 2 provides guidance on dose setting for biologics administered via parenteral routes, and the general premise of this document is also applicable to inhaled biologics, for example, mAbs and their derivatives and Fabs. The inhaled biologic should be tested in at least 1 pharmacologically relevant species, that is, one for which there is a comparable affinity and potency against the native target (typically less than 10-fold difference from the human affinity/potency). 2 For mAbs, mAb derivatives, and Fabs, testing in a second species should only be conducted if it is also pharmacologically relevant. 2 Doses can be set using saturation of target binding and/or PKPD approaches (eg, simple exposure–response relationships or more complex modeling and simulation approaches). These assist in high dose selection by identifying doses which provide the maximum intended pharmacological effect in the animal species and an acceptable exposure multiple over the maximum exposure intended in the clinic. Although this can sometimes be determined via PKPD modeling of data from other species or can be estimated based on other data, in general this is often not known at the time the first toxicology studies are initiated.
In the industry survey, the criteria for setting the maximum dose was answered in one of the 4 ways: (1) the maximum duration and aerosol concentration for the species; (2) a multiple of the clinical dose as stated in ICH S6 (R1) 2011
2
; (3) formulation and/or compound availability; or (4) unanswered. For 5 of 11 case studies, the maximum dose was set using ICH S6 (R1) 2011
2
criteria for clinical dose multiples (Table 1). B. The maximum daily dose duration
When considering the daily duration of the exposure to a novel biologic in any given study, the first priority should be animal welfare. All CROs and companies that partake in animal testing are regulated by local Government, which has policies to ensure the health and welfare of animals used in such studies. These designs are also reviewed by internal animal welfare ethical review boards or local animal welfare groups, such as the Institutional Animal Care and Use Committee in the United States. In general, it is not expected that administering a novel biologic would have any special impact on animal welfare, as it is usually delivered in a similar format and for a similar dosing duration as small molecules. However, it could become problematic if the dosing duration needs to be extended beyond the normal limit for the testing facility in an attempt to achieve a higher delivered dose when the aerosol concentration has already been maximized.
Rodents are typically dosed nose only (also described as snout only), while restrained in a tube directly attached to the central aerosol chamber. With minimal training in the restraint tubes, the animals adapt to the restraint and often sleep during longer duration exposures. For primates, the animals are typically dosed while restrained in chairs. Aerosol exposure is provided via a face mask attached by tubing to a central aerosol chamber. Consideration of the number of staff, the number of animals being dosed simultaneously, animal distraction techniques, chair restraint methodology, laboratory equipment and facilities, and number of acclimatization sessions, all influence the success of the dosing. For these reasons, dosing of rats for small molecules can be extended to as long as 4 hours daily (in line with TG412 and TG413 13,14 ) at some exposure facilities, whereas for primates, a maximum duration of 1 to 2 hours is more typical. 4 Particularly for sexually mature primates that are much larger and much stronger, this duration may need to be shortened as they are less likely to tolerate restraint in the dosing apparatus for longer than 30 minutes, particularly if daily dosing is involved (see Special considerations for use of sexually mature primates below). If large powder doses are administered to the animal, it can result in increased drinking and grooming, or for nonrodents, it can induce responses such as coughing, inspiratory paroxysmal respiration, or vomiting. Any restriction or deviation to an animal’s natural behavior, such as eating, drinking, urination, and defecation, can cause distress and may compromise the data obtained, compromising interpretation of data or erroneously being considered a treatment-related effect.
In the industry survey, the maximum dosing durations were typically less than 1 hour for both rodents and monkeys and were used as the rationale for maximum dose setting in 5 of 11 case studies. C. Maximum technically feasible dose
In contrast to recommendations within OECD TG 412 and OCED TG 413 13,14 but in line with biologics administered via other routes, administration of the highest possible dose is not necessary if appropriate safety margins can be achieved at lower doses as per ICH S6(R1) 2011. 2 However, in some cases, the optimum maximum dose level to provide the required safety margins cannot be achieved and, in such cases, a maximum technically feasible dose may be administered, in line with ICH M3(R2) 2009, 2 where the maximum duration of exposure and maximum aerosol concentration per dosing occasion have been reached. If a maximum feasible dose is selected, a scientific justification for the dose selection and projected multiples of human exposure should be provided as per ICH S6 (R1) 2011. 2 This dose should not be an arbitrary high multiple of clinical doses or a dose so high that it will yield toxicity related to high doses of protein/particle and/or subsequent saturation of homeostatic mechanisms such as particle clearance, as such doses will yield toxicity with little or no clinical relevance and confound interpretation of the study. One potential way to achieve a higher dose, especially if the inhaled drug is intended to act systemically, is to supplement the inhalation dose with concurrent systemic dosing. However, this also means toxicities cannot be attributed to specific dose routes and it may be more informative to perform separate dose groups within the study or as separate systemic toxicity studies on the biologic if this is necessary.
One case study of the 11 who provided this information cited formulation and compound availability as the criteria for setting the maximum dose. This is an atypical justification for the maximum dose—this molecule had been submitted to health authorities for clinical use but it is not known whether concerns were raised regarding maximum dose assessed.
Pharmacological activity
For any proteinaceous material delivered via inhalation, it is important to demonstrate that the biologic remains active and respirable during the aerosol generation and dosing process to confirm the animal is administered an active drug. Proteinaceous materials may be less tolerant of the harsh conditions associated with aerosol generation. For example, the physical methods used to generate dry powder aerosols, such as rotating brush generators which scrape the surface of tightly packed material to flick particles into the air to make an aerosol; or either jet nebulizers, which apply high shear forces and recirculate liquid, or vibrating mesh nebulizers, where the plate used to generate the aerosol can be become warm and thus potentially denature the protein. It is also important to determine whether the particle becomes aggregated during aerosol generation, as these may cause it to lose potency, potentially promote immunogenicity and/or become larger than the generally accepted mean respirable diameter range or MMAD.
In the 12 case study responses to a question on pharmacological activity, 9 out of the 18 individual inhalation studies conducted included an assessment of pharmacological activity (2 using a potency assay, 2 using a binding assay, and 5 indicating pharmacological activity were assessed but no information on how this was determined). The remaining 4 studies did not measure pharmacological activity of the molecule itself following aerosol generation/administration, of which 2 confirmed activity of the molecule by observing biomarker changes related to the pharmacology of the molecule. The other 2 studies did not state how pharmacological activity was confirmed. It is not known whether pharmacological activity following aerosol generation had been confirmed in previous studies was not reported or was not assessed at all.
Question 2: What Study Design Aspects Are Specific to Inhaled Biologics Studies?
The key aspects that should be considered before finalizing the study design for an inhalation toxicity study on a novel biologic and where these may differ from that for a small molecule are discussed below.
Regulatory guidelines/guidance
There are no guidelines or guidance, either local or international, that specifically address the development of inhaled biologics of any modality. Rather, other established guidelines relevant to biologics or to the end point under consideration are considered and specific elements applied if considered relevant. 16
The main guideline relevant to development of an inhaled biopharmaceutical is ICH S6(R1): Preclinical Safety Evaluation of Biotechnology-derived Pharmaceuticals, issued in 2011. 2 This guideline is commonly used in conjunction with ICH M3(R2): Nonclinical Safety Studies for the Conduct of Human Clinical Trials (for study timing; 2009), ICH S5: Detection of Toxicity to Reproduction for Medicinal Products (2005), ICH S7A: Safety Pharmacology Studies for Human Pharmaceuticals (2000), and ICH Q5E: Comparability of Biotechnological/Biological Products Subject to Changes in their Manufacturing Process (2004). 2 There are also 2 OECD TGs, TG412 and TG413, issued in 2018, 13,14 which describe the study design of 28- and 90-day inhalation toxicity studies, respectively, but these guidelines have little relevance as they only describe the technical aspects of running an inhalation toxicity study, based solely on experience gained from small molecule chemicals. There is no reference to study design modifications for assessing an inhaled biologic or any strategic guidance for interpreting the outcome as a part of the totality of the data available on the molecule.
The lack of an internationally approved guidance to serve as the framework for development of a novel inhaled biologic has meant that decisions on study design have been made based on the expertise of the person designing the study on a case-by-case basis to suit the molecule in question. Advice can be sought at regulatory authority meetings, such as an FDA pre-investigational new drug meeting, but these may not coincide with the optimal time to discuss specifics of study design. As a result, uncertainty remains about acceptance of any variation in study design by different health authorities globally.
Species selection
Regulatory guidelines for nonclinical safety evaluation of small molecules specify the use of 2 species, rodent (rat, mouse) and nonrodent (dog, minipig, NHP), to support dosing of healthy volunteers or patients, where appropriate. Occasionally rabbits may be used as a nonrodent species, particularly for ophthalmology indications. However, toxicity studies on biologics should only be conducted in pharmacologically relevant species, that is, one in which the test material is pharmacologically active due to the expression of the receptor or an epitope and that demonstrate a similar tissue cross-reactivity profile as for human tissues, ICH S6(R1) 2011. 2 If there are 2 pharmacologically relevant species for a biologic (1 rodent and 1 nonrodent), then both species should be used for short-term studies, although this could be reduced to a single species for longer-term studies if the findings from both species are similar or 1 species shows clear evidence of being the most sensitive to any adverse effects. If there is only 1 pharmacological relevant species, then only 1 is required throughout testing. Toxicology testing in nonpharmacologically active species is discouraged, although it is generally expected for assessment of chemical class effects with oligonucleotides. Selection of the toxicology species requires an understanding of target sequence homology, target expression and distribution, relative target binding affinity, and pharmacodynamic effects, including any Fc-related effects. 11 Due to the high target specificity, many biopharmaceuticals are only cross-reactive in the NHP. Potential immunogenicity can occur with administration of most humanized biologics to nonclinical species, even when the cross-species homology is identical, and a high incidence of immunogenicity that impacts exposure to the drug can exclude some species from use in toxicology studies. Inhalation toxicity studies in the dog or minipig require additional considerations of the large amounts of biologic that will have to be produced early in drug development but also the potential for significant immunogenicity which has been observed with mAbs in the dog. In general terms, immunogenicity is often lowest with the NHP as the molecules are humanized and the sequence homology for the target is often very similar. However, potential immunogenicity is species and molecule specific and needs to be determined as part of the toxicology studies conducted. As is commonly seen for biologics via other routes of administration, the NHP was used as the only toxicology species for 8 of 12 case study molecules and as one of the 2 species used for a further 2 case study molecules (Table 1). For the remaining 2 molecules, the rat only and the rabbit only were used—the reason for selection of these single species was not provided in the survey.
As biologics are catabolized into small peptides by proteases and peptidases after nonspecific endocytosis by endothelial cells, consideration of human metabolite profiles and their coverage is not required, regardless of the route of administration.
Number of dose groups and group sizes
Once the species has been selected, the next consideration is the overall study design in terms of the number of dose groups in the study and the number of animals per group. There are several factors that need to be considered when designing a nonclinical inhalation toxicity study of a biologic: A. Compound availability
Manufacturing sufficient material to conduct an inhalation toxicity study in NHPs, typically weighing approximately 3 kg, likely with daily dosing, is a major consideration when it is remembered that the majority of the material used is wasted during dosing. Compound requirements are primarily determined by the amount of airflow into the exposure system in order to deliver the aerosol to the animals, which is, in turn, based on the total ventilation required for the number of animals being simultaneously dosed on the system. As group sizes increase, often to accommodate the additional requirements of the studies conducted later in development, the airflow required to deliver the targeted doses, and therefore the amount of compound, will also have to increase. Difficulties with compound supply alone cannot be used to justify a lower dose or smaller group size as it would compromise the integrity of the study, although this was reported as the justification for the maximum dose in one of the case studies. As a result, supply needs to be carefully planned ahead of the study and then managed throughout so that sufficient material is available at study start or can be resupplied during the study, even if this means the use of several different but comparable batches. B. Historical data on the vehicle/carrier
If a novel or inadequately characterized excipient is used as one or more components of the inhaled biologic drug product formulation, it may be necessary to include 2 “negative” control groups, 1 consisting of air only or physiological saline (for nebulized products) and 1 of either the formulation only or the novel excipient only. The novel excipient aerosol concentration is normally matched to the aerosol concentration of the highest dose of biologic used in the study. By incorporating this into the main inhalation toxicity study, these data could support the clinical use of an excipient that is novel via the inhalation route but qualified via other routes, using fewer animals than a stand-alone inhalation toxicity study on the excipient only. The study needs to be of sufficient duration, usually 6 months for chronic use, although shorter studies may be possible following negotiation with the health authorities. If the excipient is completely novel, it is likely additional characterization would be needed, as per FDA Guidance for Industry: Nonclinical Studies for the Safety Evaluation of Pharmaceutical Excipients (2005). C. Number of dose groups, exposure multiples, and anticipated clinical doses
For inhalation toxicity studies on small molecules, 3 dose levels are typically selected to provide satisfactory multiples above the anticipated maximum clinical dose and a similar approach could be applied to biologics. The feasibility of having 3 dose levels, with sufficient margins over the proposed high clinical dose, may be difficult to achieve especially when comparing the pulmonary deposited dose in animals to the nominal dose (dose in the device) in humans as requested by FDA. 4 The difficulty comes primarily from 2 sources. First, it may not be technically feasible to generate an aerosol concentration high enough to achieve sufficient multiples. Second, such dose multiples may be unachievable due to exaggerated pharmacology or conversely have saturated the target making higher doses either unlikely to yield additional information or result in nonrelevant toxicity. Thus, the need for 3 dose levels when assessing the potential toxicity of a biologic has been questioned, 16,17 particularly if toxicity and/or a dose–response is considered unlikely based on knowledge of the target or from previous toxicity studies. 18 If this is the case, use of 3 dose levels would be illogical and would represent unnecessary animal use. It is acknowledged that conducting an inhalation toxicity study with only 2 active dose groups carries a degree of risk if a dose relationship (or lack thereof) in any findings cannot be demonstrated, limiting data interpretation.
Generally, 2 or 3 dose levels are chosen. The maximum dose selected does not have to be the maximum feasible dose and this should not be considered the default position. Provided the maximum dose assessed can be achieved without compromising the molecule, animals, or technical aspects of the study and provided at least the minimum safety margin for the highest intended clinical dose with a reasonable excess, there is little additional relevant information gained from pushing to achieve larger multiples. Usually the lowest dose is one that will support the lowest clinical dose predicted to show sufficient pharmacological activity and thus still allow progress into the clinic if it becomes the NOAEL. For later development studies intended to support increasing duration of clinical use (eg, 13-week or chronic studies), the choice of the dose range is generally dictated by findings in shorter duration toxicity studies and by clinical information related to adverse events and the proposed market dose. If none of these are relevant, then the same criteria as applied to FIH studies apply for long-term inhalation exposures.
Taking all the above into account, the maximum dose assessed in an inhalation study with a novel biotherapeutic must be selected following careful consideration of the above factors plus any others relevant to the specific molecule under examination.
Of the 18 toxicology studies, use of 3 dose groups was most common (11 of 18), while 4 studies used 2 dose groups, 1 used 4, and 1 study used only 1 dose group (Table 1). D. Number of animals per dose group and duration of the study
Typically, rodent and nonrodent studies of less than 13 weeks duration comprise 10 main study animals/sex/group and 3 animals/sex/group, respectively.
16,18
This is reflected in the majority of the case studies whereby the 2 rat studies had group sizes of 10/sex/group and 11 of the 14 primate studies had group sizes of 3/sex/group regardless of the duration. For the only mouse study, of 13-week duration, the animal numbers were increased to 12 animals/sex/group and, in two 26-week inhalation studies, the number of NHPs was increased to 4 animals/sex/group. The reason for an increase in the number of animals/sex/group on long-term studies was not captured as part of the survey but may be related to blood sampling volumes (mice) or be influenced by other factors, such as the incidence of circulating ADA, which could reduce exposure to the drug in some animals on study requiring a large initial group size to provide sufficient numbers of animals exposed to the biologic at the end of the study. One primate study used only 1/sex/group, but this was a 7-day dose-range finding study conducted as a preliminary to a 14 day FIH-enabling inhalation toxicity study. The duration of the studies ranged from 7 days to 26 weeks, which likely reflected the development stage and clinical requirements of each molecule, with 26 weeks typically the longest duration of toxicology study of any species required for novel biologics. E. Study end points
For rodent studies, satellite animals may be required to accommodate demand for blood sampling when end points such as toxicokinetic evaluation, ADA and cytokine analyses, immunophenotyping and target engagement, and so on are included. As microsampling of blood becomes more routine, the need for satellite animals should reduce. The number of satellite animals required is driven by the total amount of blood needed during the study and is generally only an issue with rodents due to their smaller blood volume, as oversampling can result in an erythropoietic response or worse, failure to thrive. Satellite animals may also be needed to provide sufficient tissue(s) for multiple end point evaluations during or at the end of the study and/or for serial or terminal bronchoalveolar lavage (BAL). For larger nonrodents, typically NHPs, large body sizes mean these blood samples can usually be obtained from main study animals. F. Recovery
For many inhalation toxicity studies, it will be necessary to include a recovery group of animals to evaluate the reversibility of possible compound-related effects—their use to assess for delayed toxicities has been questioned. 19,20 Typically, recovery groups comprise 5 animals per sex in rodent studies or 2 animals per sex in nonrodent studies and are generally restricted to the control and high dose groups only. From the 18 toxicology studies, only 9 included a recovery group and this was more likely to be in the longer duration studies. Despite recent recommendations to only include recovery groups in only one of the pivotal toxicology studies, 17 there was no evidence in this set of case studies of a trend to include recovery in only one of the studies conducted if more than one study had been conducted on the same molecule (Table 1).
Special considerations for use of sexually mature NHPs
Since NHPs are commonly the only pharmacologically relevant species for toxicology studies on biologics, it is worth considering some of the unique technical challenges posed when sexually mature NHPs are needed for inhalation toxicology studies. The standard age for conducting inhalation toxicity studies on small molecules and the short-term studies on biologics is 2 to 3 years, at which age the animals are sexually immature or on the cusp of maturity and weigh approximately 2.5 to 4 kg. However, long-term studies on biologics typically require the use of sexually mature NHPs to allow the assessment of a potential effect on fertility, ICH S6(R1) 2011,
2
via reproductive tract histopathology and physiological and hormonal parameters. These are not needed for small molecules as fertility assessments are conducted in the rat and rabbit. Sexually mature NHPs are typically at least 4 to 5 years of age for males, although females often mature slightly younger. The general challenges associated with the use of sexually mature NHPs in inhalation toxicity studies are discussed below: A. Suppliers/animal specification
The average body weight of a mature NHP (7-12 kg for males and 5-7 kg for females) is approximately double that of a standard aged NHP. Due to limited availability of mature NHPs, there is generally less flexibility to specify animals based on body weight, resulting in a wide range of body weights within groups. The intragroup variability in body weight can be further compounded as, ideally, the social groups the animals belong to prior to transfer to the test facility should be maintained to ensure animals are calm and compliant on arrival. Although it has been suggested that NHPs are sexually mature between the ages of 4 and 7 years,
21
in the experience of one CRO, this is more likely at 5 years and above. Body weight and age alone cannot guarantee sexual maturity, and it is recommended this is proven (repeated production of viable semen for males and a regular menstrual cycle for females provide the strongest evidence). B. Animal arrival and housing
Mature NHPs are typically received into the laboratory considerably earlier than immature animals and may arrive 5 months in advance of the study to allow confirmation of a regular menstrual cycle in the female animals (normally measured over 3-4 months). Owing to their size, animals are often transferred directly into the animal facility rather than being held in a separate holding unit while tuberculosis and hepatitis B tests confirm the absence of these diseases (approximately 3-4 days).
Generally, the smaller immature NHPs of both sexes and sexually mature females are housed in groups of 2 or 3, but mature male animals are housed at maximum of 2 irrespective of the cage size (groups of 3 males may regularly fight to establish dominance over the course of the study). C. Animal training
All the animals undergo a positive reinforcement training program of at least 5 weeks to facilitate acclimatization to the laboratory environment and procedures, starting on arrival, and this training period can be significantly longer for sexually mature NHPs. Although more difficult to complete, well trained animals exhibit less stress and are more compliant to the required study procedures. In addition, sexually mature females are trained to present their tail to allow vaginal swabs to be taken to assess menstruation. D. Handling/standard assessments
Table 2 illustrates the difference in animal technician resource required for the normal daily handling of immature and mature NHPs on toxicology studies, particularly for the very large and strong males that can weigh as much as 12 kg. Use of additional technicians improves the ease of handling, reducing stress, and ensuring easier recognition of any treatment-related clinical signs. Blood sampling can be conducted while the animal is still restrained in their chairs but if not, 4 technicians are needed to restrain the animal (1 per limb), while a fifth technician collects the sample.
Technical Resource Requirements for Handling Immature and Mature NHPs.

Abbreviation: NHP, nonhuman primate.
E. Additional assessments
Semen collection and testicular measurements are conducted monthly on mature males, and vaginal swabs are collected from mature females to confirm menstruation. In females, circulating hormone levels can be analyzed as required, for example, progesterone and estradiol.
Specific technical challenges with inhalation studies in sexually mature primates
A. Primate masks
The facial shape of older primates is different to younger ones, necessitating different sized exposure masks depending on the individual monkey. Masks have evolved significantly in recent years, and some are now modeled on mature primate skulls and produced by 3-dimensional (3D) printing. The development of the 3D printed masks has been discussed in the study by Moore 22 and examples of mature NHP masks are shown in Figure 1.

Examples of mature primate masks—Illustration of the variety of face mask sizes required for sexually mature primates on an inhalation toxicity study. Face masks were custom made. Picture courtesy of S. Moore.
A suitably sized primate restraint chair for inhalation dosing is not commercially available for larger animals and thus the normal restraint chairs have to be adapted and modified, including enlarging the chest plate and attaching padding around the neck plate. Furthermore, toys and television are used during the inhalation dosing procedure as environmental enrichment to further improve animal welfare and dosing compliance, and enhance the scientific integrity of the study. B. Respiratory minute volume challenges
The airflow for animal exposure systems has historically been based on the sum of the RMV of all animals attached to the exposure system and are calculated using known algorithms with an additional excess to ensure animal welfare.
3
However, this approach has been shown to be unsuitable for mature animals owing to the following reasons: The established RMV algorithms do not account for the use of large mature primates, as the mean body weights of mature NHPs are higher than the body weights for standard aged NHPs on which the algorithm was based.
23
This is confirmed by data discussed below. The wide body weight range, particularly for males, means airflow cannot be based on the mean body weight, as that would result in insufficient air reaching the largest animals. Rather the airflow must be calculated based on the body weight of the heaviest animal of that group. This inevitably increases the amount of test material required.
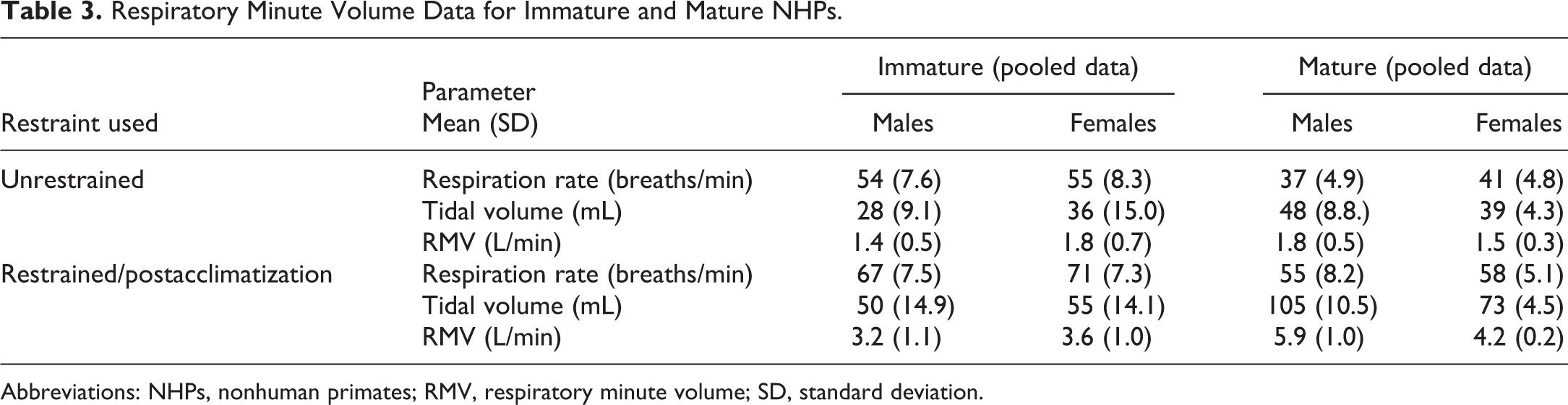
To address this, body weights should be determined prior to dosing so that accurate RMVs can be calculated for each animal per group, and the optimal airflows for exposure determined. Table 3 shows data for both immature and mature primates collected with RIP jackets and shows data collected over multiple time points from the sham dosing phase of various studies (S. Moore, PhD - personal communication, July 2020). These include data from both unrestrained animals collected at least 30 minutes prior to sham dosing (air only) and restrained/acclimatized animals collected from 1 to 24 hours postsham dosing.
Respiratory Minute Volume Data for Immature and Mature NHPs.
Abbreviations: NHPs, nonhuman primates; RMV, respiratory minute volume; SD, standard deviation.
For unrestrained animals, the respiration rate was considerably lower in mature animals than immature. Conversely, the tidal volumes were considerably greater for mature animals than immature animals, especially males. Consequently, the RMVs in unrestrained animals were quite similar in immature versus mature NHPs, which is important as these are not adjusted for body weight. For restrained animals, the respiration rate for mature animals was lower than immature animals as for the unrestrained animals, but the tidal volumes increased approximately 2-fold for mature males and females. This increase in tidal volume then resulted in RMVs in restrained mature animals almost double that of unrestrained mature animals or of restrained immature animals.
Using the recognized RMV algorithm for inhalation dosing, 3 the RMVs calculated were similar to that of the unrestrained immature animals. However, for mature male and female NHPs of an example 9 kg and 6 kg body weight, respectively, the calculated RMVs were 3.95 and 2.80L/min, respectively, which was significantly lower than the measured values of 5.9 and 4.2L/min. If mature primates are used in inhalation toxicology studies, the actual RMVs should be determined and, as more data are gathered, it may become necessary to generate a separate RMV algorithm for mature primates.
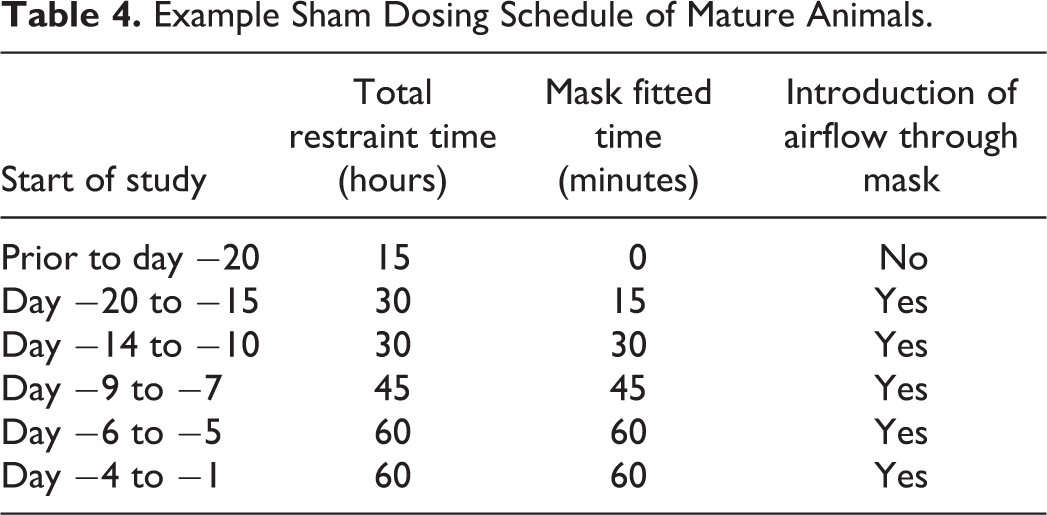
C. Sham dosing schedules
It is important that the animals are sufficiently acclimatized to the dosing equipment and procedures prior to the start of the study and this includes sham dosing, where air is circulated through the mask to simulate the airflows. Immature animals normally only require 10 to 11 consecutive days for acclimatization. For the mature animals, this is usually longer (an example of a sham dosing schedule is shown in Table 4).
Example Sham Dosing Schedule of Mature Animals.
In summary, the following technical considerations must be considered when using sexually mature primates for inhalation dosing of novel biologics. These can have significant impact on the study conduct and the time needed for prestudy activities:
Prestudy procedures are likely to take considerably more time.
More staff are required for safe handling, with minimal stress to the animal.
Additional assessments are required to confirm the sexual maturity of the animals on study.
Consideration should be given to the timing of study procedures as many are easier to conduct while the animals remain restrained in their chairs.
Longer sham dosing is required to train the animals to the dosing procedure and equipment.
Modifications are required for chair and mask designs, some of which may need specific adjustment for the individual animal(s).
The exposure system airflow should be based on the heaviest animal of the group, not the average body weight owing to the larger body weight range.
The RMV should ideally be measured in the actual study animals or be based on representative data from animals of similar size and maturity and not standard algorithms.
Question 3: What Are the Characteristic Responses to Inhaled Biologics and How Can Exposure be Demonstrated at the Site of Administration?
As a foreign proteinaceous material is being administered to the lung, possibly at high aerosol concentrations and for a substantial period of time, it is likely that some of the findings observed will solely be related to exceeding physiological responses to clearing a high protein load or to the foreign nature of the molecule triggering an ADA response. It is well established that ADA responses to drugs in nonclinical species are not predictive of ADA formation in humans, ICH S6(R1), 2011. 2 This is particularly true with molecules such as mAbs, Fabs, and Dabs, where the complementarity determining regions (CDRs) are deliberately designed to be as close to a normal human sequence as possible, which in turn is likely to mean they are further from the normal sequence in the nonclinical species and thus appear more “foreign.” For mAbs, the majority of ADAs have been shown to be against the CDRs of the molecule, rather than the human framework regions, where this has been investigated. The same may also apply to recombinant replacement proteins and peptides, although generally cross-species sequence homology in humans, monkeys, and rodents is more common for naturally occurring proteins. Antidrug antibody formation with inhaled biologics is discussed later. Typical pathology findings in inhalation studies are discussed in greater detail in the accompanying publication by Hall et al 6 and so will not be discussed further here.
Clinical observations
Reproducible inhalation dosing is dependent on calm animals undergoing passive breathing to keep the pattern of respiration consistent, such that tachypnea, favoring more pronounced upper airway deposition, or bradypnea, which will enhance deep lung penetration, do not occur. These could lead to large discrepancies in exposure between dosing days and interanimal differences in lung pathology and/or toxicokinetics.
To assess whether there are common clinical signs observed during inhalation toxicity studies with inhaled biologics, an experienced CRO analyzed the clinical observation data for 9 studies in NHPs between 5 days and 26 weeks of duration. The most commonly recorded clinical observations, defined as occurring in more than 10% of animals, are shown in Table 5 for control and treated animals.
Most Common Clinical Observations (Seen in >10% Animals) in 9 NHP Studies With Biologics Conducted at a Selected CRO Between 2014 and 2019.
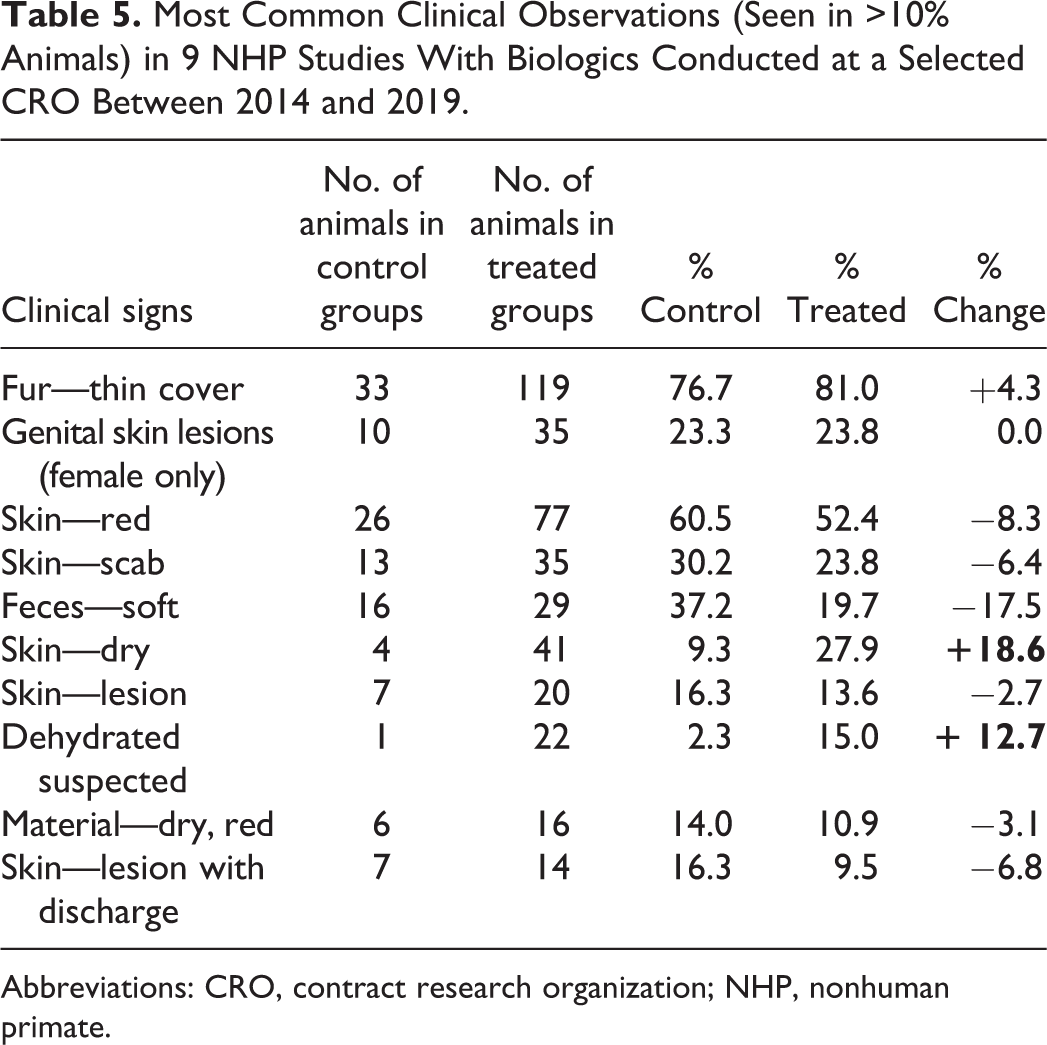
Abbreviations: CRO, contract research organization; NHP, nonhuman primate.
Of the 10 clinical observations listed, only dry skin and suspected dehydration occurred at a higher incidence in treated animals versus controls (bold numbers in Table 5). The clinical observations occurring at an incidence below 10% but at a higher incidence in treated animals included skin pallor, thin appearance, and inappetence (data not shown). All these signs could easily be seen in an inhalation toxicity study with a small molecule compound and are unlikely to be specific to biologics.
In the case studies collated in this survey, clinical signs were only observed in 5 of 18 toxicology studies. Two case studies had single animals with respiratory changes (1 mouse and 1 NHP) that were due to a respiratory tract infection and the last case study had lethargy and edema in a single NHP that was subsequently ascribed as secondary to immune complex (IC) disease. The first 2 are likely to either be coincidental or due to the pharmacology of the biologics and the third is an uncommon but known sequelae of ADA formation (see The impact of ADAs below). The final 2 case studies showed clinical signs possibly related to the administration of a biologic, namely ungroomed/wet fur and decreased food consumption (not pharmacology-related findings). Ungroomed/wet fur and decreased food consumption would be in line with those reported in Table 5 and so are likely to be a general finding associated with inhalation administration of any molecule type.
To bronchoalveolar lavage or not to bronchoalveolar lavage?
During the evaluation of inhaled drugs, or drugs that either act in the lung or accumulate there, bronchoalveolar lavage (BAL) can serve as a powerful technique to help understand target engagement and PD, efficacy, injury, and exposure to the lung. Bronchoalveolar lavage in larger nonclinical species is a relatively noninvasive procedure in which the airways and alveoli are washed with fluid, which is then extracted to obtain information on cell types and various analytes/biomarkers in the epithelial lining fluid (ELF). The BAL technique can be performed in animals as small as mice either at study termination or with some experience, repeatedly during the course of the study. 24 Analysis of the BAL fluid (BALF) may provide more specific and relevant information on the dose–exposure relationship, the time course of ADA formation in the lung compared to that from conventional systemic assessments, and/or for harvesting cells from the lung for mechanistic evaluations. The BAL procedure is described in more detail in Poitout-Belissent et al . 25
Analysis of BALF cellular and/or soluble components is clearly not a substitute for pathology, as, although many changes in the ELF have corresponded with pathological changes in the lung, this is not true for all. Normally, approximately 90% of cells in the BALF are macrophages. Four patterns of cellular inflammatory influx are discernible, although mixed responses can also occur. 26 Increased permeability of the air–blood barrier results in leakage of protein (primarily albumin) into the ELF, which is a sensitive measurement of lung injury. Useful markers of cell damage or inflammation include N-acetylglucosaminidase, γ-glutamyltransferase, lysozyme, and alkaline phosphatase activity, although lactate dehydrogenase is most commonly used. Other facets of pulmonary damage can be monitored in BALF, such as excess mucus secretion, oxidative stress, aberrant coagulation parameters, as well as changes in phospholipid content of surfactant. 27 Often, in more mechanistic and animal model studies, inflammatory mediators, such as cytokines, chemokines, and arachidonic acid metabolites, are investigated in BALF. Unfortunately, none of the BALF biomarkers of toxicity correlated with the findings in the lung in the case studies, when this was analyzed for, despite well documented publications demonstrating the utility of this approach. 27,28
Bronchoalveolar lavage may also be a better marker of lung exposure than measurement of drug in the blood. However, the data are more difficult to obtain for a kinetic evaluation and more difficult to extrapolate to human exposure as BAL is rarely performed in clinical trials. In a recent study with an inhaled oligonucleotide, the concentration of oligo in the BALF and lung tissue in rats highly correlated and, on average, was only 3% different immediately postexposure (J. Tepper, PhD, personal communication, December 2020). At 24 hours postexposure, oligo concentrations were lower in the BALF than lung tissue but still highly correlated with lung concentration. In either case, there was no correlation of the concentration of oligo in the BALF or lung tissue with that in the blood. Antidrug antibodies can be observed in the BAL and are more likely to be relevant to local effects than serum ADAs, as antibodies are produced locally in the lung but may also enter the lung from the circulation. 29
From 18 toxicology studies, ADAs were assessed in the BALF for 3 biologics—2 protein/peptides and 1 Dab. In 1 long-term study of 26 weeks duration, all NHPs except 1 were positive for ADA in the BALF (11/12 NHPs), whereas in the 3 shorter-term studies of 2 or 4 week duration, only a low number of animals were positive for ADA regardless of species (individual incidences of 4% in rats, 1/60 rats [2%] and 1/12 NHPs [8%]). This is similar to the data observed with biologics administered systemically, where ADA formation in the plasma generally tends to be low in studies of 2- to 4-week duration. Of the case studies presented in which BALF was obtained, several showed good correlation between BALF and blood drug concentration, but others showed no correlation (further details are available in the accompanying publication by Hall et al 6 ).
The impact of ADAs
As for any biologic administered via other routes of administration, inhalation carries the risk of inducing ADA. The lung mucosal membrane makes up the primary barrier to pathogens. In the lung, antigen presenting cells (APCs) such as dendritic cells, monocytes, and macrophages, are found near or protruding between airway epithelial cells in the mucosa. Upon encountering antigenic material such a biologic drug, the APCs interact with T-cells in the lymph node resulting in the production of ADA by plasma cells. Generally, drug administration involving mucosa has a higher potential to produce ADAs than administration via a parental route. 10
Both the induction of ADA and any sequelae from the presence of ADA are highly variable between individuals, both in nonclinical species and in humans, and cannot be predicted. Typically, only some individuals will generate ADA against a biologic following administration and, in those that do, the clinical manifestation can range from no discernible effect, through simple changes in PKPD (from increased or decreased clearance of the drug), to hypersensitivity reactions, which can also range from mild to anaphylaxis. In nonclinical animal species, the likelihood of ADA can be influenced by various factors such as the presence and percentage of non-native amino acid sequences (as the biologic was developed to be homologous to the human rather than homologous to the nonclinical species), the mechanism of action (immune activation), target expression on immune cells, the ability to be internalized following binding to the target, altered glycosylation patterns, post-translational modifications, and aggregation. 2,30 Antidrug antibodies can enhance clearance of biologics although, in molecules with a short half-life such as Fabs, ADAs can have the opposite effect whereby the biologic-ADA complexes take on the half-life of the ADA Ig and so are cleared more slowly than the noncomplexed biologic. If ADA enhances clearance resulting in the loss of systemic exposure to the inhaled biologic in a small percentage of animals, then this is unlikely to impact the overall study interpretation. However, if the drug target is systemic and a large percentage of animals lose all or a significant portion of their systemic exposure, this could render the study invalid, as potential effects arising from target engagement over the duration of dosing may not have been properly assessed. It also makes interpretation of the TK parameters very complicated. Most of the knowledge on the impact of ADA is based on systemic exposure and, although there are little data on ADA in the lung, the same concerns would apply regarding possible loss of exposure and the resulting loss of target engagement in this tissue. 2,30 This is of particular concern for NHP studies where typical group sizes are only 3 animals/sex/dose group and, if less than half of those are still exposed at the end of the study, this would severely compromise the data obtained. For this reason, it may be prudent to increase the number of NHPs per group to at least 4 animals/sex/dose group in longer term studies, especially if ADA incidence is very high in the shorter-term studies with the molecule. Increased group sizes for NHPs above this may be considered case-by-case but would be difficult to justify ethically.
It is critical to remember that the incidence and severity of immunogenic responses to drugs induced in nonclinical species are not predictive of the incidence and severity of ADA formation in humans, ICH S6(R1) 2011, 2 and thus a molecule which is highly immunogenic in preclinical species may show very low or no immunogenicity in humans. Nonetheless, it is important to understand the impact of ADA in any nonclinical toxicology study if, for example, changes in TK and/or PKPD are observed, ADA-related toxicity is observed or suspected (eg, hypersensitivity or IC disease; see below) or to provide supporting information if neutralization of a nonredundant target via ADA is suspected (as can be seen for recombinant proteins).
Only a few studies have been reported where ADA incidence was assessed after inhalation and compared to parenteral injection both nonclinically and clinically. Exubera has now been withdrawn from the market but is the only approved recombinant human insulin that was administered by the inhaled route in nonclinical studies in rats and cynomolgus monkeys and which also included assessment for ADA. In rats, Exubera was administered up to 40 times the human starting dose. A weak ADA response was observed that was similar following inhalation or subcutaneous administration with no effect on exposure or safety. However, ADA was not induced in 1- and 6-month inhalation toxicity studies in either serum or BAL of the monkey. The small difference in ADA response between species was attributed to a slight difference in insulin sequence in rats compared to humans and monkeys. 31 In humans, the binding ADA incidence increased from 3% to 4% predose to 29.0% postdose via inhalation, while the ADA incidence remained relatively unchanged when dosed via the subcutaneous route. 32 For Afrezza (another inhaled insulin), ADAs were only assessed in early toxicology studies but not in later studies (J Tepper, PhD, personal communication, December 2020).
Antidrug antibodies have also been associated with localized hypersensitivity in the lung in nonclinical species. In addition, information on the incidence of ADA following administration is also available. Inhalation toxicology studies of 1- or 6-month duration were conducted with recombinant human(rh) DNase (Pulmozyme), developed for the treatment of cystic fibrosis, in both rats and monkeys at doses up to 69-fold the expected daily clinical dose (reviewed by Wolff 1 ). In the 1-month studies, mild-to-moderate alveolitis was observed in rats at the highest dose and bronchiolitis was observed in 1 of 8 monkeys, also at the highest dose. These findings were not seen after a 4-week recovery period in either species. After 6 months of dosing duration, bronchiolitis was observed in the rat but at a lower incidence, suggesting that the findings had not progressed with longer dosing duration. Increased perivascular lymphocytic cuffing, peribronchial lymphoid hyperplasia, terminal airway-related bronchiolitis/alveolitis with eosinophilic infiltrates, and increased siderophages were observed in the monkey. Antidrug antibodies to rhDNase were observed from week 4 and persisted throughout treatment, with the ADA titer appearing to correlate with the severity of the pulmonary lesions. In the conclusions of the review, 1 these lesions were considered to be consistent with an allergic or hypersensitivity (type I) response to a foreign protein, perhaps not unexpected when there is a relatively low homology between human, monkey, and rat DNases. It is not unexpected then that the ADA incidence in humans is much lower at less than 5% and has little effect on the safety profile of the drug. 8
Case study data on ADA incidence in serum and/or BAL were collected from 12 preclinical studies on 9 different molecules (see Table 6). There were 9 studies using NHPs, 2 studies using rats, and 1 study using mice. Overall, ADAs were observed in all studies with the incidence of ADA increasing with the increase in duration of the study, a known phenomenon with biologics in nonclinical species. 30 Studies of 4 weeks or less tended to have only a small percentage of animals with ADA, whereas the longer-term studies tended to have 100% or close to 100% of animals with ADA. However, the 100% incidence of ADA observed in the longer-term studies is significantly higher than normally seen with a systemically administered biologic, where such an incidence is very uncommon. It is difficult to comment on whether rodents were more likely to mount an ADA response than the NHP from the case study data, as only a small number of rat studies were conducted and all were relatively short in duration (4 weeks or less). However, for the single Fab molecule where inhalation toxicity studies of 13 weeks duration were conducted on the same molecule for both NHPs and mice (case studies 11a and b), the ADA incidence appeared similar in both species. There were no clear trends based on modality or on whether the target was an immunomodulatory drug but the numbers of each types of studies were too small to draw definitive conclusions.
In the majority of cases, accompanying pathology findings were also noted in the respiratory tract. The pathology associated with inhaled biologics and any association with the concomitant presence of ADA are discussed in depth in the accompanying publication by Hall et al. 6 Briefly, there appears to be an association between the presence of ADA and the presence of lymphocyte infiltration into the lung and bronchus-associated lymphoid tissue (BALT) hypercellularity, whereby 75% of NHPs that developed perivascular/peribronchiolar (PV/PB) mononuclear cell infiltrates also developed ADA and 95% of NHPs that developed PV/PB mononuclear cell infiltrates presented with enlarged tracheobronchial lymph nodes. Similar analysis also showed a very high correlation between alveolar mixed inflammatory cell infiltrate and ADA when present (20% of NHPs). However, while likely from a weight of evidence perspective given the morphologic features, kinetics of the response, high frequency of ADA, and similarity to published lung models of immune responses, no definitive conclusion can be made as to whether the pathology findings are attributable to ADA induction, owing to the lack of accompanying mechanistic end points/studies.
Summary of the Inhalation Toxicology Studies—Ordered by Increasing Study Duration.
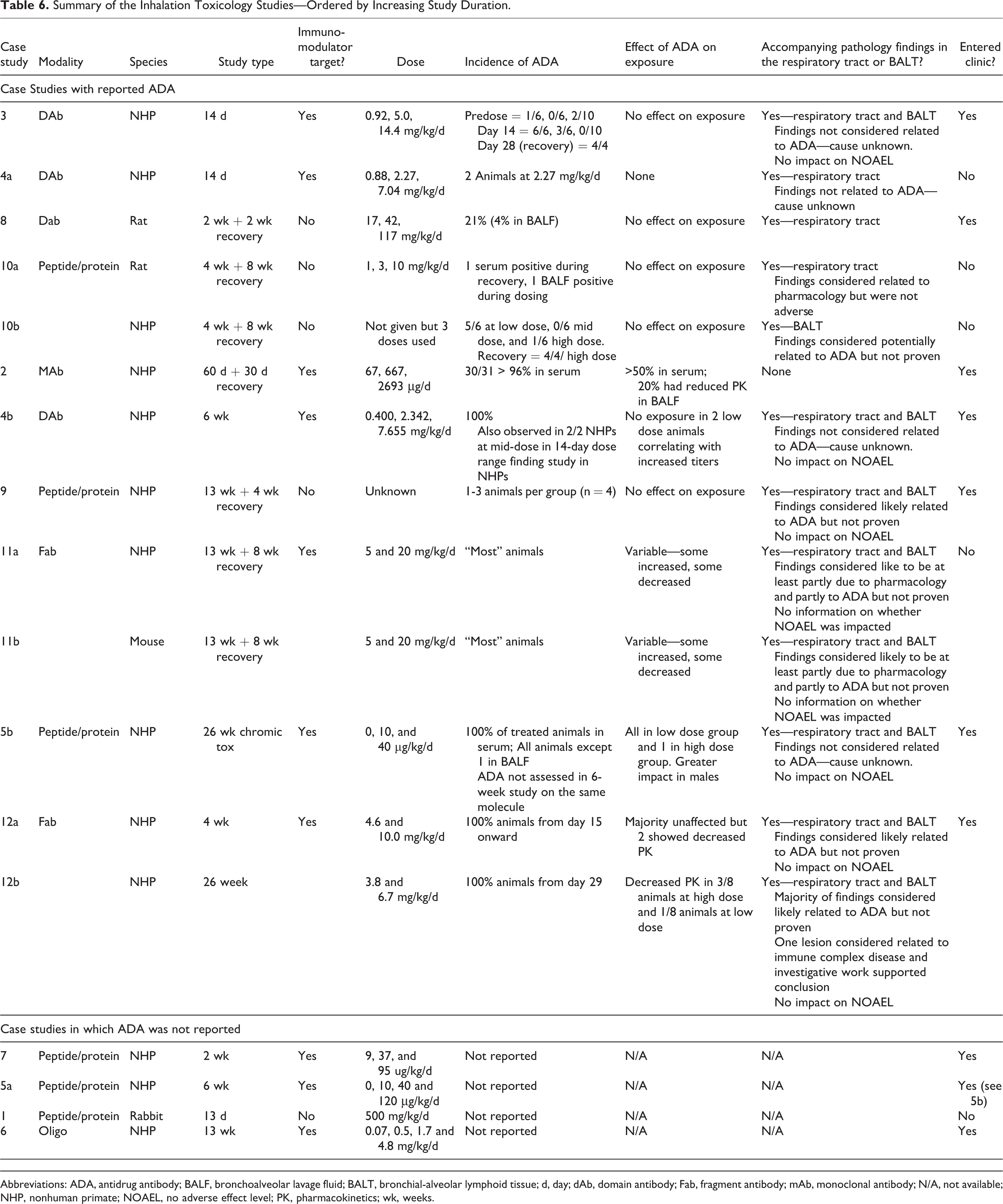
Abbreviations: ADA, antidrug antibody; BALF, bronchoalveolar lavage fluid; BALT, bronchial-alveolar lymphoid tissue; d, day; dAb, domain antibody; Fab, fragment antibody; mAb, monoclonal antibody; N/A, not available; NHP, nonhuman primate; NOAEL, no adverse effect level; PK, pharmacokinetics; wk, weeks.
The high incidence of ADA did not appear to prevent any of these molecules from progressing into clinical trials, even when titers were very high and the ADA impacted the PK in at least some of the animals on the study. It is reassuring that health authorities have taken into consideration that ADAs in preclinical species do not predict immunogenicity in humans, 2,33,34 rather they are used to aid interpretation of toxicity study and derivation of the NOAEL.
One obvious additional consideration would be whether there is any way to reduce or prevent the ADA from being induced in the first place, thus reducing the likelihood that exposure is altered during the study. Although there are medications that can aid in post-treatment occurrence of infusion reactions, such as diphenhydramine (reviewed by Mease et al, 2017) 35 , and anti-inflammatory medications, such as dexamethasone, these are unlikely to prevent or reduce ADA incidence. None of the case studies collated for this article had attempted to reduce the incidence of ADA.
Finally, there is one other potential sequelae of ADA to be considered when unexpected clinical signs or pathology become apparent during the inhalation toxicity study. Immune complex deposition (ICD), sometimes also referred to as immune complex disease, is a relatively uncommon and idiosyncratic sequelae arising from the formation of biotherapeutic drug/ADA ICs leading to a type I hypersensitivity reaction and thus is different to the type III hypersensitivity reactions and anaphylaxis mentioned previously. Immune complexes are normally formed during an immune response but are generally cleared without inducing any tissue damage. Immune complex deposition occurs when the drug/ADA complex either form directly within the tissues or form in the circulation and deposit within tissues and cause localized damage, facilitated by release of vasoactive amines, cytokines, and complement degradation products 36,37 but require a specific set of factors to occur simultaneously for this to occur. Immunohistochemistry (IHC) can be used to assess for ICD by determining the colocalization of human IgG; IgG fragments or any other biologic (drug); primate IgG, IgM, and IgA (ADA); and complement such as C3 and sC5b-9. Immune complex depositions have been observed as pathology changes and/or as hypersensitivity reactions in some repeat-dose toxicology studies with novel biologics in the primate via other routes of administration (reviewed by Rojko et al, 2014), 37 but it is not possible to predict when and if ICD will occur in nonclinical studies. Many factors drive the likelihood of ICD occurring, including the ratio of the drug to its ADA, the quantity, size, and affinity of the ADA for the drug under assessment and the equilibrium between IC formation and clearance rates in the tissues. Immune complexes form naturally in tissues and are typically cleared without incident, but ICD can occur when the clearance pathway becomes saturated and the ICs deposit in tissues leading to localized inflammation and tissue damage. All these factors differ between individuals both within and between studies, as the ADA response is highly individualistic. Immune complexes have been observed in kidney glomeruli, blood vessels, synovium, lung, liver, skin, eye, choroid plexus, and other tissues or bound to neutrophils, monocytes/macrophages, or platelets. 37 In a review of IC-related findings occurring in primates after administration of biotherapeutics drugs, the clinical signs ranged from clinically apparent reactions, which were sometimes fatal, to subclinical reactions, which showed no overt clinical signs and subsequently were only uncovered on microscopic evaluation, for example, inflammation of vessels, joints, heart valves, choroid plexus, skin, or other sites. 37 If ICD is suspected, a weight of evidence evaluation should be conducted using all relevant data, such as clinical signs, their temporal relationship to dose administration, PK/PD and ADA data, hematology changes, dose relationship, plausible alternative diagnoses, and the outcome of IHC analyses, as well as any other specialized investigative assays. 35 Importantly, the known lack of predictivity of ADA incidence and severity in the nonclinical species for ADA incidence and severity in the human (ICH S6[R1], 2011) 2 means the same lack of predictivity will apply to ICD, as the first step is ADA induction.
One of the case studies (case study 12a) had findings in the NHP 26-week study that were believed to be due to ICD in the lung, based on IHC investigations and a weight of evidence approach which concluded the findings were not related to the pharmacology.
Target occupancy and target engagement
Target occupancy and/or target engagement are intrinsically linked to the ability of the biologic to demonstrate the potential consequences of target modulation. Target occupancy or target engagement can be measured by analyzing the presence of the free or bound target or of a pharmacodynamic biomarker associated with its modulation. Monoclonal antibodies and related formats are highly potent, with affinity in the pM range, and highly selective for the target, such that any toxicity is usually due to exaggerated pharmacology-related effects on the target. 11 Assessing target occupancy and/or engagement poses specific issues for inhaled biologics, owing to the relative difficulty of sampling the site of administration and often the site of highest target occupancy/engagement—the lung. For 10 of the case studies, the target was in the respiratory tract. Potentially, repeat measurements of BALF could allow longitudinal assessments of the amount of biologic and target occupancy and engagement. However, in reality, this is not possible as BALF should only be sampled infrequently, it is a complex matrix which complicates analyses of individual components and BALF is only a surrogate for the actual tissue of interest, the lung. The more common route is to measure the concentration of the biologic, target, or biomarker in samples of the lung tissue taken at terminal necropsy, although this provides only 1 assessment time point. Although administered to the lung, systemic exposure does occur (and is desirable in some cases), and it is possible a systemic pharmacodynamic biomarker can be analyzed, as a surrogate.
In the 12 case studies, only 2, and possibly a third, specifically evaluated biomarkers in the lung and/or BALF to assess target occupancy/target engagement. It is possible that additional biomarker analyses were conducted in other studies, as lung tissue and/or BALF were taken from 6 of the 12 case studies, but no data were provided. The majority of the biologics assessed had respiratory system targets—10 of 12 molecules—and it may have been assumed that there would be full target occupancy in the lung, particularly if it is already known that the target is expressed at low levels in the lung. Of the 12 molecules in the case studies, 8 had immunomodulatory targets and it is known that molecules such as cytokines are often expressed at very low levels in healthy animals, meaning target saturation at the site of administration is easily achieved even at low doses.
One difficulty that must be acknowledged is the technical feasibility of measuring the target occupancy or target engagement, particularly if the free target is to be analyzed. As mentioned above, molecules such as cytokines often have very low levels in healthy animals, typically in the pg/mL or ng/mL range, which makes developing an assay with sufficient sensitivity to detect free target particularly difficult. This is even more difficult if the biologic is intended to inhibit the target, as this would further reduce the free concentrations of these. Where feasible, a total target assay can be used as a surrogate, as the free target would be expected to increase in the relevant matrices as it will adopt the half-life of the biologic binding to it. In addition, BALF from each dose group is usually pooled, especially for the smaller nonclinical species, and the potential dilution of the intended analyte could alter the equilibrium between bound and free target. For many such biologics, it is not technically feasible to develop an assay that can detect very low concentrations of the free target.
An alternative approach is to use the systemic PK analysis to indirectly inform on target occupancy in the lung, but this can only be conducted if a free PK assay is available. This relies on the premise that the biologic would first bind to all the available targets in the lung and thus is no longer “free.” If free biologic is then detected in the serum, it would be excess to the bound drug in the lung, showing that the biologic was administered in excess of the amount needed to saturate the target in the lung. Drug already bound to target in the lung would be assumed to pass into the systemic circulation still bound to the target, if not cleared by other mechanisms. The challenges of PK analysis of inhaled biologics are discussed further under Question 5.
Measuring target occupancy or target engagement can also provide data on the impact of ADA on animals on study, as not all ADAs affect PK and thus could leave the target binding site free. Whether this can be determined is related to the PK and ADA assay format used. If available, the PK of an IgG or IgG derivative biologic can be measured using a generic detection agent against the human Fc region or light chain, but this has the downside of providing no information on whether the biologic has bound to the target. Thus, in the majority of cases, a free PK assay is preferred. More commonly, ADA assays are simple bridging ligand-binding assays, that is, the ADA binds to the drug, forming a bridge to the detection agents, but the site of binding is unknown, and it is not known whether the ADA impacts the ability of the drug to bind to its target. Neutralizing ADA assays, which can determine this, are rarely developed for nonclinical studies. In addition, ADAs are often assessed in the systemic circulation rather than in the respiratory tract, and the number of animals that are ADA positive in the lung may differ from that in the systemic circulation. This has been shown in a number of the case studies where ADA was measured in both serum and BALF and in all cases, the incidence of ADA-positive animals was lower in the BALF.
In general, an assay that can either directly or indirectly assess for target occupancy/engagement in the tissue of interest is a useful addition to an inhalation toxicity study. However, the feasibility of developing such an assay for any given target should be considered on a case-by-case basis.
Question 4: How Can We Differentiate Between a Physiological Response to Foreign Biological Material and True, Human Translatable Toxicity?
Treatment-related effects—Are these related to the drug or to the administration of a foreign proteinaceous material?
One of the key factors to evaluate when assessing the potential translatability of any findings in an inhalation toxicity study with a biologic is whether any findings are related to the expected pharmacology, off-target effects, or are subsequent to administering a foreign proteinaceous material to the lung over a substantial period of time and, possibly, at high aerosol concentrations considerably in excess of the intended clinical exposure. The latter effects would be common to many inhaled biologics and would include effects attributed to innate immune system activation and/or ADA. The pathology findings commonly observed with the case studies collated for this publication are discussed in detail in the accompanying publication by Hall et al. 6 Briefly, comparison of the common changes induced by inhaled biologics, compared to inhaled small molecules, highlighted some similarities in the pathology induced in the respiratory tract but also some differences in the frequency of induced changes, particularly in the nasal cavity, trachea, and larynx. The most common pathology findings were microscopic changes in the terminal airways/alveoli, characterized by perivascular/peribronchiolar (PV/PB) mononuclear and alveolar inflammatory cell infiltrates ranging from simple (“uncomplicated”) increases in alveolar macrophages without other changes through to alveolar inflammation with more severe macrophage infiltration accompanied by other changes such as type II pneumocyte/bronchoalveolar hyperplasia and alveolar septal thickening.
In the 12 case studies, 8 reported alveolar inflammatory cell infiltrates that involved macrophages, of which 3 consisted of minimal alveolar macrophage accumulation only without accompanying inflammation. It is likely that phagocytosis of inhaled material was the principal driver of simple macrophage accumulation and, in the absence of damage, these findings are likely to be considered nonadverse. This is in line with the conclusion for simple increases in macrophages in the lung observed in inhalation toxicology studies with small molecules, which are also considered a nonadverse clearance mechanism. 38,39 These findings and their relevance to the clinic have been extensively discussed in other publications, including a white paper by the Society of Toxicologic Pathologists and the proceedings of a meeting held by the Academy of Pharmaceutical Science of Great Britain “Drugs in the Lungs Network” in collaboration with HESI (See Nikula et al 38 and Forbes et al 39 , respectively). Both these publications generally concluded that an increase in the size or number of foamy alveolar macrophages in the absence of other infiltrates is consistent with a nonspecific adaptive physiological response to the delivery of particles to the alveolus and may be considered nonadverse.
Other microscopic changes in the airways after inhaled biologics consisting of predominantly lymphoid PV/PB mononuclear inflammatory cell (MIC) infiltration (75% of the case studies) were associated with lymphoid hyperplasia of the draining lymph nodes, likely secondary to ADA induction. 6 Findings related to ADA induction would not translate to clinical risk. More complex findings were also observed with some of the molecules. However, understanding the relevance and/or adversity of the infiltration of these findings is more complex and would need further consideration.
Question 5: What Particular PKPD Aspects Are Important for and/or Peculiar to Inhaled Biologics?
When considering the lung as a target organ, there is an assumption that the drug distributes evenly throughout and 100% is absorbed, which is unlikely in reality given its complex structure. As the primary goal of PKPD is to determine the efficacy of the therapeutic, the key concern is the true exposure at the site of action which is difficult to determine. The site of action is typically very difficult to access or even is unmeasurable, for example, the interstitial fluid, and thus the BALF is often used as a surrogate to approximate exposure in the ELF. The BALF measurements can provide an approximation of lung tissue concentrations, but limited sampling opportunities prevent their use for kinetic analysis. In addition, BALF is prone to inherent dilution effects from the administration of a liquid volume to wash out the lungs, extracting some of the ELF when it is aspirated and thus altering the interface between the lung and the air. As a result of these difficulties, pharmacokinetic profiles of inhaled products are typically analyzed from measured concentrations in the serum or plasma. It should be noted that as absorption into the systemic circulation may be relatively low, sensitive methods are required to detect drug especially for potent drugs that may be administered at very low doses. As a result, serum concentrations can be 1000-fold lower than an equivalent dose administered by systemic routes. Pharmacokinetic analysis of BALF from a single animal is not sufficient owing to high variability between animals. If sampled, BALF should be extracted from each dose group using a composite design to allow modeling of the expected exposures based on a range of time points.
For 10 of the 18 studies (6 of the 12 case studies), exposure was measured in either BALF and/or lung tissue. Unfortunately, details of which matrices were evaluated, how the concentrations obtained compared to serum concentrations, or whether a composite design was used were not provided. However, it is unlikely a composite design was used for many of these, as they were part of multidose studies in NHPs where group sizes are too small.
In rare cases, sufficient data are generated to allow the PK profile in the serum to be correlated to the pharmacokinetics in the lung, allowing serum PK to be used to model the PK in the BALF. This has been performed for 1 inhaled biologic by one of the authors, where modeling of the pharmacokinetics revealed 2 absorption rates from the lung to the systemic circulation with a mix of fast and slow absorption. This is likely related to the complexity of the transport mechanisms across the lung epithelium and revealed that there was potential for the lung to be exposed for a longer period of time than initially apparent. Although a seemingly ideal route of administration, distribution of any drug within the lung is complex, variable, and cannot be easily measured directly.
Exposure in the lung can also be affected by ADA. Although ADAs are routinely measured in the systemic circulation, it is often not known whether ADA affects exposure in the lung directly as it is not routinely measured in BALF. However, when both were measured, the incidence of ADA in the BALF appears to be lower. Whether the apparent lower incidence could be an artifact arising from the BALF sampling procedure resulting in dilution of ADA present below the limit of quantification is not known. Analysis of lung tissue homogenate may also be used to demonstrate the presence of ADA in the lung, but tissue ADA doesn’t indicate whether exposure is affected at the surface of the lung.
An important aspect of translation of animal studies to human is the ability to predict a suitable therapeutically active dose. The PKPD relationship helps to define the dose, the frequency of dosing required by relating the exposure to target turnover, expression levels, accessibility, and other aspects to establish the maximum interval between doses required to maintain target interaction. Often a biomarker is used to establish PD but this PD biomarker can be even more challenging than measuring the target itself, especially in the clinical setting, where BAL samples are rarely taken. This increases the reliance on clinical effects as a readout, leading to long clinical trials in patients for most indications such as idiopathic pulmonary fibrosis or chronic obstructive pulmonary disease.
Most animal disease models, especially inflammatory diseases, are imperfect, but such models have been successfully used for proof-of-concept evaluations to demonstrate that a mechanism of action has an effect on the disease outcome and to understand the relationship between dose to the respiratory tract and the effect. It is important to study the most appropriate phase, where the drug target is present, and to understand the reliance on the model to demonstrate the entire disease course. Animal models can also aid understanding of the relationship between exposure and effect, including changes in target turnover, delay in effect, delay to return to baseline, and changes in expression. These models are very helpful when developing a PKPD mathematical model specific to the disease, target, and dosing regimen to understand the efficacious dose in patients, help identify the therapeutic window to guide dosing in the clinic and the likelihood of achieving an efficacious dose in light of any nonclinical toxicity observed.
The development of inhaled biologics is still a relatively new concept, with only 3 molecules having progressed to market, and as a result, the accompanying PKPD concepts for inhaled biologics are also not fully developed. There are many considerations around understanding exposure and development of future PKPD concepts that mean inhalation of biologics remains an ongoing field of discussion, with much progress yet to be made.
Question 6: How Are NOAELs Defined for Inhaled Biologics (vs Small Molecules)
As for any pivotal toxicology study, one of the primary goals is to determine the NOAEL for the study and thus allow calculation of the safety margins for the intended clinical doses. This is a complex evaluation that requires careful consideration of all the data gathered on the study in question, as well as previous inhalation toxicity studies on the same or similar molecules, and any other supporting data that would aid in providing context to the findings in the study. As such, it is not possible to establish a decision tree or overall framework that would apply generically to any biologic administered via the inhalation route. Rather each biologic needs to be considered on a case-by-case basis. However, there are various points that should be considered when determining an NOAEL for any biologic, even if a particular point can be readily dismissed for the specific biologic in question. The pathology findings commonly found in the case studies are discussed in depth in the Pathology Review accompanying this article
6
and should be referred to when microscopic findings are observed. These findings primarily apply to the respiratory tract, as this is the most likely place for findings in these studies. Should airway/alveolar macrophage infiltration be considered adverse?
The most common pathology findings that were attributable to high protein or particle load in the case study cohort were microscopic changes in the terminal airways/alveoli, characterized by inflammatory cell infiltrates ranging from simple (“uncomplicated”) increases in macrophages to mixed inflammatory cell infiltration and inflammation. The simple increases in alveolar macrophages were most likely secondary to clearance mechanisms and, in the absence of damage, these findings were considered nonadverse. This is in line with the conclusion for simple increases in macrophages in the lung observed in inhalation toxicology studies with small molecules, also considered a clearance mechanism and not adverse.
38,39
No conclusion on adversity could be made for the other findings.
6
Should lymphoid perivascular/peribronchiolar MIC infiltration be considered adverse?
In the case studies, there appeared to be an association between the presence of ADA and the presence of lymphocyte infiltration into the lung and BALT hypercellularity. It is possible this finding represents recruitment of cells to produce ADA during the period the animal is exposed to the drug. Lymphoid PV/PB MIC infiltration was observed in 75% of the case studies and was associated with lymphoid hyperplasia of the draining lymph nodes, likely secondary to ADA induction. These animals were ADA positive and the targets for the biologic were not known to induce immune activation. More complex findings were also observed with some of the molecules. However, understanding the relevance and/or adversity of the lymphocyte infiltration is more complex and would need further consideration.
Can the physicochemical properties of the molecule impact any pathology findings?
As for any inhaled small molecule, physicochemical properties of the inhaled biologic formulation/aerosol such as pH, osmolality, particle size, and shape could impact the pathology findings. Aggregates formed on aerosol generation could alter deposition in the respiratory tract and/or could lead to inactivation of the molecule, meaning that the toxicity of the molecule had not been adequately assessed.
Are any findings attributed to exaggerated pharmacology sufficient to be considered adverse?
Owing to the very high specificity of biologics for their target, regardless of the route of administration, observed toxicities are frequently the result of exaggerated pharmacology 11 or the downstream effects of pharmacology. For example, the inhaled insulin Afrezza induced substantial decreases in serum glucose which became the dose-limiting toxicity. However, decreased glucose is the expected pharmacology, and high doses would lead to exaggerated effects compared to the intended clinical dose. In cases such as these, a thorough understanding of the pharmacology and PD of the biologic is vital to provide context for any findings. Additional investigations may be required to demonstrate this and support the intended clinical therapeutic dose.
Should intended or unintended immune system changes be considered adverse?
Four of the case studies were associated with some form of damage or inflammation in the respiratory tract. Three of these were immunomodulators and it is possible that modulation of target pharmacology resulted in alveolar infiltration and progression to alveolar inflammation. Such findings need to be evaluated on a case-by-case basis based on the observed findings and the risk–benefit to the patient. In the fourth case study, pathology changes in the respiratory tract were attributed to a type III hypersensitivity reaction consistent with immune complex deposition (ICD), secondary to ADA induction. Immune complex deposition would be considered adverse to the animal and thus would contribute to the derivation of the NOAEL. However, as previously discussed, ICD in the NHP is not predictive of ICD in the clinic and thus should not drive clinical dose setting.
Is discussion of potential adversity with health authorities warranted?
For some studies, adverse findings in the inhalation toxicity study will be considered attributable to nondrug related issues, such as clearance of particulates, ADA or ICD, but the resulting NOAEL does not provide a margin of exposure of sufficient magnitude to proceed into the clinic. In such cases, it may be useful to take advantage of any presubmission meetings with the health authorities who will receive clinical trial applications for clinical use of these molecules, to discuss the data and hypotheses supporting the conclusion on the NOAEL, and why these are not translatable to the clinical situation. Of the 12 inhaled biologics in the case studies, 8 have proceeded into the clinic and all except 1 molecule had toxicology findings in the respiratory tract and frequently in the BALT in one or more studies. These often presented as inflammatory cell infiltrates and increased macrophages, and for some molecules, this was attributed to the pharmacology. Where comment was provided (5/12 case studies), the findings did not impact the NOAEL.
Only once all of the above have been considered, and all relevant data on the biologic have been assessed, can an NOAEL be assigned. There has been some recent discussion in the industry regarding whether the NOAEL in any toxicology study with a biologic is assigned based solely on adverse effects to the animal, and the human relevance of these findings is then discussed fully in the regulatory documents on submission to the health authorities, or whether the NOAEL should be assigned including the potential relevance to the human as part of the decision-making process. There is a strong preference for the former among the authors of this publication, particularly as knowledge of the molecule expands over the drug development process and some of the considerations regarding human relevance evolve over time. Any conclusions in a GLP-compliant toxicology study cannot be changed after the report is issued, which would preclude any updates in evaluation of the likely human relevance. Whichever approach is used, it should be made clear in the study report.
Conclusions
Although the first inhaled protein-based drug was approved several decades ago (Pulmozyme in 1993), it is clear that the inhaled route is still a relatively novel route for delivering biologics. Inhaled biologics pose both some of the same and additional challenges as those encountered in the development of inhaled small molecules. These additional challenges have further complicated the design and interpretation of toxicology studies with inhaled biologics, further confounded by the paucity of published data describing common methods or common outcomes in previous inhalation toxicology studies with biologics. This in turn made it difficult to convince health authorities that some findings may be peculiar to biologics and were not sufficient to prevent these molecules from entering clinical development. Following discussion at several recent scientific conferences, a working group was formed to author this review, which summarizes the current state of knowledge across the pharmaceutical and CRO industries on inhaled biologics. It also presents the conclusions gained from an anonymized cross-industry survey, which collated data from 12 inhaled biologic case studies assessed in 18 individual inhalation toxicity studies. The 12 molecules comprised 5 peptide/proteins, 3 Dabs, 2 Fabs, 1 mAb, and 1 oligonucleotide and represents a reasonable spectrum of the types of biologics typically found in current drug development. The survey gathered information from each of these studies regarding the technical, toxicological, and PKPD aspects of the nonclinical studies conducted to support clinical studies. An accompanying paper, focusing on the pathology, likely mechanistic origin and immunogenicity-related findings after inhalation of various biologics, has also been published. 6
Data obtained from the collated case studies have provided important new information on best practices in inhalation toxicology studies and have shed new light on issues that occur with some regularity in inhalation studies with novel biologics. To our knowledge, although the number of case studies is relatively small and some questions were not answered for all case studies, the outcome of this industry survey is the largest collation of technical data from unpublished inhalation toxicology studies on biologics to date and consequently provides a rich data source to investigate biologics-specific aspects of conducting inhalation toxicology studies.
A total of 6 key questions were considered for this review and these have been discussed in detail throughout this publication. The key conclusions that can be drawn from this review are summarized below: 1. What technical difficulties are associated with biologics inhalation studies?
Numerous technical difficulties are associated with inhaled biologics, primarily due to their proteinaceous nature. Some are similar to those associated with small molecules but others were specific for biologics, for example, maintaining molecular integrity and pharmacological potency, and minimizing aggregation throughout dosing, to ensure the molecule remains pharmacologically active with unaltered potency.
2. What study design aspects are specific to inhaled biologics studies?
In common with all toxicology studies on biologics, toxicology studies only need to be conducted in pharmacologically relevant species and frequently this is the NHP only. Use of the NHP can have an impact on study design as (1) acceptable dosing durations are shorter for this species compared to rodents and other nonrodents, potentially limiting doses; (2) large amounts of test material are required to dose NHPs; (3) larger sexually mature NHPs may be required, which brings a number of specific issues related to their body size and strength and further compounds the previous 2 factors; and (4) ADA production may alter exposure and/or efficacy in a large percentage of animals in longer studies, which may require increased group sizes.
3. What are the characteristic responses to inhaled biologics and how can exposure be demonstrated at the site of administration?
There are no clinical responses characteristic to inhaled biologics that differ to those frequently observed with small molecule inhalation toxicology studies. As pharmacodynamic responses to the novel biologic are often not clinically evident, exposure or effects on a PD biomarker using lung tissue or BALF are recommended, wherever possible, as well as the relationship to any ADA response.
4. How can we differentiate between a physiological response to foreign biological material and true, human translatable, toxicity?
The case studies suggest there is an association between the presence of ADA and the presence of PV/PB lymphocyte infiltration into the lung and BALT hypercellularity. 6 Other changes that might also be associated with immune stimulation include alveolar inflammatory cell infiltrates and inflammation. Simple increases in alveolar macrophages were also noted and considered likely secondary to clearance mechanisms and, in the absence of damage, were considered nonadverse. This is in line with similar findings with small molecules. 38,39 Other histopathology findings would require further investigation on a case-by-case basis before adversity could be decided upon.
5. What PKPD aspects are important for and/or peculiar to inhaled biologics?
The PKPD analysis of biologics in the lung is complex, owing to the inability to obtain longitudinal PK analysis at the site of administration, the lung. Serum PK is often used as a substitute but there are no established PKPD models that define the relationship between lung and serum concentrations and whether this changes with different biologic formats. In addition, inhaled biologics are frequently associated with a very high incidence (at or close to 100%) of ADA in case studies of over 4 weeks duration and this may require changes to study design to allow sufficient numbers of animals to reach the end of the study still exposed to the biologic. However, as with parenterally administered biologics, the high ADA incidence does not necessarily translate to a high incidence of ADA in humans.
6. How are NOAELs defined for inhaled biologics (vs small molecules)?
Assigning the maximum dose and subsequent determination of an NOAEL for the inhalation toxicity study of a novel biologic is challenging. Many biologics are not toxic per se, as they are highly specific for their target, although effects attributable to exaggerated pharmacology may be observed. It is not advised that doses are increased beyond a reasonable multiple of the intended clinical dose solely in an attempt to induce toxicity or to reach the maximum feasible dose, as information obtained from excessively large dose multiples is unlikely to be clinically relevant. Rather, the maximum dose should be assigned based on factors including species-specific tolerable dosing durations, maximum aerosol concentrations over the required dosing duration, and acceptable safety margins for the intended clinical studies.
Clearly, the analysis of study data from 18 different inhalation studies on 12 different biologics has provided valuable insights into some of the issues faced when conducting toxicology studies with inhaled biologics. This article has collated valuable information on technical matters including aerosol generation issues, the use of young and sexually mature NHPs, PKPD modeling, exposure and immunogenicity assessment, maximum dose setting, and NOAEL determination, in a single reference. Although the current data set is too small to allow firm conclusions, testing of novel biologics remains an active area of research and is likely to remain so for molecules where delivery via the inhaled route has a particular benefit. In the future, it is hoped that others will continue to share their experiences and build on the conclusions of this review to further improve our understanding of these complex issues and, ultimately, facilitate the safe introduction of inhaled biologics into clinical use.
Footnotes
Acknowledgments
The authors would like to thank the following people for their critical review of this article: Graham Paul and Tim McGovern. The authors would like to thank the pharmaceutical companies who donated case studies and all the companies that donated time and resource to the preparation and review of the article via the contributing authors.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.