
Abstract
Drug responses are often unpredictable in juvenile animal toxicity studies; hence, optimizing dosages is challenging. Renal functional differences based on age of development will often result in vastly different toxicologic responses. Developmental changes in renal function can alter plasma clearance of compounds with extensive renal elimination. Absorption, distribution, metabolism, and excretion of drugs vary depending on animal age and kidney maturation. Toxicity can result in malformations or renal degeneration. Although renal morphologic development in humans generally occurs in utero, maximal levels of tubular secretion, acid–base equilibrium, concentrating ability, or glomerular filtration rate (GFR) are reached postnatally in humans and animals and subject to drug effects. Maturation of renal metabolism and transporters occurs postnatally and plays a critical role in detoxification and excretion. Maturation times must be considered when designing juvenile toxicity studies and may require cohorts of animals of specific ages to achieve optimal dosing schemes and toxicokinetics. In recent years, critical end points and windows of susceptibility have been established comparatively between species to better model pharmacokinetics and understand pediatric nephrotoxicity. There are examples of agents where toxicity is enhanced in neonates, others where it is diminished, and others where rat nephrotoxicity is expressed as juvenile toxicity, but in humans as gestational toxicity.
Introduction
Drug-induced kidney injury (DIKI) is an important cause of hospitalization in human patients, with as much as 20% of cases of acute kidney injury (AKI) related to pharmaceutical agents. 1 In humans, renal disorders related to drug overdose or chemical exposure can result in AKI, chronic renal disease (CKD), or nephrotic syndrome and may lead to dialysis dependency or necessitate kidney transplantation. While the majority of cases occur in adults, DIKI may also affect infants or neonates. As regulatory authorities have strived to make pharmaceutical advancements available to pediatric patients, drug research into pediatric medicine has blossomed in the past decade. This has led to enhanced drug labeling for infants and young children, yet today over 50% of drugs used in pediatric patients remain unlicensed or prescribed off-label. 2 Hence, juvenile toxicity studies remain critical to drug development efforts and provide clinicians with relevant safety information in the pediatric population. Juvenile studies in rodents are the cornerstone of pharmaceutical safety assessment decisions regarding pediatric patients, and juvenile studies in rats have been particularly valuable in detecting and predicting nephrotoxicity. Multiple studies have demonstrated there is a high concordance for predictivity of kidney toxicity in rats as compared to human and suggest the rat is a good toxicologic model for human renal hazard. 3,4 However, the efficient design of juvenile toxicity studies faces multiple challenges regarding proper dose, exposure, the timing of critical windows of morphologic and functional maturation, duration of studies, and translatability. With regard to toxicity in the kidney in juvenile rodent studies, it is not uncommon to find unexpected toxicologic renal findings that are not present in adult rodents given similar doses. This is because the kidney undergoes both morphologic (anatomic) and functional maturation during the postnatal period when rodent juvenile studies are performed, and immature kidneys may have vastly different toxicologic responses than mature counterparts. Renal functional differences based on age and stage of functional development will often result in vastly different toxicologic susceptibility. This review explains some of the mechanistic basis of these age-dependent responses in nephrotoxicity.
Xenobiotic Effects on Anatomical Maturation in Rodent Kidneys
Embryologic ontological development of the kidney has been described in detail and involves regression of the pronephros and then mesonephros, to ultimately form the metanephros in both rodents and humans. 5- 7 The ureteric bud of the mesonephric duct undergoes a series of inductive interactions with adjacent mesenchymal cells, which results in significant growth, branching, and differentiation of buds of epithelium. The ureteric bud dilates to form the pelvis, ureter, and collecting duct system and is covered by a cap of metanephric tissue, which will form the nephron. Mesenchymal cells condense around these elongating metanephric buds, which will eventually form tubules and glomeruli. Infolding of primitive epithelium forms comma-shaped structures and then these subsequently form S-shaped bodies, which elongate and differentiate into proximal and distal tubules. 6- 11 Endothelial cells trapped within folds of S-shaped bodies will form glomerular capillary loops and with the patterning of other surrounding epithelial and mesenchymal tissue, eventually form the mature glomerular tufts. Genes responsible for critical developmental pathways during embryogenesis involved in patterning and morphogenesis can be disrupted by drug or chemical exposure, resulting in renal malformations and kidney aplasia. 12- 14 Further, the ureteric bud and the metanephric blastema progenitor cells require correct biochemical signaling in the proper sequence for normal morphogenesis and tubule development. 15 Due to their importance in regulating normal nephrogenesis and tubule differentiation, dysregulated proliferation and apoptosis can lead to the various phenotypic malformations of congenital renal disease, depending on timing. Nephrotoxic effects in the fetal kidney are dependent on delivery of the teratogen or toxin into the fetal circulation, where it then is exposed to the developing renal vasculature. Placental passage of a xenobiotic agent depends on its lipid solubility, size, and the concentration gradient between maternal and fetal circulations. 1,8,16 In humans, congenital anomalies of the kidney and urinary tract (CAKUT) represent a spectrum of structural and functional malformations that include renal agenesis, aplasia, hypoplasia, dysplasia, and a variety of other defects. There are many examples, including colchicine, allopurinol, retinol, and azathioprine, in which drugs or chemicals have induced renal congenital malformations, but the spectrum of disorders is often phenotypically similar despite the variety of inciting agents. For instance, retinol is a demonstrated and potent renal morphogen and can severely disrupt the development of the metanephros and, therefore, has been associated with many cases of kidney aplasia or hypoplasia. Azathioprine given to a pregnant mother results in fetal kidney toxicity but does not alter nephron structure, and instead only delays it. 16 Despite these differences in specific target and pathogenesis, azathioprine may still induce renal hypoplasia, albeit of lesser magnitude than retinol.
Because human renal morphogenesis, tubulogenesis, and tubule differentiation occur during gestation, it is extremely uncommon for CAKUT lesions to represent effects of juvenile nephrotoxicity related to neonatal exposure. Nephrotoxicity associated with drug or chemical exposure in humans after birth is instead more likely to be associated with nephron degeneration, necrosis, or functional renal disorders rather than structural defects, since the kidney is already anatomically formed at the time of birth. Prenatal exposure to a nephrotoxin can result in degeneration, necrosis, or atrophy of specific renal elements without pronounced structural defects. Instead of aplasia or dysplasia, drugs or chemicals that are nephrotoxic in adults and are administered during pregnancy may alternatively result in tubule degeneration in the fetus after transfer across the placenta, especially if late in the third trimester. Significant injury to segments of the nephron can result in severe renal compromise in the developing fetus just as they do in adult humans or adult animals. Examples of nephrotoxic agents that result in degeneration or necrosis of renal tubules in metanephric kidneys include doxorubicin, cisplatin, bleomycin, ifosfamide, and dacarbazine. 8,17 In animal toxicity studies, when severe nephrotoxicosis occurs in the earliest stages of development, there is a high likelihood of abortion and or fetal resorption, and therefore, evidence for extent and character of nephrotoxicity (and hence the mechanism of abortion) may be missing.
In contrast to humans, renal malformations may occur with early neonatal drug or chemical exposure in rodents, particularly in rats. 8 Drug or chemical treatment during neonatal and juvenile toxicity studies may result in altered nephrogenesis and maturation, resulting in either or both abnormal renal morphology or function. This is because tubulogenesis, tubule differentiation, and terminal fusions of significant structures such as the ureteropelvic junction occur postnatally in rodents. 2 As noted with prenatal human exposure, degenerative lesions in the kidney may also occur in rat or mouse kidneys as a result of neonatal xenobiotic exposure, either as a primary lesion or as a secondary degenerative, cystic, or atrophic process as sequelae to the original malformation. 14
Nephron maturation occurs proximally to distally in an orderly process in which the proximal tubules form initially and distal medullary structures form last during kidney development. 2 Nephrogenesis is completed in humans in utero by gestational week (GW) 36 with a full complement of nephrons in each kidney at birth, 2 while it continues postnatally in rats until at least postnatal day (PND) 11 (Table 1). 2,8,9,17 In mice, histomorphologic examination of kidneys suggests tubule formation is completed at birth, 18 but based on elegant molecular experiments in neonatal mice, it is clear that nephrogenesis actually continues for a few days postnatally. Investigators have found that labeled mouse renal tubule progenitor cells do not disappear until PND 2 to PND 4, indicating that nephrogenesis is not fully completed in mice until this time. 7,19- 21 Additionally, embryonic gene expression experiments have shown that branching of the tips of renal tubules is not completed until PND 2 in mice. 2,19,21 Although nephrogenesis in human kidneys is completed prior to birth, there is still some postnatal nephron maturation and tubule elongation that occur until approximately 1 year of age, accompanied by a large increase in tubular volume. Tubule differentiation occurs up to PND 21 in rats but is completed just after birth in mice. 8,19- 21 Tubule elongation in rodents may occur for several weeks after birth in rodents, but slows considerably after PND 28. Vasculogenesis, the process of blood vessel maturation, is completed by PND 17 to PND 19 in rats and by PND 7 in mice but is already completed at birth (before GW 36) in humans. 6- 9,18,22 These comparative timings of kidney structural development can be incredibly important for the toxicologist or pathologist to understand if findings in juvenile toxicity studies are to be put in proper context and clinical risk assessment best interpreted and most fully explained. There are multiple examples of juvenile toxicity in rodents that have gestational clinical implications based on differential renal maturation.
Comparative Age at Completion of Anatomical Maturation in Kidney.
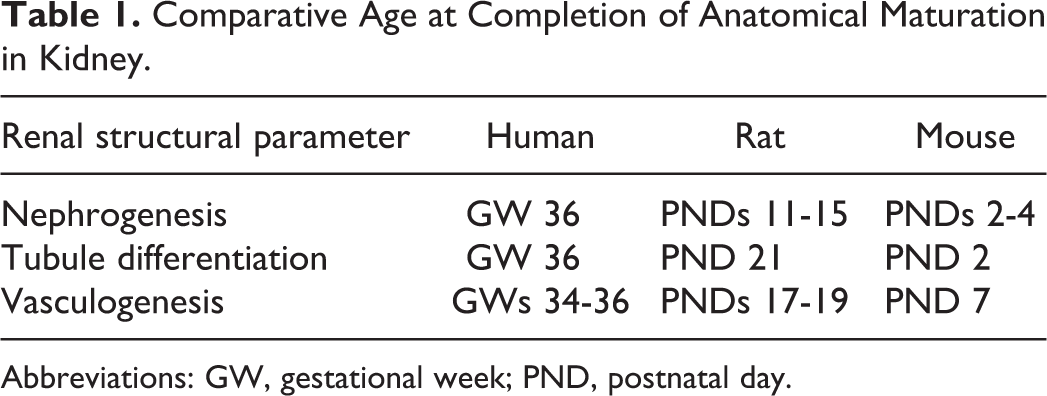
Abbreviations: GW, gestational week; PND, postnatal day.
When angiotensin-converting enzyme (ACE) inhibitors such as enalapril are given during pregnancy in a variety of species, teratogenic kidney effects can occur such as tubule dilation, acute tubular necrosis, renal developmental abnormalities, and immature glomeruli as well as pronounced effects on amniotic fluid volume and neonatal renal failure. 23,24 The ACE inhibitors are, therefore, contraindicated throughout pregnancy. In both humans and monkeys, these significant and devastating effects on renal morphogenesis occur when ACE inhibitors are given in the past few weeks of pregnancy, but not when administered after birth. The ACE inhibitors are routinely given to human infants older than 1 year for hypertension and are considered safe in pediatric patients after 12 months. The molecular pathogenesis for teratogenic effects during pregnancy has been elucidated and involves tubule dysmorphogenesis, but ACE inhibitors may also induce fetal hypotension, decrease renal blood flow (RBF), and disrupt the maternal and fetal renin–angiotensin axis. 23,24 In fact, angiotensin II (AT II) activity is required for multiple steps of renal morphogenesis, and the angiotensin I receptor is involved in the development of the renal pelvis. 24,25 In AT IIr–mutant mice, an association was noted with congenital renal anomalies. A similar gene mutation in AT II receptor was noted in some humans with CAKUT. 26 Thus, the molecular basis and pathogenesis of both congenital renal disease and teratogenic drug intoxication were similar involving interference with tubule development, in conjunction with hemodynamic compromise. 24- 27 In contrast, the critical window for renal developmental effects in rats is different and generally limited to fetal exposure 5 days prior to birth and continuing postnatally until about PND 14. 27 Postnatal ACE inhibitor administration in rats results in similar renal morphogenic effects to those noted when other species such as monkeys or rabbits are given the same drugs during pregnancy. Species differences in the timing of nephrogenesis are largely responsible for these windows of sensitivity. Since renal development and nephrogenesis in humans and monkeys are completed before birth, postnatal exposure does not result in renal dysmorphogenesis or renal degeneration. Conversely in rats, since nephrogenesis extends into the postnatal period, rat neonatal exposure to enalapril can result in juvenile toxicity. Nephrogenesis, tubule differentiation, and branching in rat kidneys are all completed well before PNW 4 (tubulogenesis as early as PND 15 and differentiation by PND 21) and the formation of nephrons has already ended by PND 10. 2,8,28 Hence, susceptibility to ACE inhibitor toxicosis will be very limited after 3 weeks of age in rats. 8,22- 27
2,3,7,8-Tetrachlorodibenzo-p-dioxin (dioxin) is an environmental toxin that is teratogenic and capable of inducing renal dysmorphogenesis, including renal dysplasia and increased incidence of polycystic kidney disease (PCKD). The mechanism for many renal teratogenic effects is related to direct damage to renal tubules and importantly effects on cilia, which control fluid flow and regulation of apoptosis during nephrogenesis. 29 The cystic effects produced by dioxin in multifocal tubules are specifically associated with alterations in cilia function. The pathogenesis of many inherited cystic renal diseases including autosomal dominant PCKD involves gene mutations encoding polycystin-1 and polycystin-2, which are critical to ciliary function. 30 It appears that some cases of PCKD may be related to drugs or chemical exposure in utero with compounds that have similar effects to dioxin on ciliary function. However, an interesting but separate toxic phenomenon occurs postnatally in rodents exposed to dioxin and represented as juvenile toxicity. Rather than multifocal cystic tubules found in the cortex, there is marked dilation of the renal pelvis with radiating bands of dilated tubules extending through the medulla into the cortex. Hydronephrosis is encountered in juvenile rodents with increased frequency associated with a handling artifact 31 but may also occur as an induced toxic drug response to dioxin and other agents. 32- 36 Hydronephrosis induces compression atrophy and other degenerative changes in tubules of the renal cortex and may even result in neonatal mortality. Many cases of drug-related hydronephrosis, such as those associated with sulfonamide antibiotics, are related to the formation of crystals from drug precipitation that fill and obstruct the distal tubules or the ureters. 35 However, chemicals such as dinoseb, methyl salicylate, and ethylenethiourea can induce hydronephrosis, independent of crystalluria and obstructive nephropathy. 36,37 Hydronephrosis has been observed in fetal and neonatal rats exposed to dioxin during gestation and the neonatal period and is attributable to anatomical and functional blockage. The molecular pathogenesis for these changes is based on activity of the aryl hydrocarbon receptor (AHR), which is involved in ureteropelvic junction maturation. 33 The mechanism further is complicated by additional AHR-mediated effects on renal fluid transporters such as aquaporin-2 (AQP-2), causing electrolyte and water imbalances and fluid overload in juvenile rodents. 32,33 Proof of this concept has been demonstrated in both AHR and AQP-2 transgenic mice who exhibit hydronephrosis of varying degrees. 32,33,38 As noted later in this manuscript, tubule transporter maturation also tends to occur in rats in the first 4 weeks postnatally, so rodent kidney development takes a double hit with dioxin toxicosis compromising both structural and functional development in kidneys. The specific pathogenesis of hydronephrosis associated with dioxin is related to effects on later processes of renal morphogenesis. The ureteropelvic junction, which is responsible for initiating peristaltic movement along the lower urinary tract, must be patent and mature for proper fluid dynamics within the nephron. If fusion of this structure is perturbed by disrupted development, alterations in fluid flow result in obstruction and hence dilation of the renal pelvis. Secondary ureteral epithelial hyperplasia further impedes filtrate flow and potentiates obstructive uropathy. Anatomical maturity and structural integrity of the ureteropelvic junction develop in parallel with distal medullary tubule nephrogenesis. 33 This case illustrates how renal maturation differences can influence clinical risk assessment. Rat ureteropelvic junction maturation occurs much later (up to PND 18) comparatively than in humans where tubule nephrogenesis is completed entirely in utero and the ureteropelvic junction structurally matures just before or around the time of birth. Hence, if AHR-mediated effects on morphogenesis are a suspected mechanism for hydronephrosis for a drug, there may be a greater gestational clinical risk instead of a pediatric risk of nephrotoxicity based on species differences in the timing of developmental differentiation.
Xenobiotic Effects on Functional Maturation in Rodent Kidneys
As noted above, disruption of anatomical maturation in the human kidney is much more likely to be a gestational, rather than juvenile, toxicity issue. In contrast to the rare examples of structural kidney maturation mechanisms of clinical pediatric nephrotoxicosis, defects in functional renal maturation can be a major factor in both human and animal susceptibility to renal toxicants in the neonatal period. Functional development lags behind anatomic or structural maturation in all species. 2,5 Because of this extended period of functional development, the kidney remains particularly vulnerable to toxic insult in the early postnatal time frame. For example, developmental changes in renal function in the period following birth can alter plasma clearance of compounds with extensive renal elimination, resulting in massive swings in pharmacokinetics of a particular xenobiotic agent over a few weeks after birth in rodents. Absorption, distribution, metabolism, and excretion (ADME) of drugs vary depending on the age of the animal and organ maturation stage, which will lead to differences in toxicity and efficacy. Many functional developmental end points do not reach maximal levels until well after birth, and like anatomical maturation, these processes do not exactly match-up between humans and rodents in order or sequence. Specific developmental events such as maximal levels of tubular secretion, acid–base equilibrium, concentrating ability, or GFR are reached postnatally in both humans and animals, and these processes either affect xenobiotic concentrations in the kidney and/or may be subject to perturbation by the same agents. Maturation of GFR, RBF, renal transporter function, and metabolic pathways will have a huge impact on renal clearance and local concentration of drugs and chemicals in proximal tubules. In recent years, critical end points and windows of susceptibility have been established comparatively between species to better model pharmacokinetics and understand potential pediatric or gestational drug toxicity in the kidney, and these are discussed in more detail below.
Renal Blood Flow and Glomerular Filtration Rate and Effect on Nephrotoxicity
In human embryos prior to GW 34, there are minimal functional demands placed on the fetal kidney, but functional burden increases dramatically toward the latter part of the third trimester and after birth. The adult human kidney receives 20% to 25% of cardiac output, while the fetal kidney receives only 4% of cardiac output. This value increases rapidly to 10% by PND 7 in human neonates. In concert with blood flow, the contribution of the kidney toward maintaining the body’s homeostatic fluid balance also starts to emerge in the third trimester in humans. 39 Renal blood flow progressively increases during gestation to reach maximal levels by GW 32 to GW 35 in humans (Table 2). Paradoxically, RBF values at birth are actually less than those in adults even when corrected for size or weight, due to the requirements for transition of homeostasis postgestation. This transition is characterized by marked physiologic functional changes that facilitate adaptation to life outside the uterus. The increase in RBF is accompanied by markedly elevated glomerular filtration pressure, resulting in a rise in glomerular filtration rate (GFR). Historically in rodents, RBF was measured indirectly using para-aminohippurate (PAH) clearance and, more recently, has involved actual measurements using techniques such as flow probes, transducers, and now magnetic resonance imaging. Laboratory animals all tend to have different trends for alterations in blood flow during the perinatal period. In rats, RBF decreases from PND 1 to PND 3, but then increases gradually to peak in the week prior to PND 24. 2,40,41 Mice appear to follow a slightly different pattern, with a rapid increase in renal perfusion from birth to PND 3 and PND 7, then values tend to level off with peak levels between PND 16 and PND 22, although one author noted a peak in RBF in CD-1 mice at PND 13. 2,42 In nonhuman primates, RBF is similar at birth and the early neonatal period, and in rhesus monkeys, it has been shown to only double from PND 1 to adulthood. The changes in blood flow in rodents can result in decreased amounts of drug reaching the kidney in the same span of time as compared to adults, resulting in marked shifts in local concentration and exposure. Based on RBF alone, one would predict that all drugs would be better tolerated in infants and pediatric patients than in adults since drug exposure is low. This is in fact the case for a variety of agents such as vancomycin, gentamicin, cisplatin, and mercuric chloride. Renal toxicity associated with these agents is less severe in neonatal animals as compared to those reaching maturity. 43 This difference in susceptibility has been shown to be due to markedly different maximal drug concentrations (C max), which increase proportionally with age and are really a reflection of RBF maturity. Thus, the renal tubule epithelial cells of neonates are receiving lower local drug concentrations of these particular agents than older animals given the same dosage. Unfortunately, there are a whole host of other factors (described below) that also affect local renal concentrations in the tubules and cause drug concentrations to increase in proximal tubules and/or urine filtrate. This makes predicting and modeling drug exposures in pediatric patients and neonatal rodent kidneys very difficult. This gap in knowledge often necessitates initial dose ranging cohort experiments in neonatal animals of different ages to better predict optimal drug exposures in definitive good laboratory practice juvenile toxicity studies.
Comparative Age of Full Maturation of Renal Functional Development.
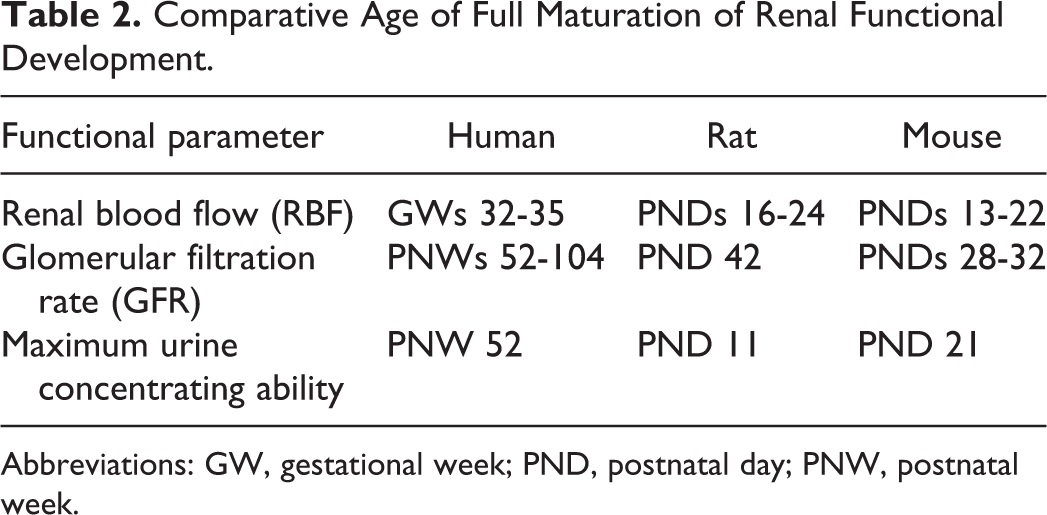
Abbreviations: GW, gestational week; PND, postnatal day; PNW, postnatal week.
During gestation in humans, GFR remains relatively low, but rises rapidly in the first few weeks of the postnatal period, after which it increases steadily and reaches adult levels at 1 to 2 years of age after correcting for body weight or size. 10,21 The variety of methodologies used for determining experimental values of GFR in rodents has resulted in widely variable standard reference values for GFR in rats and mice. It is, therefore, difficult to compare between studies, but values appear to increase in rats between PND 3 and PND 18. 2 Peak levels for GFR are not reached until PND 28 to PND 42 in rats. 10,44 Because of logistical problems in obtaining values for GFR in neonatal mice, standard ranges are even more variable than in the rat, but trends tend to be similar and values or standard ranges in mice of various ages are available. 18 Maximal GFR is reached in mice around PND 28 to PND 32. 2
Rodent fetuses and early neonates have inefficient urine concentrating ability, as a result of short proximal tubules and inefficient salt gradients. Tubular function related to maximal concentrating ability is dependent on the length of the loop of Henle and hence the depth of the medulla. In human kidneys, maximal concentrating ability is not comparable to adults until at least 12 months of age and in rat kidney until about PND 11 (Table 2).2.7,10 Maximum concentrating capacity in mice kidneys is achieved at around PND 21.
Renal Transporter Maturation and Effect on Nephrotoxicity
The identification of renal transporters in the kidneys of animals and humans has become increasingly important in pharmacokinetic modeling, prediction of drug–drug interactions, and for elucidating the mechanism of nephrotoxicity. 45,46 A giant leap in understanding these processes in pediatric patients and how juvenile kidney studies could model these interactions came with recent investigational reviews of transporter maturation across species. 2,47,48 Renal transporters play a critical role in moving substances from tubules to the urine filtrate for excretion and transporters develop at different times in different species. Most renal transporters are localized to the proximal tubules. 49 There are both species differences in expression patterns and differences in maturation timing between rodents and expression can further be influenced by gender. Renal transporters are generally immature at birth across species, with maturity occurring alongside other functional parameters in the kidney. 50,51 Uptake transporters occur on both basolateral and apical surfaces of proximal tubules, and function to bring substances into the cell from either blood or urine, respectively. Inhibition or dysfunction of the transporters can reduce the efficiency of removal of a xenobiotic agent from the blood or increase the intracellular accumulation of drugs or chemicals within proximal tubules, dependent on their location and function. Drug–drug interactions at the level of basolateral transporters typically decrease the clearance of an affected drug, causing higher systemic exposure and potentially increased systemic extrarenal organ toxicity. Conversely, interactions at the apical surface can also lower drug clearance, but may be associated with even higher renal toxicity, due to intracellular accumulation within tubules and failure of secretion into the urinary filtrate. Proximal tubule reuptake transporters include organic anion transporters (OAT), organic cation transporters (OCT), the organic cation/carnitine transporters (OCTN), and the organic anion transporting polypeptides (OATP; Table 3). 52 They can be critical to the mechanism of some types of juvenile renal toxicity in rodents, in addition to effects on pharmacokinetics related to drug–drug interactions. Organic anions, including a variety of drugs and chemicals, generally tend to be transported by OAT1 and OAT3 on the basolateral side of proximal tubules and multidrug-resistance protein (MRP) 2, MRP4, OATP1A2, and breast cancer–resistance protein (BCRP) on the opposing apical or brush border surface. Other specific transporters found in the kidney include multidrug and toxin extrusion 1 (MATE1), uric acid transporter 1 (URAT1), and peptide transporter 2 (Pept2; Table 3). The secretion of PAH is often used as a surrogate for OAT1 secretion in investigative experiments. Antibiotics and other drugs such as cephaloridine inhibit PAH transport and are thus considered substrates of OAT1, but other OAT1 substrates such as probenecid and cimetidine are also often used in drug coadministration studies to investigate OAT1 activity. Cephalosporins may be transported across the apical cell border by PEPT transporters, while other drugs utilize a variety of other transporters. Many organic cations are transported by OCT2 on the basolateral border at the blood–epithelial interface. 2,53- 55 For instance, cisplatin uptake into proximal tubules is dependent on OCT2, among others, and its nephrotoxicity can be ameliorated by inhibiting renal OCT2 transporter activity with quinidine. There are some differences between human and rodent expression patterns in transporters. The OATP4, which transports statins and cardiac glycosides, is highly expressed in human renal tubules, but different isotypes appear to function in its place in rodents. 52,53
Comparative Development of Selected Renal Transporters Across Species.
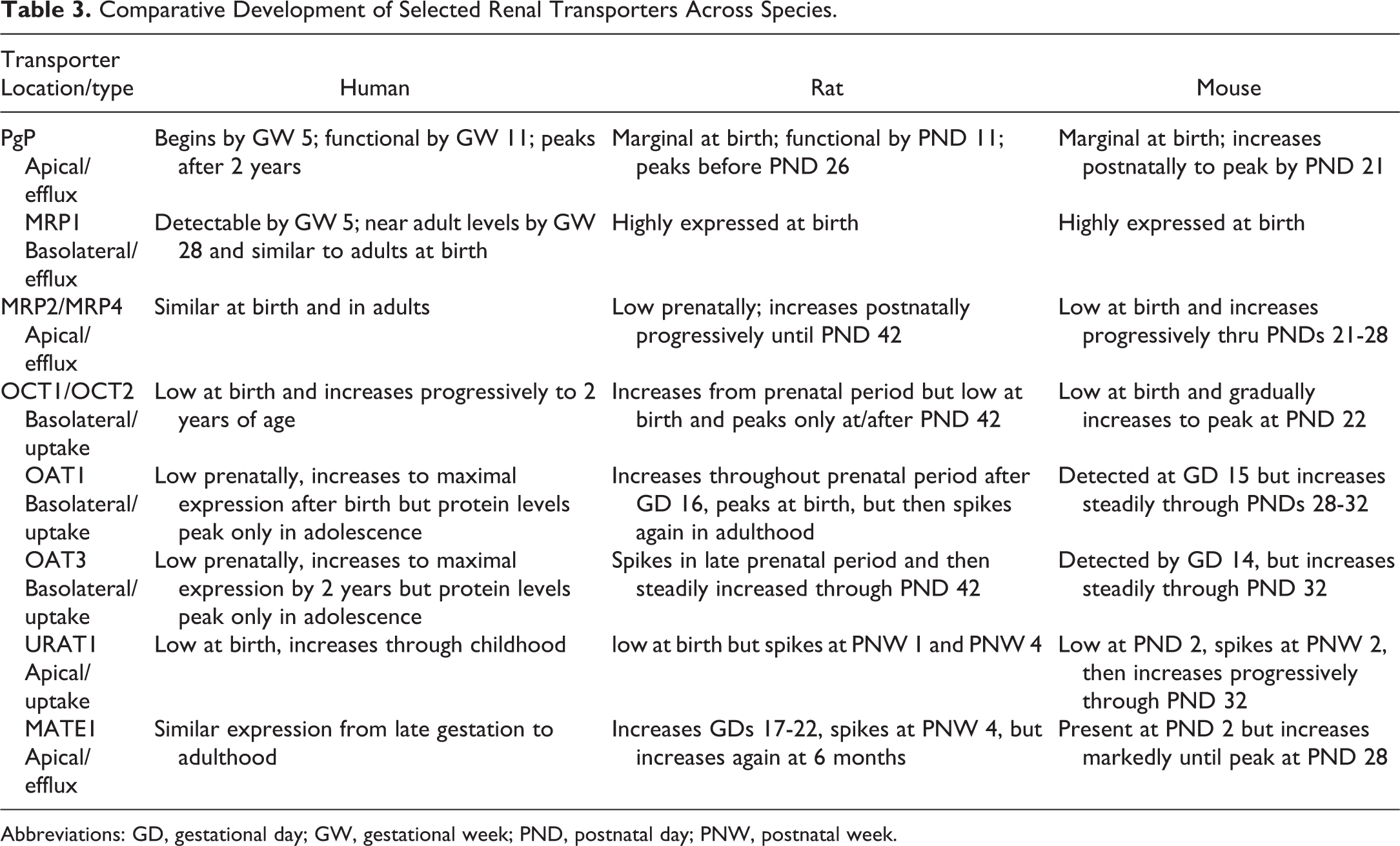
Abbreviations: GD, gestational day; GW, gestational week; PND, postnatal day; PNW, postnatal week.
Completion of maturation of renal transporters occurs differentially but occurs almost universally after birth across species. Kidney transporters are thus immature at birth in humans and as a rule of thumb in rats and mice mature around PNW 3 to PND 6, with some variation between transporter classes. P-glycoprotein (PgP) is a widely dispersed multifunctional drug transporter that is expressed in utero in human kidneys by GW 5.5 and is pronounced by GW 11 and throughout gestation. 2,56 However, peak PgP expression and protein levels are not reached until preadolescent childhood. 2,47 The PgP expression in fetal rat kidneys is low but increases during the length of gestation and steadily increases from birth, with maximum levels achieved between PND 11 and PND 26. In mice, peak levels are achieved by PND 21, but a dramatic decrease has been noted in male mice at PND 45, which remains unexplained. 57,58 In humans, OAT1 is low in infants and then increases through childhood. 2,47 In rodents, OAT expression is similarly low during gestation but increases substantially within days after birth. 52 Maturation of selected renal transporters is listed in Table 3. Expression of OAT1 and OAT3 are noted as early as day 16 of gestation in rodents but increase dramatically between birth and PND 21 and are considered mature by PND 28 with a steady rise in expression until adulthood. 50 Like the OATs, other transporters such as MATE 1, OCT1, OCT2, OCTn1, OCTn2, and URAT are present during gestation at low levels in rodents and increase after birth. 51,59 Gender differences in the expression of some transporters have been noted in mice, especially after PND 30. 60 The OCT2 shows gender differences in rats, with female expression levels peaking at PND 21 and then dropping slightly over time as compared to much later peaks in males. 2 Such gender differences (and maturation times) are often only noted in specific carriers, not in related transporters of the same class. For example, despite similarities in function, OCT3 reaches maximal levels by PND 35 to PND 40 in both sexes of rats. 54 It should be noted that messenger RNA (mRNA) levels do not always equate with actual transporter protein abundance. For instance, OAT3 mRNA expression is low in human infants and reaches adult levels prior to 2 years of age, but peak protein levels are only reached in adolescence. 48
The vast majority of protein in the urine filtrate is actively reabsorbed in animals and humans by tubules. Proteins small enough to filter through the glomerulus, including intact and especially fragmented albumin, bind to the brush border of proximal tubules and are reabsorbed via endocytosis into endosomal vacuoles, which then fuse with lysosomes for catabolism. Protein transport depends on size, charge, and shape of the particular protein or amino acid, and in the rat, there is a huge difference based on gender, with female rats reabsorbing much more protein than males. 61,62 It has only been recently realized that protein reuptake in the proximal tubules may also be age dependent and reflect renal maturation. The endocytic receptors megalin and cubilin are essential receptors responsible for the reabsorption of proteins. 63 Megalin is a 600-kDa glycosylated receptor on the apical surface of proximal tubules that regulates receptor trafficking and endocytosis, whereas cubilin is a 460-kDa glycosylated protein that forms a complex with megalin to promote internalization of proteins and the multimeric complex depends on other proteins such as amnionless (AMN) for proper function. Both megalin and cubilin receptors are abundant in the mesonephros and ureteric bud at GD 11 and GD 14 to D 15 in rodent embryos, respectively, but expression disappears as the nephron develops. 62,64 It is believed that these transporters are necessary for proper nephrogenesis. They begin to be expressed later in gestation in rodents as the proximal tubules are formed and are largely confined to this location in the kidney, but with minor expression also noted in glomerular podocytes. Maturation of these transporter complexes within proximal tubules appears to occur near or just after birth, at least in rodents, although the precise date has not been determined with certainty. Megalin may bind and mediate proximal tubule uptake of several potentially nephrotoxic drugs and chemicals, including several aminoglycoside antibiotics. 65 It may also be important in uptake of some antisense oligonucleotides (ASOs) when bound to fragmented albumin (free ASOs are generally absorbed in tubules via clathrin- and caveolin-mediated transporters independent of megalin). 66 Hence, there is the possibility that protein transporter maturation could affect drug tolerability in very young neonates, but drug interaction with this receptor complex is more likely to involve in utero fetal toxicity. Fortunately, ASOs do not tend to cross the placenta. 66 With tubule damage or immaturity, excessive protein can build up in the urine, leading to further potentiation of tubular injury, interstitial inflammation, and/or fibrosis. Once proteins are broken down in phagolysosomes, free amino acids are released and transported across the basolateral membrane by specific amino acid transporters back into blood, but no developmental data are available on these transporters in the kidney of humans or animals.
When applying ADME principles to help predict exposures in juvenile toxicity studies or to help explain toxicity related to highly unexpected blood concentrations or pharmacokinetics in neonatal rodents, it is helpful if transporter (drug–drug interaction) data have already been performed in rat livers with a given agent. This is most often the case with pharmaceutical products and is fortuitous based on typical timings of most drug development programs where juvenile toxicity studies are often postponed until phase III clinical trials are underway. Although transporter data are not always freely applicable or transferable from liver to kidney, it is quite common to discover that a candidate drug is a substrate for a particular OAT or OCT transporter in hepatocytes, and it can be assumed that it is also a substrate in kidney. There is also often early information from in vitro receptor screening paradigms (which may contain information on hundreds of extracellular and intracellular receptors and transporters) demonstrating whether a candidate has the potential to inhibit or bind to a particular transporter at physiologic concentrations. This information is important, but also needs to be put into context regarding whether the transporter is an uptake or efflux type, on which cellular surface (basolateral or apical) it resides, binding coefficient at physiologic concentration and whether the drug or its metabolite is present in the urine. Pharmacokinetic profiles are also important to determine whether there may be a very high maximum blood concentration (C max) or if drug exposures remain high for prolonged periods, which can point to critical windows of susceptibility. The most relevant and helpful investigative studies to elucidate a potential transporter as a mechanism for juvenile nephrotoxicity are coadministration studies using cimetidine or probenecid for OATs and coadministration of other compounds (eg, quinidine for OCTs), which are substrates for other transporter classes in a competitive inhibition-type assay. The demonstration of amelioration, potentiation (or no change) in toxicity based on comparative histopathology is reflective of transporter influence or lack of involvement depending on outcome. 46,47 Novel renal biomarkers such as urinary albumin, kidney injury marker-1, neutrophil-associated gelatinase lipocalin, as well as standard analytes such as urinary protein, serum creatinine, and urea nitrogen have been successfully utilized in juvenile toxicity investigative studies to monitor for renal toxicity in rodent neonates as young as 9 days of age. 45,46 Reference ranges for normal values appear similar to adults, but must be normalized to urine creatinine (and not specific gravity due to dilute urine).
Metabolism Maturation in the Kidney and Effect on Nephrotoxicity
Maturation of enzymes such as the Cytochrome P450 (CYP450) metabolism systems can potentiate or degrade the toxic potential of a xenobiotic agent within the kidney, as well as have a profound effect on local parent drug concentrations. The kidney plays a less prominent role in drug metabolism than the liver, and only a small portion of recognized metabolizing pathways have been identified in renal tissue, especially in pediatric human patients or in neonatal animals. The CYP450 content of the kidney is only 10% to 20% of liver, but the concentrations in tubule epithelial cells in the pars recta (S3 segments) in the outer stripe of the medulla are comparable to hepatocytes. 1 A xenobiotic may be metabolized in the kidney to a reactive intermediate in this region by a variety of local enzymatic systems. Phase I reactions involve oxidation, reduction, or hydrolysis to produce more water-soluble metabolites and facilitate excretion in urine. In phase II metabolism, a reactive intermediate can conjugate with glutathione to form a stable inactive metabolite, or in some cases, certain xenobiotics are metabolized to sulfur-containing intermediates by conjugation with glutathione, which after further metabolism eventually forms reactive thiols that can injure the surrounding tubules. 62 Among the CYP450 family, only CYP2B6, CYP3A5, and members of the CYP4 family have been found in human kidney in significant levels and closely related homologues appear to be present in rodent kidneys as well. Maturation of renal metabolism can be extremely confusing, because expression levels of enzymes do not increase linearly and instead may go up and then disappear. Neonatal animal data on metabolism maturation in general are sparse, and the juvenile animal data to-date do not demonstrate any clear patterns or allow comparison between enzymatic classes or between species regarding timing. Evidence in mice indicated CYP2B9 is mature by PNW3 and present at relatively high concentrations until PNW 10. CYP2B9 then declines between PNW 10 and PNW 10 months of age in females, but not male mice. 67 In humans, CYP2B9 is expressed in renal tubules in utero from GW 5 to GW 20, with expression in adults declining as compared to the neonatal period. CYP3A2 is present and has been shown to peak by PNW 2 in neonatal rat kidneys. 68 However, no comparable data are available for humans or mouse kidneys. CYP3A4/5 enzyme metabolizes a variety of drugs, including statins, benzodiazepines, calcium channel blockers, and viral protease inhibitors, and has been demonstrated in the human fetal kidney by GW 28, but in mice the predominant isoform CYP3A11 was undetectable at any time point between PNW 3 and PNW 10 months of age. CYP3A25 was noted in female mice, but not in males, at PNW 9 to PNW 10. 68- 71 CYP4 family members, which metabolize arachidonic acid, are found in adult human kidneys, but expression in pediatric patients or juvenile animals is largely unknown. CYP4F4 expression was evident as early as PNW 2 in rats, peaked by PNW 8, then declined between PNW 12 and PNW 16. 69 CYP4F5 showed a similar pattern in rats, while CYP4F6 expression increased steadily from PNW 2 to PNW 12. By PNW 18, CYP4F6 levels were reduced to their 4-week peak levels. 72 CYP2C8 enzyme metabolizes drugs such as chloroquine, paclitaxel, and rosiglitazone and, in humans, is expressed in the first trimester and stably expressed throughout pregnancy. Expression increases in the postnatal period to adulthood, but no comparable animal data have been generated. 73,74 Similarly in humans, CYP2C9 is expressed in the first trimester (GWs 5-12) and throughout in utero development in the kidney, with peak expression only in adulthood. 69,72 In rats, CYP2C23 expression increased progressively from birth until declining at PNW 3 around weaning. 68,73,74 In mice, CYP2C37 is expressed by PNW 3 to PNW 4 with expression increasing until adulthood, with a peak in females at PNW 9 to PNW 10. 67,68 CYP2C29 has been identified in male mice at PNW 3 to PNW 4 at very low levels until PNW 9 to PNW 10, but in female mice, it has only been found at PNW 9 to PNW 10. 67
Another important route of metabolism in adult kidneys involves the uridine glucuronyl transferase (UGT) catalyzed conjugation reactions, but few have been described regarding ontogeny or maturation in kidney. UGT1A1 was detected in both the mesonephros and metanephros stages of human renal development. 75 UGT2B7 is found in even greater concentration in fetal kidneys as compared to liver, and expression continues postnatally. 76 Unfortunately, no rodent data are available at this time. Sulfotransferases (SULT) are present and active in the kidneys, but ontogeny data are scarce. 77 SULT1A1 is detectable in humans at PNW 15 and is steadily expressed until 18 months of age, 78 while SULT1A3 expression is higher in human fetal kidneys compared with perinatal or adult kidneys. 79,80 SULT1C2 is also expressed highly in human fetal kidney, while SULT2A1 is expressed beginning in the second half of gestation and achieves levels comparable to adults in the neonatal period. 81 In rats, SULT1C1 is minimally expressed during gestation, while SULT1C2 expression has been shown to be highly expressed in the kidney during gestation. 82,83 In mice, many SULT enzymes have been shown to be extremely low or absent during gestation, but SULT1C1 and SULT1D1 were found from 2 days before birth peaking at PND 45. SULT1C2 increased in expression in mouse kidneys from 2 days before parturition up until birth and remained stable up to PND 10, after which it declined. Among the glutathione S-transferase (GST) family of enzymes, GSTα protein is detectable in human kidneys from GW 8 onward, with peak activity at 2 years. 84,85 GSTµ enzyme is also detectable during gestation and remains constant after birth through adulthood. 84,85 GSTπ is highly expressed during gestation in humans, but declines after birth. Comparative data from animal species are extremely limited. 2 In rats, GSTα is present by PNW 1, then increases between PNW 1 and PNW 4, while GSTπ levels are relatively stable between PNW 1 and PNW 4. 86
Based on renal metabolism data accumulated to date in animals, it is apparent that no real patterns emerge and it is very difficult to compare enzyme maturation periods from one class to another or from one subclass to another related subclass, much less between species. However, maturation of a given enzyme pathway could play a major determinant in renal toxicity, if metabolism of a drug is dependent on that enzymatic function for detoxification and/or renal elimination and a given enzyme system is not functionally mature. Certainly, metabolism of a xenobiotic agent can effectively diminish the local or systemic concentrations of a parent drug or chemical, and this will have a profound effect on toxic potential in the kidney, depending on whether the metabolite has greater or lesser nephrotoxicity. For instance, 3,4,5-trichloroaniline is a chemical used in a variety of commercial and industrial products and is a recognized nephrotoxicant. When kidneys are pretreated with the CYP450 inhibitor piperonyl butoxide, the cyclooxygenase inhibitor indomethacin, or a peroxidase inhibitor mercaptosuccinate, renal tubule necrosis usually associated with exposure to this agent is significantly reduced in rodents. This indicates that bioactivation to toxic metabolites contributes to the mechanism of nephrotoxicity. 87 These types of coadministration studies can help pinpoint a responsible metabolic cascade, but metabolism must first be suspected as a potential mechanism in a juvenile study. A fortunate clue for the nephropathologist in establishing mechanism is that the majority of xenobiotics requiring metabolic transformation to exert nephrotoxic injury preferentially affect the pars recta, where the metabolic activity of the nephron is localized. Many chemicals requiring metabolic activation will, therefore, often result in greater degeneration or necrosis in the S3 segments of proximal tubules (or distally into the medulla) as compared to other segments. Examples of metabolic activation-related nephrotoxicity in juvenile animals are much harder to reference than transporter mechanisms, since ontogeny of metabolic pathways has just recently been investigated in neonates. However, there are a few previously published examples of juvenile toxicity related to functional maturation activity that colorfully illustrate the potential impact of metabolism on drug development and one of these case studies is listed below.
Dabrafenib is an anticancer drug, which demonstrated unexpected renal toxicity in juvenile rat studies. 88 Kidney lesions were not noted in preclinical toxicity studies in adult rats given the drug. Microscopic findings in the kidney of juvenile rats included mineralized crystals within tubule lumina, associated with an increased incidence of cysts, tubular dilation, and tubular basophilia. These kidney adverse findings were present when dosing was given between PND 7 to PND 35, but not afterward. 10,88 Subsequent investigative studies found changes consistent with obstructive nephropathy and crystalline intraluminal deposits in the medullary collecting ducts, and secondary degenerative cortical tubule lesions. The exposure was similar to adult rats and did not change significantly from PND 13 through PND 35, excluding unexpectedly high systemic parent drug concentrations as a causal factor. Using tissue matrix-assisted laser desorption/ionization and liquid chromatography–mass spectrometry, dabrafenib and its metabolites were detected diffusely localized within medulla, with the highest concentration in rats dosed from PND 7 to PND 13. 88 Of interest, the predominant metabolite with highest concentration in this age group was carboxy-metabolite following by a hydroxy-metabolite, both of which are lacking in adults. The renal tubular deposits were identified as heterogeneous mixtures containing calcium, phosphate, potassium, and lipids and did not contain the parent drug or metabolite. As calcium or phosphate is not reabsorbed in the collecting duct, the mineralization was considered dystrophic. 36 It was concluded that specific metabolites of this anticancer drug more common in juvenile rats than in adults were resulted in toxic effects on medullary collecting ducts and thus epithelial degeneration and necrosis. Dystrophic mineralization resulted from damage to collecting duct epithelium and was visualized as luminal crystallization, aggravated by disturbed local calcium–phosphorus tubule concentrations related to collecting duct degeneration. Calculi, once formed, are very slow to resorb and hence were still present at PND 35. This was in spite of the fact that the crystals were likely only formed during the critical window from PND 7 to PND 15. 88 Maturation of metabolism can partly explain this window of age sensitivity since decreased conversion to less active (adult) metabolites was noted and the metabolites were concentrated in distal nephron segments (suggesting local renal, not hepatic, metabolism). Extrarenal clearance may also have been a factor, as increased kidney concentrations may have been related to lack of developed biliary excretion (important for this drug) in pups less than 2 weeks of age. In discussions with regulatory authorities, it was recognized that there is a theoretical nephrotoxic risk with dabrafenib for human infants younger than 1 year when the metabolite/clearance profiles and renal functional maturation rates are compared between juvenile human and rat kidneys, but overall clinical risk in pediatric patients is minimal, given the implicated metabolites are not present in significant concentrations in either human adults or infants. This toxic principle is, therefore, both age- and species-specific.
Maturation of Other Enzyme Systems and Effect on Nephrotoxicity
In addition to transporters and metabolic enzymes, there is differential timing of expression of other enzymatic systems before or after birth that may interact with a candidate drug. For example, the effect of cyclooxygenase-2 (COX2) inhibition on prostaglandin control of vasodilation in the renal medulla and papilla is well established. Nonsteroidal anti-inflammatory drugs (NSAIDs) are known to affect this axis and result in renal papillary necrosis in multiple species, including rodents, dogs, monkeys, and humans. 36 COX2 expression in the macula densa and thick ascending limb of immature and developing nephrons of fetal and newborn kidneys has been found to be significantly higher than that in mature kidneys in rodents. 8 Because of the high expression levels, COX2 in immature kidney is considered to play a role in nephron development and maintenance of RBF through effects on the renin–angiotensin system. Both COX2 knockout mice and human infants born from mothers given NSAIDs in the third trimester have exhibited renal abnormalities. Neonatal animals are more sensitive to some NSAIDs than adult animals, resulting in glomerular lesions if given to rodents prior to PND 21. 1 While the COX2 enzyme pathway is likely already mature at the time of birth, greater functional demand for COX2 in neonates as compared to adults is the most likely factor to explain the difference in susceptibility to this class of drugs in pediatric patients. Other enzymatic systems may show similar differential timing of expression or reflect specific neonatal requirements for development that predispose the pathway involved to nephrotoxicity. Hence, if toxicities are described in adult animals and the mechanistic target is already defined, it may be possible to determine that functional demand of targeted enzymes in the early neonatal period may differ from adults and result in increased susceptibility to toxicity during this time frame.
Conclusion
Differences in renal functional parameters based on age and maturation stage may result in vastly different toxicologic responses between neonates and adult rodents. Developmental changes in renal function can alter plasma clearance of compounds with extensive renal elimination. The ADME of drugs all vary depending on the age of the animal and renal maturation stage, which will lead to differences in toxicity and efficacy. In rodents, a portion of both renal morphologic and functional development occurs postnatally and any perturbation of processes related to ontogeny can result in malformations, degeneration, or functional disruption. While nephrogenesis and tubule differentiation in humans generally occur in utero, specific developmental events such as maximal tubular secretion, acid–base equilibrium, and concentrating ability occur postnatally as they do in rodents. Maturation of RBF, transporter function, or functional metabolic pathways may be required to excrete toxins from the kidney, and these functions develop at different times in different species, resulting in differences in toxic potential. Critical end points and windows of susceptibility have been established comparatively between species to better model pharmacokinetics and understand potential pediatric or gestational drug toxicity in the kidney. The differential effects of ADME may confuse, and confound pharmacokinetics in juvenile toxicity studies such that drug exposure can vary markedly from PND 1 to PND 36 in rodents and require separating animals of different ages into cohorts to reach optimal dosing schemes. Effects of drugs and chemicals are susceptible to functional maturation, and dosages have to be carefully considered and modeled appropriately. This may involve giving different doses to juvenile animals in different age groups to equilibrate pharmacokinetics and optimize exposure without decreasing tolerability. There are examples where juvenile animals are resistant to nephrotoxicity (eg, aminoglycosides and acetaminophen), other examples where toxicity is enhanced in infants (eg, VEGF inhibitors or other antineoplastic agents), and yet other examples where nephrotoxicity in rodents is expressed as juvenile toxicity, but in humans only as a gestational toxicity (eg, ACE inhibitors or AHR agonists). Many of these kidney toxicities are directly a result of immature ADME functions and/or incomplete functional or anatomical development. A thorough understanding of the factors involved will help refine juvenile safety assessment studies in the future.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.