
Abstract
The International Committee for Classification of Corneal Dystrophies (IC3D) categorized corneal dystrophies in humans using anatomic, genotypic, and clinicopathologic phenotypic features. Relative to the IC3D classification, a review of the veterinary literature confirmed that corneal dystrophy is imprecisely applied to any corneal opacity and to multiple poorly characterized histologic abnormalities of the cornea in animals. True corneal dystrophy occurs in mice with targeted mutations and spontaneously in pet dogs and cats and in Dutch belted (DB) rabbits, but these instances lack complete phenotyping or genotyping. Corneal dystrophy in DB rabbits can be an important confounding finding in ocular toxicology studies but has only been described once. Therefore, the ophthalmology and pathology of corneal dystrophy in 13 DB rabbits were characterized to determine whether the findings were consistent with or a possible model of any corneal dystrophy subtypes in humans. Slit lamp and optical coherence tomography (OCT) imaging were used to characterize corneal dystrophy over 4 months in young DB rabbits. The hyperechoic OCT changes correlated with light microscopic findings in the anterior stroma, consisting of highly disordered collagen fibers and enlarged keratocytes. Histochemical stains did not reveal abnormal deposits. Small clusters of 8 to 16 nm diameter curly fibers identified by transmission electron microscopy were consistent with Thiel-Behnke (TBCD) subtype of epithelial-stromal transforming growth factor β-induced dystrophies. Sporadic corneal dystrophy in DB rabbits appears to be a potential animal model of TBCD, but genotypic characterization will be required to confirm this categorization.
Keywords
Introduction
The cornea is a hydrophobic, highly innervated but avascular transparent membrane covering the anterior globe. In order, the layers of the cornea include the anterior epithelium (nonkeratinized stratified squamous, wing and regenerative cells), an epithelial basement membrane, a subepithelial Bowman layer (anterior limiting lamina) that is only identifiable in some mammals, stroma (keratocytes and regularly arranged collagen fibers), Descemet membrane (posterior limiting lamina), and endothelium. Detailed comparative anatomy and histomorphology of the cornea have been published. 1 -6 Corneal shape and transparency are maintained in the stroma by orderly and parallel aligned collagen fibrils (lamellae), which becomes random and interweaving near the anterior corneal surface. Collagen is mostly type I and V fibrils of approximately 24 nm diameter, with lesser amounts of XII and XIV fibrils and nonfibrillar type VI. The lamellae contain small leucine-rich proteoglycans, including decorin, lumican, keratocan, and dermatan and keratan sulfates. Keratocytes are specialized fibroblasts that maintain the extracellular matrix and are primarily located in the anterior stromal layers. The avascular cornea receives nutrients from the aqueous humor. 7 -9
In nonclinical studies, corneal opacities, often reported as corneal dystrophy, are the most frequently reported spontaneous ocular finding in rabbits 10 and Sprague Dawley rats 11 and reported at various incidences depending on strain and age of rats and mice, 12 -18 Syrian hamster, 12 Guinea pigs, 19 and beagle dogs 12,20,21 but are seldom reported in micromini- and mini-pigs 5,22,23 and cynomolgus monkeys. 24 These clinically reported opacities are sometimes associated with a variety of traumatic, environmental (light-induced), procedural (perianesthetic), metabolic/systemic (lipid), age-related or unknown events, and some may be a misdiagnosis or occasionally from drug-induced (phospholipidosis) or genetic-induced abnormalities including corneal dystrophy. Corneal opacities are found in mice with induced mutations in connective tissue and microfilaments 25,26 and occur secondary to mutational effects such as perinatal failure to close eyelids. 27 Microscopically, spontaneous corneal opacities frequently translate to corneal mineralization, degeneration, edema, erosions, fibrosis, lipid deposits, or vacuolation, and opacities can be age-, procedure-, or treatment-related or a postmortem artifact. 13,17,20,28,29 The existing veterinary literature frequently categorizes any corneal opacity as corneal dystrophy, and even microscopic evaluations refer to corneal dystrophy rather than to specific morphological findings such as corneal edema or mineralization.
Corneal dystrophy is a term that has been indiscriminately applied in both the veterinary and human literature as a disease moniker or morphological diagnosis for corneal opacities and deposits, irrespective of etiology. Dystrophy, derived from the Greek (dys = wrong, difficult; trophe = nourishment) was initially applied to a heritable disease of the musculature, but by 1890 was also applied to diseases of the cornea. For humans, the Cornea Society formed an International Committee for Classification of Corneal Dystrophies (IC3D) and published a new classification system for corneal dystrophies in 2008. 30 A 2015 revision to the classification 31 categorized corneal dystrophy by anatomic location (epithelial and subepithelial dystrophies, epithelial-stromal transforming growth factor β-induced (TGFβI) dystrophies, stromal dystrophies, and endothelial dystrophies), phenotypic variants, morphologic features, and updated genotypic assignments. This 2015 revised classification 31 eliminated unjustified phenotypes, provided additional histopathology and transmission electron microscopy (TEM) illustrations, and some confocal microscopy and optical coherence tomography (OCT) characterization. In animals, several spontaneous and induced mutations in mice 32 -43 and spontaneously arising true (heritable) corneal dystrophies are reported in rabbits 44 and pet dogs 32,45 -52 and cats. 53 Some of these corneal dystrophies have been reported as models of or being equivalent to corneal dystrophy variants in humans, but often with little clinicopathological or genetic characterization. Now that some clarity has been brought to harmonizing the numerous reports of corneal dystrophy in humans, a review of the veterinary literature is also timely. Spontaneously occurring mutations in animals can be confounding variables in toxicology studies and defined genetic mutations can be useful for therapeutic interventional testing and help to define the genetic basis of disease in humans. Awareness of diseases with a genetic basis is also important to breeding programs for domestic and laboratory animals to prevent perpetuation of heritable diseases.
Rabbits (Oryctolagus cuniculus) are commonly used for testing of ocular and dermal therapeutics and medical devices, with the albino New Zealand white (NZW) rabbit used most frequently, but the more compact Dutch belted (DB) rabbit selected when a pigmented strain is required for testing. 54,55 Dutch belted rabbits are affected by corneal opacities described as anterior corneal dystrophy of juvenile American DB rabbits. 44 Corneal opacities that typify corneal dystrophy in DB rabbits are usually not present until several months after weaning, and affected rabbits may inadvertently be enrolled in a nonclinical ocular toxicity testing study. These sporadic corneal opacities can become a confounding lesion and falsely attributed to a test article, particularly for topical ophthalmic therapeutics, but also for studies of periocular and intraocular therapeutics and medical devices. Although corneal dystrophy is a recognized sporadic problem in DB rabbits, 55,56 only one study has attempted to characterize the microscopic and TEM changes. 44 Therefore, the purpose of this study was to (1) critically review the literature on corneal dystrophies in laboratory and domestic animals compared to the IC3D classification in humans to identify true corneal dystrophies in animals; (2) characterize the spontaneous corneal opacities known as anterior corneal dystrophy in a group of 13 DB rabbits using OCT, light, and TEM; and (3) determine whether the corneal dystrophy in DB rabbits is consistent with or a possible model of corneal dystrophy in humans.
Materials and Methods
Comparative Literature Review of Corneal Dystrophy in Animals and Humans
Pubmed (https://pubmed.ncbi.nlm.nih.gov/), Google Scholar, and journal archives for Toxicologic Pathology, American Journal of Pathology, Investigative Ophthalmology & Visual Science, Comparative Medicine, and International Journal of Toxicology were searched for the terms “corneal opacity” and “corneal dystrophy” in animals to identify the veterinary medical literature included in this review. The clinicopathologic and genotypic data in these publications was reviewed (by JCLS) and compared to the 2015 IC3D classification edition 2 31 and secondarily to the original 2008 IC3D classification of corneal dystrophy in humans. 30 References were segregated to a multiclass system of confirmed, unconfirmed, probable, or possible examples of corneal dystrophy, based on the amount of available genotypic and phenotypic information (classes are defined in Table 1). These examples of corneal dystrophy in animals were then correlated to the 2015 IC3D classification for humans. 31 The National Center for Biotechnology Information Gene database (https://www.ncbi.nlm.nih.gov/gene) was also searched for gene information associated with corneal dystrophies in humans and animals.
Anatomic Category, Subtypes, and Variants of Corneal Dystrophy and Known Genetic Defects Reported in Humans (Weiss et al 31 ) Compared to the Published Reports That Likely Represent True Corneal Dystrophy in Animals.
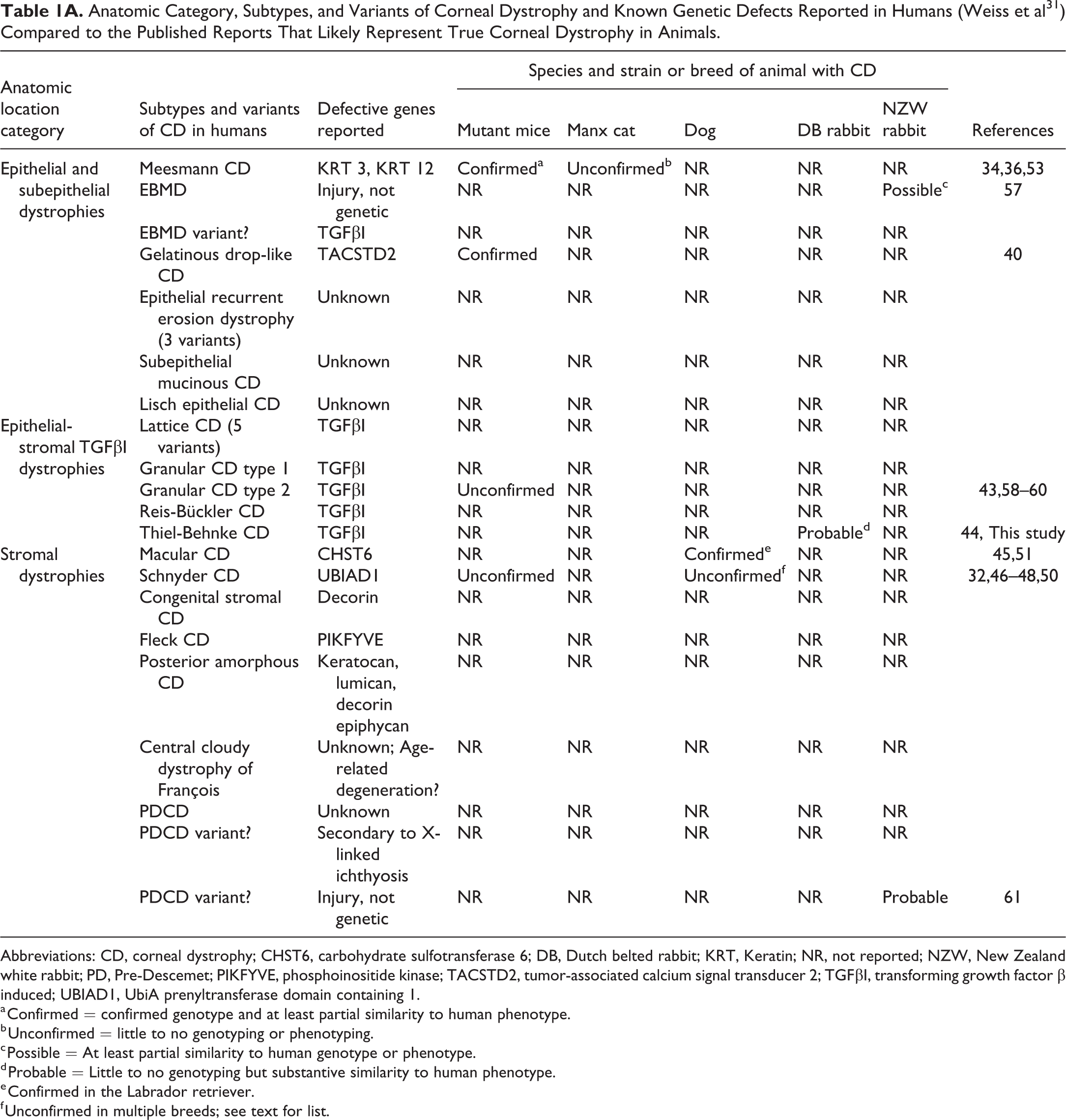
Abbreviations: CD, corneal dystrophy; CHST6, carbohydrate sulfotransferase 6; DB, Dutch belted rabbit; KRT, Keratin; NR, not reported; NZW, New Zealand white rabbit; PD, Pre-Descemet; PIKFYVE, phosphoinositide kinase; TACSTD2, tumor-associated calcium signal transducer 2; TGFβI, transforming growth factor β induced; UBIAD1, UbiA prenyltransferase domain containing 1.
a Confirmed = confirmed genotype and at least partial similarity to human phenotype.
b Unconfirmed = little to no genotyping or phenotyping.
c Possible = At least partial similarity to human genotype or phenotype.
d Probable = Little to no genotyping but substantive similarity to human phenotype.
e Confirmed in the Labrador retriever.
f Unconfirmed in multiple breeds; see text for list.
Subtypes and Variants of Corneal Dystrophy and Known Genetic Defects Reported in Humans (Weiss et al 31 ) Compared With the Most Likely Published Reports That Represent True Corneal Dystrophy in Animals.
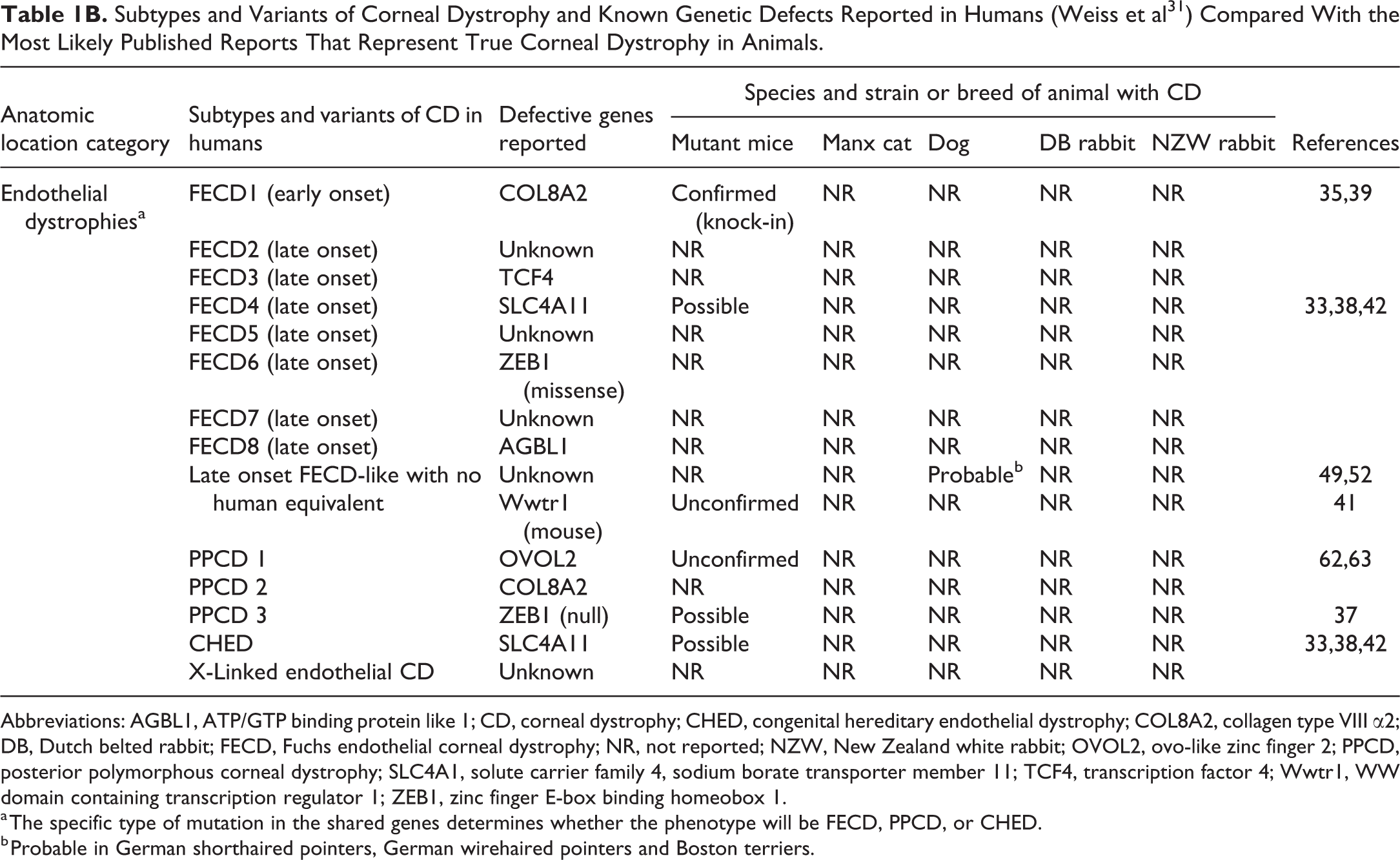
Abbreviations: AGBL1, ATP/GTP binding protein like 1; CD, corneal dystrophy; CHED, congenital hereditary endothelial dystrophy; COL8A2, collagen type VIII α2; DB, Dutch belted rabbit; FECD, Fuchs endothelial corneal dystrophy; NR, not reported; NZW, New Zealand white rabbit; OVOL2, ovo-like zinc finger 2; PPCD, posterior polymorphous corneal dystrophy; SLC4A1, solute carrier family 4, sodium borate transporter member 11; TCF4, transcription factor 4; Wwtr1, WW domain containing transcription regulator 1; ZEB1, zinc finger E-box binding homeobox 1.
a The specific type of mutation in the shared genes determines whether the phenotype will be FECD, PPCD, or CHED.
b Probable in German shorthaired pointers, German wirehaired pointers and Boston terriers.
Prospective Characterization of Corneal Dystrophy in Dutch Belted Rabbits
Otherwise healthy DB rabbits that were rejected from use in ocular toxicity studies due to corneal opacities were sourced for this study. Rabbits were individually housed in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited laboratory animal facility that was in compliance with the Guide for the Care and Use of Laboratory Animals. 64 Dutch belted rabbits were selected from a subset of DB rabbits identified in a large survey of spontaneous ocular lesions in 1840 NZW, NZW × New Zealand (NZ) red cross (WRF1), and DB rabbits at 6 to 16 weeks of age. 10 In this retrospective and prospective survey, 177 (9.7%) rabbits were eliminated from use in ocular studies over a 2-year period due to spontaneous ocular lesions, particularly of the cornea (5.7%). The incidence of corneal opacities was also dependent on supplier and was more frequent in male (6.3%) than in female (0.5%) DB rabbits. In our follow-on investigation, 15 of 197 male DB rabbits from a single shipment of rabbits (Myrtle Rabbitry) were rejected for use in preclinical ocular studies due to corneal lesions. One rabbit with bilaterally affected corneas was euthanized for unrelated reasons prior to study start, and 1 animal with a unilateral opacity was ophthalmologically recategorized as a scar. These 2 animals were lost to the study leaving 13 animals with corneal dystrophy-like opacities for detailed ophthalmic, microscopic, and ultrastructural investigations.
Starting at 5.5 months of age (juveniles), the 13 rabbits were examined every 4 weeks with a complete ophthalmic examination including direct pupillary reflexes prior to mydriasis with topical 0.5% tropicamide (Mydriacyl, Alcon Pharmaceuticals), slit lamp biomicroscopy (LS15, Kowa Optimed) with fluorescein staining strips (Phoenix Pharmaceutical, Inc) and digital photodocumentation, indirect ophthalmoscopy, anterior segment OCT (Optovue, Inc) with photodocumentation, intraocular pressure (IOP) tested by applanation tonometry (Tono-Pen, Reichert Technologies), and serum screened at study start for fasted total cholesterol and triglycerides (Antech Diagnostics). The presence and progression of the corneal opacities were recorded and digital photographs captured.
At necropsy, a 3 mm disposable biopsy punch (Miltex) was used to collect isolated samples of the affected and unaffected corneas from both eyes. For histopathology, 21 corneal biopsy samples from 13 animals were successfully collected into 10% neutral buffered formalin. The corneal lesions in remaining biopsies were deemed too small to reliably section and were not processed or examined microscopically. Corneal biopsies were paraffin-embedded, sectioned at 4 to 6 µm, and stained with hematoxylin and eosin (HE), hematoxylin, phloxine and saffron (HPS), and Masson trichrome. As histochemical staining of the cornea is an important but not exclusive or consistent diagnostic feature for corneal dystrophies located in epithelium and stroma of humans, an algorithm (Figure 1) was created from the IC3D classifications 30,31 to select and apply the commonly used and most useful histochemical stains to rabbit corneas. Small corneal lesions were lost with serial sectioning and not all ocular lesions were captured in sections subsequently stained with periodic acid Schiff (PAS; no diastase applied), Congo red, and Alcian blue. Digital photomicrographs were acquired to a desktop computer using a Canon T2i camera (Canon USA, Inc., Melville, NY, USA) mounted on an Olympus B50 microscope (Olympus America Inc., PA, USA). Four biopsy samples of cornea from 2 affected rabbits and 1 unaffected rabbit, and the normal cornea from a rabbit not included in our study, were collected in 4% formaldehyde:1% glutaraldehyde for TEM. The TEM samples were postfixed in 1% osmium tetroxide, processed, embedded in Polybed 812 resin, sectioned at 1 µm, stained with toluidine blue, and examined microscopically to identify representative affected and unaffected areas. Trimmed resin blocks were sectioned at 60 nm, and sections were placed on copper grids and stained with uranyl acetate and lead citrate. The grids were examined at 10 to 40,000× direct magnification and digital images acquired on a FEI Tecnai 120 KV electron microscope (Ultrapath Imaging).
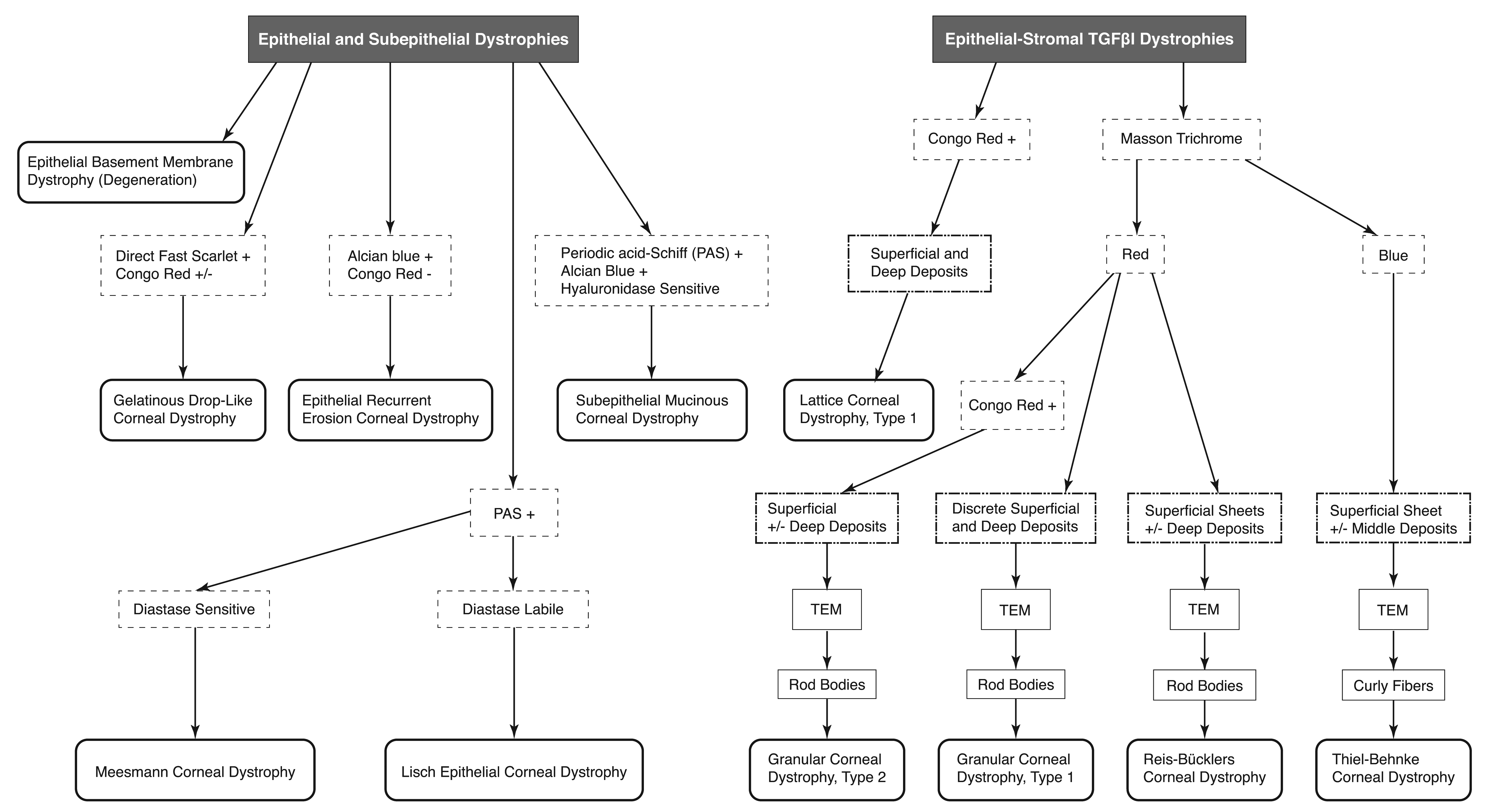
Algorithm for commonly used and the most useful diagnostic histochemical stains (results: + = positive; − = negative; ± = depends on variant and stage of disease) for anatomically categorized epithelial and subepithelial or epithelial-stromal transforming growth factor β-induced (TGFβI) corneal dystrophies reported in humans (extracted from Weiss et al 31 ). Transmission electron microscopy (TEM) on abnormal stromal collagen fibrils is an important differentiating feature for some epithelial-stromal TGFβI corneal dystrophies. TGFβI indicates transforming growth factor β induced.
Results
Comparative Literature Review of Corneal Dystrophy in Animals and Humans
The IC3D has attempted to restrict corneal dystrophy in humans to “a group of inherited corneal diseases that are typically bilateral, symmetric, slowly progressive, and without relationship to environmental or systemic factors,” but frequent exceptions to this rigid categorization were identified. The 2015 IC3D classification concluded that the term corneal dystrophy has more historical than practical significance, and their classification still incorporates other corneal abnormalities that have been traditionally considered corneal dystrophy. 31 In the veterinary medical literature, corneal dystrophy has been used to describe a variety of corneal opacities with degenerative and inflammatory changes, recurrent erosions and indolent ulcers, and mineralization that are not consistent with heritable corneal dystrophy. After reviewing the veterinary literature, the majority of publications titled corneal dystrophy were found to be descriptions of corneal opacification that were nonspecific (no histopathology) or microscopic degeneration/erosions, mineralization, edema, fibrosis, lipid deposits, mineralization, or vacuolation such as drug-induced phospholipidosis and also included inflammation and systemic abnormalities (eg, mucopolysaccharide storage disease) that affect the cornea. The remaining literature included a few examples of nonsystemic, noninflammatory, and usually bilateral, symmetric, slowly progressive cornea-specific abnormalities with morphological features and possible established heritability, consistent with true corneal dystrophy. These publications of apparently heritable corneal dystrophies in laboratory rabbits, induced mutations in mice, pet dogs, and Manx cats were reviewed and compared to the 2015 IC3D, 31 to determine if they fit and how well they fit (confirmed, unconfirmed, probable, or possible) the phenotypic and genotypic characteristics described for the anatomical categories, subtypes, and variants of corneal dystrophy in humans, with the results summarized in Table 1A and B. The histochemical and TEM findings most commonly applied and useful for diagnostic identification of the corneal deposits in the first 2 categories of the epithelial and subepithelial, and the epithelial-stromal TGFβI dystrophies are summarized in Figure 1.
Epithelial and subepithelial dystrophies
Meesmann corneal dystrophy (MECD) has been reported in keratin 3 and 12 deficient mice. 34,36 The primary finding is a thickened and disorganized corneal epithelium with intraepithelial cysts containing PAS positive and fluorescing debris and PAS diastase-sensitive glycogen. A corneal dystrophy described in inbred Manx cats 53 is of uncertain relationship to MECD as there has been no additional reports in this breed. In NZW rabbits, peripheral corneal epithelial dystrophy 57 is likely the result of injury rather than a heritable dystrophy. The corneal epithelial changes appear similar to the majority of cases of epithelial basement membrane dystrophy (EBMD) in humans in which a degenerative change, rather than a gene defect, results in loosely attached basal epithelial cells that predispose to erosions; the Bowman layer is normal. One subtype of EBMD may be a mutation in TGFβI, similar to epithelial-stromal corneal dystrophies. Gelatinous drop-like dystrophy with subepithelial and stromal amyloid has been recently modeled by deletional mutation of tumor-associated calcium signal transducer 2 in mice. 40 Gelatinous drop-like dystrophy band keratopathy subtype in humans is not the same as band keratopathy or calcific band keratopathy changes of degeneration and mineralization reported in animals. 65,66
Epithelial recurrent erosion dystrophy (nocturnally induced severe erosions), subepithelial mucinous corneal dystrophy (PAS, Alcian blue, and hyaluronidase sensitive material anterior to Bowman layer), and Lisch epithelial corneal dystrophy (vacuolated basal cells and epithelial cysts containing PAS positive, diastase labile, Luxol fast blue, and Sudan black negative) have not been described in animals.
Epithelial-stromal TGFβI dystrophies
Transforming growth factor β-induced gene mutations of locus 5q31 result in abnormal protein folding and aggregation that are the genetic basis for all epithelial-stromal TGFβI corneal dystrophies in humans. Most subtypes stain immunohistochemically for TGFβI protein (keratoepithelin). There are no examples in animals of epithelial-stromal dystrophies typed as lattice corneal dystrophy (amyloid deposits between collagen lamellae) and granular corneal dystrophy (GCD) type 1 (“snowfall” opacities with Masson trichrome red deposits from epithelium to posterior stroma and rod bodies by TEM). A knock-in point mutation of Arg124His in B6-Tgfbitm1.1(TGFBI * R12H)Ccbko resulted in mild granular corneal deposits in some mice but could not be demonstrated to be amyloid. 43 This phenotype appeared to be similar to GCD type 2 in humans (often less severe than GCD type 1). However, an Arg124Leu knock-in transgenic mutation did not result in any corneal deposits in mice. 58 Overexpression of an Arg155Trp TGFβI variant transgenic mouse resulted in retinal photoreceptor atrophy, 59 but the same insertional promotor and another transgenesis protocol resulted in mice with central corneal opacities that were not microscopically phenotyped. 60 Deletion of TGFβI did not result in any ocular lesions, and the reported increased incidence of spontaneous tumors and 7,12-dimethylbenz[a]anthracene-induced skin tumors in these TGFβI−/− mice 67 is small.
Reis-Bückler corneal dystrophy (RBCD) or Thiel-Behnke corneal dystrophy (TBCD) dyscollagenesis has not been described in animals. Reis-Bückler corneal dystrophy is typified by replacement of Bowman layer by Masson trichrome red deposits, with extension into the stroma. Thiel-Behnke corneal dystrophy frequently shows a Masson trichrome stained blue avascular fibrocellular pannus with wavy stromal ridges and furrows (sawtooth) overlaid by alternating thickening and thinning of the epithelium. Reis-Bückler corneal dystrophy and TBCD can be clinically similar and they are best differentiated by TEM identification of rod/trapezoid bodies in RBCD and 9 to 15 nm diameter curly fibers in TBCD.
Anterior corneal dystrophy in American DB rabbits 44 appears to fit the localization and characteristics of epithelial-stromal TGFβI dystrophies. Moore et al 44 identified corneal opacities in the epithelium, basement membrane, and subepithelial corneal stroma in 3 of 5 DB rabbits, and a further 10 of 34 inbred animals in the originating breeding facility. Microscopically, plastic sections of the epithelium stained with PAS and Jones HE were thin and disorganized, and the basement membrane was irregular and thickened, with some detachment of basal epithelial cells. Collagen lamellae in the stroma were disorganized. They compared their findings to the descriptions of MECD, EBMD, and RBCD, but they were unable to identify a direct correlate between the findings in the rabbits and the corneal dystrophies in human reported at that time. It should be noted that these 3 anterior dystrophies in humans were anatomically, genotypically, and phenotypically recategorized twice after this report in DB rabbits, and reported cases of TBCD may still be confused with RBCD. 30,31 Heritability of this corneal dystrophy in these inbred DB rabbits was supported by finding that 6 of 9 affected females had been sired by 1 affected male in the breeder facility.
Stromal dystrophies
In Labrador Retrievers, a genotypically confirmed carbohydrate sulfotransferase 6 gene mutation results in loss of keratocytes and stromal deposition of PAS and iron positive glycosaminoglycans (GAGs) consistent with macular corneal dystrophy in humans. 45,51 In humans, Schnyder corneal dystrophy (SCD) is a mutation in UbiA prenyltransferase domain containing 1 (UBIAD1) that may result in hyperlipidemia, crystals in the cornea that stain for intracellular and extracellular phospholipids and cholesterol in epithelial cells, Bowman layer, and stroma. Secondary deposition of amyloid and GAGs may also occur. Preliminary phenotyping of a Ubiad1 point mutation in mice appears to be similar to SCD. 32 In several dog breeds, 46 -48,50 SCD has been described but the hyperlipoproteinemia that is frequently but not always found in humans may be variable or absent in these dogs, including in the Shetland Sheepdog, Siberian Husky, American Cocker Spaniel, Miniature Schnauzer and Airedale Terrier, 46 and the Beagle. 48,50 A normolipoproteinemic form of SCD has been reported in the Cavalier King Charles Spaniel and Rough Collie dogs. 47 It is not clear that these possibly heritable abnormal lipid deposits in the cornea of dogs are true corneal dystrophies as there has been no follow-up studies in over 30 years, and these publications lack substantive phenotypic and genotypic characterization.
The other 5 stromal dystrophies in humans including congenital stromal corneal dystrophy (stromal lamellae separation with amorphous material), fleck corneal dystrophy (swollen keratocytes that stain positive for lipids, and Alcian blue and iron positive GAGs), posterior amorphous corneal dystrophy (irregular stromal lamellae, thin Descemet membrane, and attenuated endothelial cells), central cloudy dystrophy of François (posterior stroma undulations and stains positive for GAGs), and pre-Descemet corneal dystrophy have not been described in animals.
In NZW rabbits, sporadic pre-Descemet corneal opacities 61 have been described as ectopic endothelial cells in the posterior stroma. These findings may or may not be different from pre-Descemet corneal dystrophy in humans, which has variants that may be genetic-based, age-related, degenerative, or secondary to X-linked ichthyosis. 31 The endothelial excursion reported in these rabbits may signify a breach in Descemet membrane with postinjury-related endothelial proliferation into the stroma. Rabbit is one species in which corneal endothelium divides throughout life and their endothelium may even form multinucleated cells. 68,69
Endothelial dystrophies (4 subtypes)
Three of the endothelial dystrophies including Fuchs endothelial corneal dystrophy (FECD), posterior polymorphous corneal dystrophy (PPCD), and congenital hereditary endothelial dystrophy (CHED) share gene mutations. The specific type of mutation often determines the severity and phenotype of the dystrophy and whether it is classified as a variant of FECD, PPCD, or CHED. Fuchs endothelial corneal dystrophy has an early onset form, FECD1, with a genetic defect in collagen type VIII α2 (COL8A2) found in Descemet membrane. The reported focal thickening of anterior Descemet membrane (guttae), progressive loss of endothelial cells, and stromal edema is also found in Col8a2 knock-in mice. 35,39 Mutations in COL8A2 are also found in PPCD variant 2 in humans, but PPCD2-like disease is not reported in animals. For late-onset FECD, with more severe guttae and endothelial loss, there are 7 variants mapped to multiple chromosomal loci (Table 1). 31,70 Mutations in solute carrier family 4, sodium borate transporter, member 11 (SLC4A11) are associated with both late-onset FECD4 and CHED. Congenital hereditary endothelial dystrophy may be asymmetric with keratoconus, and microscopically the cornea will have diffuse epithelial and stromal edema with laminated collagen deposits on the posterior Descemet membrane and irregularities of the endothelium. Mild features of late-onset FECD4 and CHED, but also with the cochlear and kidney abnormalities (Harboyan syndrome) caused by certain types of SLC4A11 mutations in humans, are modeled in Slc4a11 knockout mice. 33,38,42 The corneal changes in these mice have been related to endogenous ammonia uncoupling and toxicity. 71 Zinc finger E box-binding homeobox 1 (ZEB1) mutations associated with FECD6 are missense changes that cause mild disease, but truncation or early termination results in null mutations with more severe disease categorized as PPCD3. Targeted Zeb1 deficient mice have a corneal abnormality that is partially similar to PPCD3. 37 The PPCD1 mouse is an accidental gene targeting mutation of Zeb-related pathways, with features similar to the ovo-like zinc finger 2 gene defect associated with human PPCD1, but these mice also have iridocorneal endothelial syndrome with retinal detachment. 62,63 Heritable late-onset FECD-like disease has been phenotypically described in German shorthaired and wirehaired pointers 49 and Boston terriers 52 but not genotyped. Mice deficient in WW domain-containing transcription regulator 1 have a preliminary phenotype of late-onset FECD including decreased endothelial cells and altered Descemet membrane. 41 The final subtype of endothelial dystrophy in humans, X-linked endothelial corneal dystrophy has not been reported in animals. This congenital disease results in subepithelial granular deposits, thinning and loss of Bowman layer, thickening of Descemet membrane, and loss and discontinuities of endothelial cells.
Prospective Characterization of Corneal Dystrophy in Dutch Belted Rabbits
All juvenile rabbits were clinically normal on physical examination, except for the presence of corneal opacities that excluded them from use in preclinical studies. 10 In the 13 rabbits, corneal opacities, consistent with corneal dystrophy, were present in 24 of 26 eyes (92%), with changes bilateral in 11 (85%) and unilateral in 2 (15%) of the rabbits, with the contralateral 2 eyes unaffected. The breeding facility was unable to provide animal lineage data postshipping but reported that at least 1 buck at the breeding facility had been intensely used during production of the 13 affected, single source, single shipment, DB rabbits that we included in our investigation.
There were no abnormalities in pupillary reflexes, adnexal tissues, ocular examinations, and, except for the opacities, no other corneal changes. There was no disruption in epithelial integrity as noted by the lack of fluorescein staining. The mean IOP (17.0 ± 2.40 mm Hg; reference range 15-23 mm Hg), serum total cholesterol (15 ± 4.6 mg/dL; reference range 10-80 mg/dL), or triglycerides (35 ± 12 mg/dL; reference range 27-47 mg/dL) were normal. Slit lamp biomicroscopy identified one or more circular to oval, 2 to 10 mm diameter white, granular and avascular opacities that were occasionally multifocal and linear. These opacities were predominately located nasally and paracentral below the cornea midline, occasionally close to the corneal scleral junction.
The localized opacities illuminated by slit lamp (Figure 2A-C) were confirmed by OCT (Figure 2D-F). The OCT findings were characterized as dense and granular opacities that were sometimes elevated (Figure 2A and D) and occasionally linear (Figure 2B and E and 2C and F) and predominately located in the subepithelial (Figure 2B and E) and stromal regions, extending as deep as mid-stroma (Figure 2A and D and 2C and F), but less frequently involving thinning of the epithelium (Figure 2D). There was increased density of the basement membrane and irregularity densities of the affected stroma. Posterior stroma, Descemet membrane, and endothelium directly beneath affected areas of the cornea were normal in all rabbits. Three patterns of hyperechoic lesions were identified using OCT: (1) anterior stromal band with raised and irregular epithelium (Figure 2D); (2) subepithelial line with stroma unaffected (Figure 2E); and (3) subepithelial line merging into a hyperechoic stromal band into the midstroma (Figure 2F). Bilaterally, the corneal densities were often similar but there was variation in vertical and horizontal dimensions and depth of the opacities into the stroma in the right versus left cornea. There was gradual progression in both size and density of the corneal changes over the 4 ophthalmic examinations. During this period, no new spontaneous lesions were identified, and the 2 rabbits with unilateral unaffected eyes at study start did not develop corneal opacities.
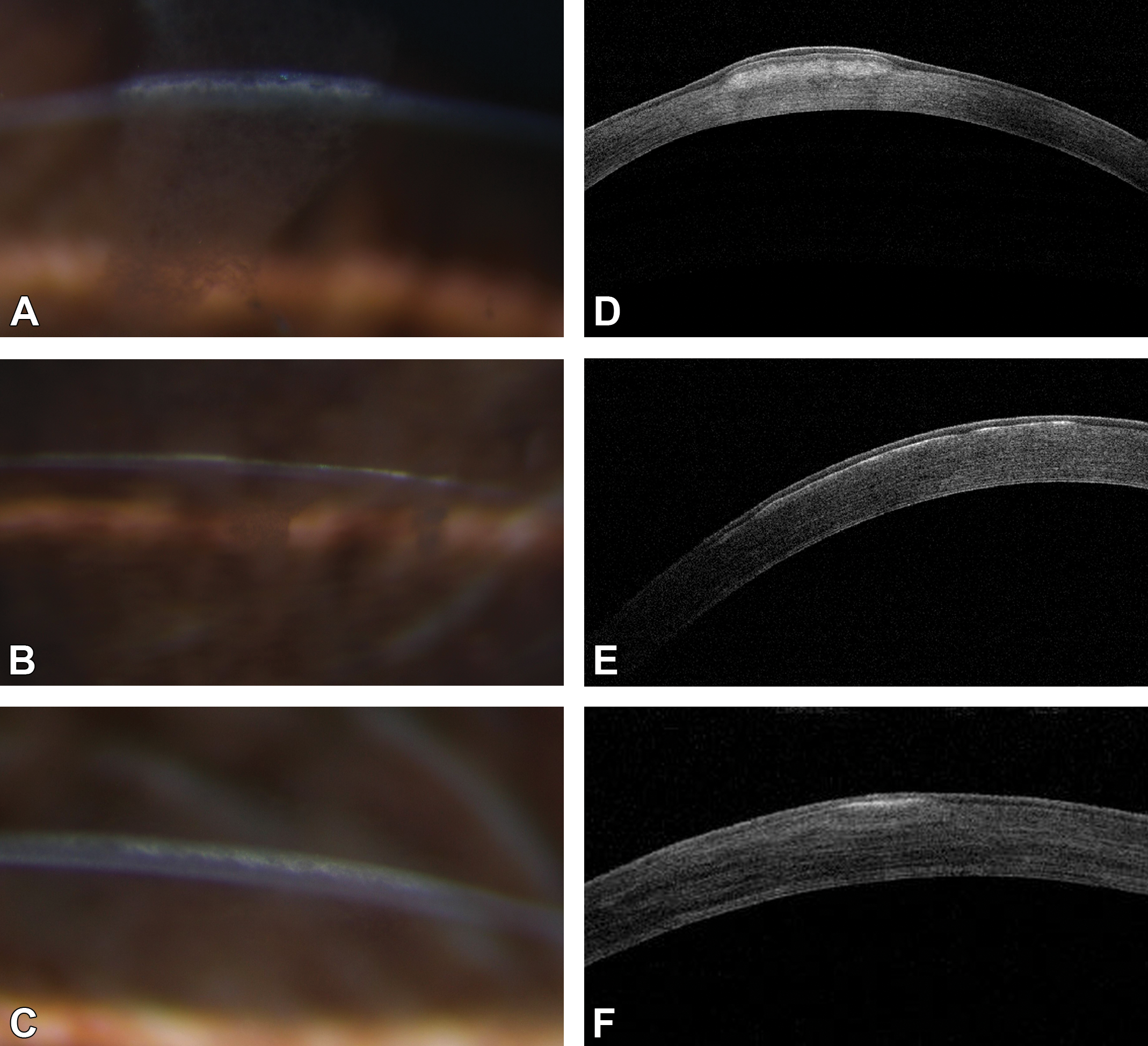
Comparison of slit lamp biomicroscopy (A-C) and respective optical coherence (OCT) images (D-F). Three patterns of corneal opacities by slit lamp biomicroscopy and hyperechoic lesions by OCT were (1) anterior stromal band with raised and irregular epithelium (A and D), (2) subepithelial line with stroma unaffected (B and E), and (3) subepithelial line merging into a hyperechoic stromal band in the mid-stroma (C and F).
Of the 21 biopsies processed, the 2 unaffected corneas were normal, and 14 of 19 biopsies from ophthalmoscopically affected eyes contained abnormal areas of epithelium and stroma. The abnormality was easier to identify in sections stained with HPS and Masson trichrome compared to HE stained sections due to the connective tissue and epithelial contrast provided by HPS and Masson trichrome stains (Figure 3A-C). With Masson trichrome, there were no red deposits in the subepithelial or stromal areas, and the affected anterior stroma was darker blue compared to the unaffected stroma (Figure 3C). Further sectioning of the blocks to apply additional histochemical stains was successful for some specimens: 10 of 14 corneas were stained for PAS (to detect mucopolysaccharides; Figure 3D), 7 of 14 corneas were stained with Alcian blue (to detect mucin), and 5 of 14 corneas were stained with Congo red (to detect amyloid). All corneas that were stained were negative for these substances.
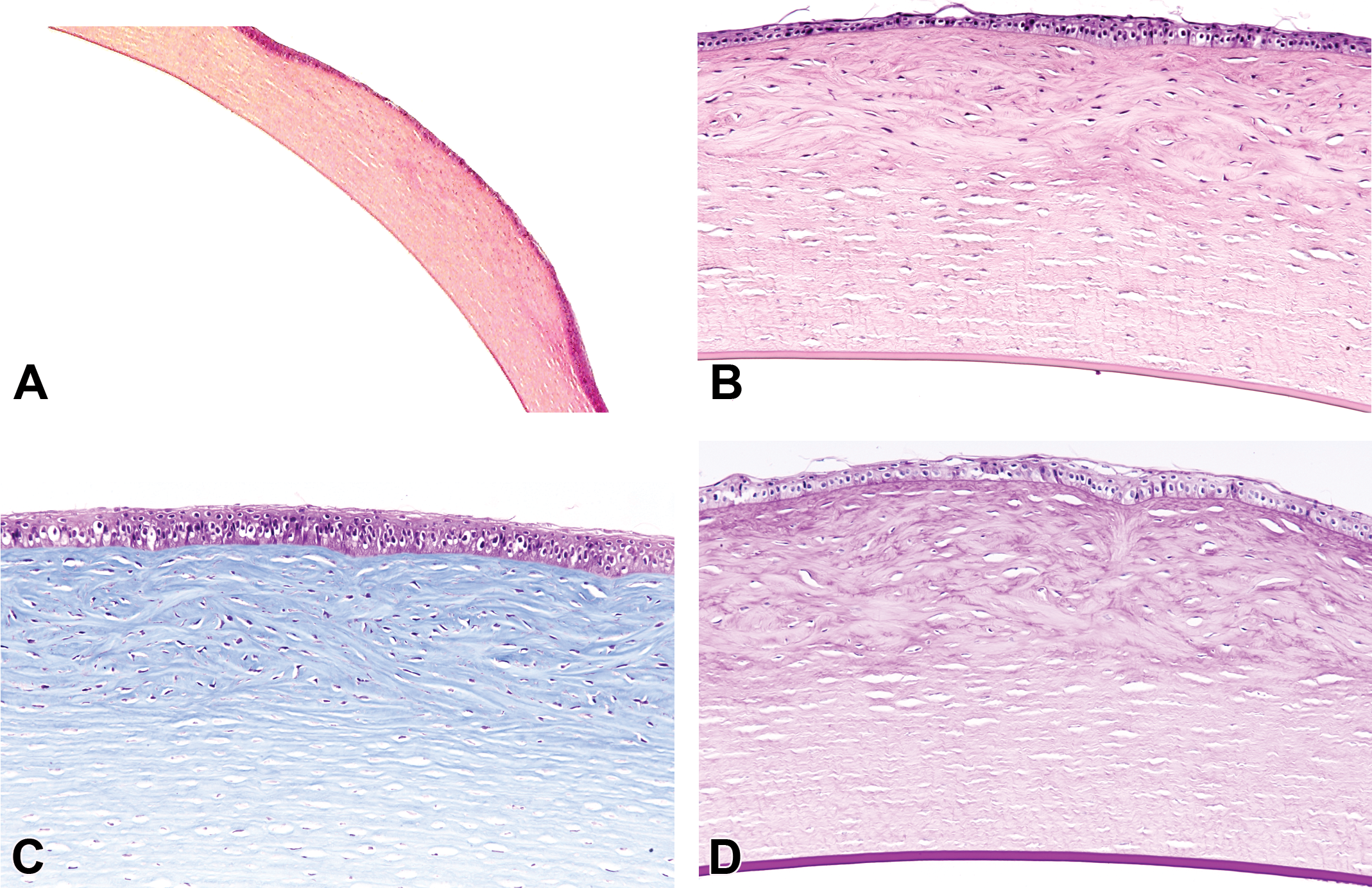
Photomicrographs of a slightly raised corneal surface with increased tinctorial density and marked disarray of the subepithelium to midstromal collagen. Epithelium was generally thin with cuboidal changes to the basal layer and intermittent thickening and irregularity of the basement membrane. There is no involvement of the posterior stroma, Descemet membrane, or endothelium (A-D). The corneal abnormality was easier to identify in hematoxylin, phloxine, and eosin (A and B) than with hematoxylin and eosin staining (not shown). All histochemical stains used to help characterize the corneal dystrophy (Figure 1) did not identify any unusual deposits including Masson trichrome (C) and periodic acid Schiff (D) and Alcian blue or Congo red (not shown).
Microscopic findings correlated to the OCT assessment of discrete corneal changes that sometimes elevated the corneal surface and primarily involved the epithelium and anterior stroma, occasionally extending to the midstroma. There was no involvement of the posterior stroma, Descemet membrane, or endothelium. Compared to unaffected corneas (Figure 4A and B), the morphological changes were typified by thin epithelium with keratinization, reduced numbers of cells and cuboidal basal cells, infrequent and irregular thickening of basement membrane, and hyperplasia of epithelium at the edges of the lesions (Figure 4C). Stromal architecture was highly irregular with increases in the size and number of keratocytes (Figure 4D) and marked disarray of the fibrils with enhanced tinctorial staining (avascular fibrocellular pannus; Figure 4C and D).
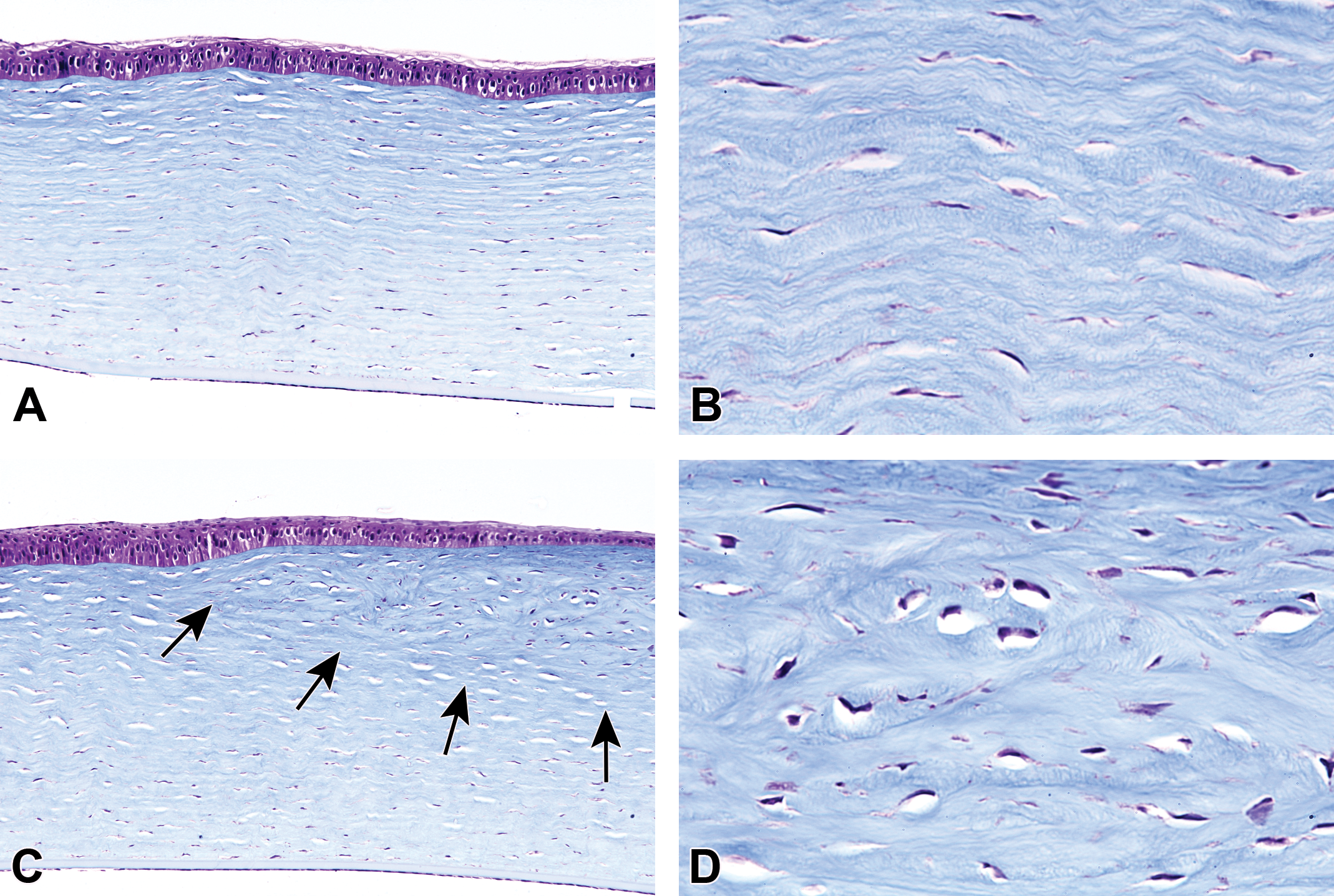
Masson trichrome stains of unaffected (A and B) and affected (C and D) rabbit corneas. Normal corneal epithelium is of regular thickness with columnar basal cells, and normal stroma has regularly oriented lamellae and regularly distributed keratocytes (A). At higher magnification, the collagen lamellae of the anterior stroma have a regular horizontal orientation with interspersed elongate keratocytes (B). Conversely, the epithelium over the affected cornea (arrows) is thinned, and epithelium at the margin of the lesion is thickened (C). The avascular fibrocellular pannus consists of highly irregular collagenous lamellae with darker blue staining compared to the normal collagen and increased numbers of enlarged keratocytes (D).
The TEM confirmed the abnormalities in the collagen architecture and keratocytes. Compared to a clinically normal cornea in which there was orderly and parallel alignment of collagen fibrils around keratocytes (Figure 5A), an affected cornea displayed disorder of the collagen fibrils and enlargement of the keratocytes. Additionally, clusters of thin curly fibrils that measured 8 to 16 nm were found only in the affected corneas (Figure 5B).
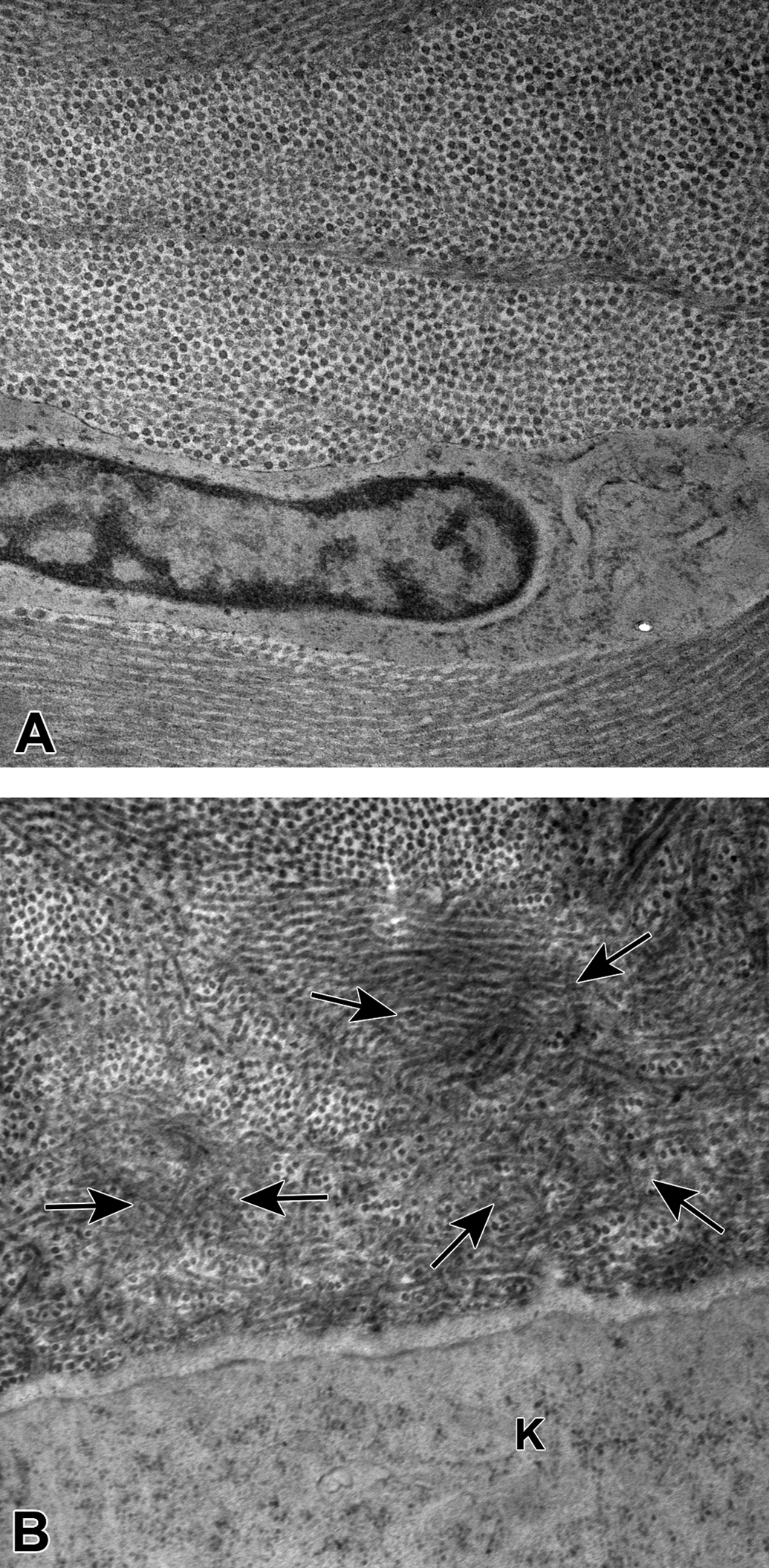
Ultrastructurally, the normal corneal stroma consists of orderly arrangements of the collagen fibers with a keratocyte (A). The cell membrane of an enlarged keratocyte (K) is adjacent to disarray of collagen fibrils, with clusters of thin curly fibrils (arrows) measuring approximately 8 to 16 nm diameter (B). Resin embedded, ultrathin sections.
Discussion
In the process of trying to phenotypically characterize corneal dystrophy in DB rabbits, we noted that a number of references in the veterinary literature used the term corneal dystrophy as a nonspecific diagnosis for any clinically apparent corneal opacity and a variety of histologic corneal findings. We conducted a comparative and critical review of the veterinary literature that confirmed that most reports of corneal opacity or dystrophy in animals are not a heritable abnormality of cornea but represent mineralization, degeneration, edema, erosions, fibrosis, vacuolation, trauma, lipid deposition, corneal manifestations of systemic disease, postmortem change, or age-, procedure-, or treatment-related changes. 13,17,20,28,29 Using the IC3D classification system for comparison, a heritable or true corneal dystrophy with phenotypic comparability and plausible or a certain genetic basis is present in targeted mutations in mice and occurs spontaneously in several breeds of dogs and probably in Manx cats and DB rabbits. Anatomically located epithelial and subepithelial corneal dystrophies were identified in induced mutations of mice 34,36,40 and possibly Manx cats. 53 Epithelial-stromal corneal dystrophy occurs in an induced mutation in a mouse 43 and probably DB rabbits. 44 Stromal dystrophies were found in an induced mutation in a mouse 32 and spontaneously in several breeds of dogs, 45 -48,50 including a genotypically verified missense mutation that results in macular corneal dystrophy in Labrador retrievers. 51 Numerous induced mutations in mice have been created as models of corneal endothelial dystrophies, 33,35,37 -39,42,62,63 and spontaneous endothelium dystrophies have occurred in dogs. 49,52 Although mice with targeted mutations for genes linked to corneal dystrophy subtypes in humans provide strong evidence for their use as animal models of human disease, the available phenotypic characterization of these mice models is weak, and the biological activity of the gene defects, particularly for phenotypic–genotypic relationships of endothelial corneal dystrophies, has not been confirmed to be totally comparable across species. Conversely, in dogs, cats, and rabbits, phenotypic characterization predominates with little genotypic characterization reported. Many of the older reports in these species are problematic as these were often single center studies with no follow-up publications, and at the time of publication, corneal dystrophies in humans also had poor phenotypic and genotypic characterization. While corneal dystrophy may be appropriate nomenclature for communication of clinically defined specific corneal disorders, pathologists should focus on providing morphological diagnoses that help to define the morphological and functional variants of such disorders. This is partially supported by the International Harmonization of Nomenclature and Diagnostic Criteria guide for nomenclature of special sense organs for rodents, 28 which indicates corneal mineralization as a preferred term over corneal dystrophy (and as a differential diagnosis for corneal edema), but does include Fuch dystrophy, with its multiple phenotypic–genotypic variants in humans, as a potential differential diagnosis for hypertrophy of Descemet membrane in rodents. Moving forward, the IC3D has advocated for more stringent criteria for publishing new and apparently unique corneal dystrophies. 31 The veterinary medical literature would similarly benefit from more complete histochemical, phenotypic, immunophenotypic, and genotypic characterization of corneal diseases in animals prior to publication.
Corneal opacities are a common finding in NZW, NZ red and DB rabbits, 10 and in DB rabbits, these opacities occur at a high incidence and are considered to be a probable heritable corneal dystrophy. 44,55,56 Although affected rabbits are usually rejected from toxicity studies at prescreening, if corneal opacities first manifest during a study, they may be falsely attributed to a test article effect and confound the study results. In the Holve et al 10 survey, the incidence of apparent corneal dystrophy in DB rabbits varied by facility with a much higher incidence in males than in females from one of the breeding facilities. In this survey, a pre-Descemet membrane corneal opacity identified in a NZW rabbit by slit lamp biomicroscopy and OCT (data not shown) was similar to those previously described. 61 These sporadic findings appear to be a degenerative or traumatic change that may be related to the lifelong proliferative capacity of corneal endothelial cells in rabbits 68,69 and rodents that is absent in most mammals. Although pre-Descemet corneal dystrophy reported in humans is still included in the IC3D classification, 31 this opacity is also of uncertain heritability in humans and may be a degenerative or age-related change similar to that reported in NZW rabbits. 61
Using 13 juvenile DB rabbits from a single breeding facility shipment that were identified with corneal dystrophy during the Holve et al survey study, 10 we extended the previously reported phenotypic observations for corneal dystrophy in DB rabbits. 44 Monthly slit lamp and OCT imaging, lipid chemistry, histopathology, histochemistry, and TEM in 13 otherwise healthy DB rabbits identified normal IOP and serum lipids, lack of evidence for trauma, inflammation, neovascularization, or other ocular disease. The multiple cross-sectional OCT images provided a high resolution and direct correlate to the histological appearance and depth of the discrete lesions at study termination, and confirmed that OCT can be an important diagnostic tool for subtyping corneal dystrophies. 72 Microscopic histochemistry is an important but not exclusive classification tool for diagnosis of corneal dystrophies involving the epithelium or stroma in humans. 31 Using an algorithm of differential histochemical staining (Figure 1), we ruled out a variety of abnormal deposits and localized the corneal dystrophy in DB rabbits to the human category of epithelial-stromal TGFβI dystrophies. The microscopic disorder of stromal lamellae was confirmed by TEM to be consistent with the curly fibers found in the Thiel-Behnke subtype of corneal dystrophy (TBCD). The source of these rabbits from a single shipment from a single production facility supported a possible diagnosis of heritable corneal dystrophy. A heritable basis could not be confirmed due to an inability to retrieve breeding records from the production facility, and we lacked the capability to genotype the rabbits. Although domestic rabbits exhibit genetic heterogeneity both within and across breeds, 73 individual breeding facilities are often semiclosed and rely on a small number of internally sourced breeders, and line breeding may result in unnoticed mutations that do not manifest clinically until the animals leave the facility.
Detracting from making a definitive diagnosis of TBCD is that we could not immunohistochemically stain for TGFβI protein (keratoepithelin) due to lack of reagents and we could not genotype these rabbits for tfgβi mutations. Additionally, we did not identify a saw-tooth undulation between the epithelial basement membrane and stroma reported for TBCD in humans in OCT and microscopy evaluations. 31,72 Rabbits along with most mammals other than humans and nonhuman primates lack a microscopically defined Bowman layer, which is a specialized acellular, nonreplicating collagenous anterior limiting lamina between epithelium and stroma. 1,74,75 As Bowman layer is heavily involved by RBCD and TBCD, the lack of this layer in the rabbit may result in a less pronounced delineation between the stromal avascular fibrocellular pannus and epithelium. The reason why corneal dystrophies begin as small discrete lesions in specific geographic segments of the cornea is unknown. Over time, the corneal damage progresses in distribution and depth, and with TBCD, the granular opacities develop a characteristic honeycomb appearance. 31 We did not observe this more advanced appearance but as we only observed our DB rabbits for 4 months up to 9.5 months of age and only saw limited lesion progression, the full extent of corneal dystrophy in aged DB rabbits needs to be evaluated.
Our findings were similar to those reported by Moore et al 44 for the juvenile status of the DB rabbits at time of identification, mostly bilateral and paracentral opacities by slit lamp imaging, and histopathological involvement of epithelium, basement membrane, and anterior stroma. With the limitations of classification of corneal dystrophy subtypes in humans at that time, they concluded that this was a unique dystrophy and not comparable to EBMD, RBCD, or MECD anterior dystrophies in man. Both EBMD and RBCD but not MECD have significant involvement of Bowman layer, which is absent in rabbits. 74,75 Epithelial basement membrane dystrophy localizes to the basement membrane and RBCD includes stromal involvement. Based on the description provided by Moore et al, 44 their findings would also be consistent with an epithelial-stromal TGFβI dystrophy, particularly RBCD or TBCD.
An important differential diagnosis for corneal dystrophy in rabbits is fibrosis or scarring. Typically, scarring is often preceded by obvious trauma and concave erosions or ulcerations highlighted by fluorescein staining and inflammation, which were not found in our study. Fibrotic lesions tend to not progress in size and depth and often become smaller during remodeling. Histologically findings may include a concave defect with permanent loss of epithelium, inflammation, loss of keratocytes, and increased collagen density that is relatively uniform and organized after healing. The ophthalmological findings and disordered collagen fibers noted histologically and by TEM in our rabbits were not consistent with scarring. By TEM, the diameter of collagen fibers in scars is generally smaller than in normal tissues but may show heterogeneity in size and shape and with a less orderly arrangement of fibrils 76 but do not exhibit the disorder and curly fibers we observed. Hyperlipidemias in the rabbit are reported as heritable, 77 high cholesterol diet induced, 78 and subcutaneously injected poloxamer-407 thermo-gel excipient induced. 79 Therefore, we screened for and found our rabbits to have normal cholesterol and triglycerides. Lack of vacuoles, foam cells, and inflammation that are usually associated with systemic dyslipidemias was also absent in our rabbits and excluded lipid keratopathy as a possible diagnosis.
The gene TGFβI is highly conserved 80 with at least 355 invertebrate and vertebrate orthologs including the rabbit (https://www.ncbi.nlm.nih.gov/gene). This gene encodes for TGFβI protein which binds to collagens I, II, and IV and is present in extracellular matrix of many tissues including the corneal stroma and retinal pigment epithelium. The TGFβI protein (keratoepithelin) is reported to affect a variety of biological processes including embryogenesis, cell adhesion, migration, inhibition and progression of tumors, apoptosis, and angiogenesis. 80,81 Over 68 autosomal dominant amino acid mutations of the 5q31 locus of TGFβI have been associated with misfolded and insoluble protein accumulations that can result in corneal dystrophy. Mutational hotspots occur at positions 124 and 555, 70,81,82 with a knock-in mutation at 124 in B6-Tgfbitm1.1(TGFBI*R124H)Ccbko mice that at least partially recapitulates the granular deposits in GCD 2 corneal dystrophy. 43 In humans, genotype–phenotype correlations are often difficult 70,81,82 as there is substantial inter- and intra- family variability in corneal dystrophy phenotypes. Overlapping subtypes of epithelial-stromal TGFβI dystrophies are often misdiagnosed, published under a variety of eponymous and geographical names or published under the old classifications, rather than the current recommended 5 subtypes. 31
In animals, there is a risk that previously described or putative models of human disease will be lost as animals with heritable mutations may not be recognized or preserved in time to save breeding stock, or founder lines removed to eliminate the abnormality. This risk of genetic loss exists for the DB rabbit as breeders have tried to eliminate this corneal opacity, which negatively affects the acceptance of animals into ocular studies 10 and has an impact on interpretation of ocular findings in toxicity studies. 55,56,83 Although our study confirms and extends the findings of Moore et al 44 regarding corneal opacities in DB rabbits classified as corneal dystrophy, additional work will be required to definitely prove a heritable basis, identify the genetic defect, and confirm a possible relationship to tgfβI mutations. If the DB rabbit corneal dystrophy is confirmed as an epithelial-stromal TGFβI corneal dystrophy similar to the Thiel-Behnke type, this may be a valuable animal model for testing proposed therapies including gene editing. 84
Footnotes
Authors’ Note
Partial results of the pathology in the prospective study were reported in a proceedings abstract (Boorman G, Crabbs TA, Kolenda-Roberts H, et al. Proceedings of the 2011 National Toxicology Program Satellite Symposium. Toxicol Pathol. 2012;40(2):321-344).
Acknowledgments
We thank Dr Connie A. Cummings of UltraPath Imaging, Durham, NC, for the electron microscopy preparation and photography.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.