
Abstract
There is a striking difference in the potential for regeneration of injured axons in the central and peripheral nervous systems, which is important in neurotoxicologic studies. In contrast to the former, there is a ready mechanism for replacement of peripheral nerve axons that have degenerated following exposure to toxins, where long-distance axon regeneration and substantial functional recovery can occur. This relates at least in part to the nature of the glial and other supporting cells of the peripheral nerve. To provide background for these events, data on regeneration following traumatic injury to peripheral nerve are reviewed. This is followed by descriptions of nerve fiber regeneration after experimental exposure to 3 peripheral nerve axonopathic toxins, organophosphate tri-ortho-tolyl phosphate, the industrial chemical carbon disulfide, and the antituberculosis drug isoniazid.
Keywords
Introduction
There is a striking difference in the potential for nerve fiber regeneration in the central and peripheral nervous systems. 1 Axon regeneration in the mature vertebrate central nervous system is extremely limited after injury, and functional deficits involving axonal disconnection persist after a number of pathologic states. This situation differs from that in the peripheral nervous system, where long-distance axon regeneration and substantial functional recovery can occur in the adult. Both extracellular molecules and the intrinsic growth capacity of the neuron influence regenerative success. This has significance regarding the consequences of neurotoxic disease, often caused by the same agent, in these 2 components of the nervous system. Hence, the inclusion of this article in this issue of Toxicologic Pathology.
This presentation is divided into several sections. Because the literature for regeneration after traumatic injury to the peripheral nervous system is so extensive compared to the more limited amount of information concerning such events after toxic injury, there is an initial review of the nature and mechanisms of the former. This is followed by a detailed consideration of selected examples of repair after toxic neuropathy. Although repair of chemically induced peripheral nerve injury may be seen in other neuropathies, such as those featuring demyelination, the focus in this article is on regeneration after axonal lesions (axonopathy). Admittedly, this is a complex reparative process, superimposed upon prior damage, and thus may be difficult for the pathologist to fully assess. Thus, an objective of this review serve as a guide to toxicologic pathologists in understanding mechanisms of recovery in the peripheral nervous system, including use of 3 examples of toxicants that elicit axonal lesions, organophosphates, carbon disulfide, and isoniazid.
In a terminology note, Hall 2 calls for use of the term axon regrowth as indicating the presence of regrowing axons in the proximal level of the distal stump (below the site of injury) and axon regeneration for such growth reaching appropriate targets carrying a potential for functional recovery. In this review, the term regeneration is used in a more general sense, to indicate repair by regrowing fibers into previously injured regions of peripheral nerve, making note of functional issues when needed.
Regeneration After Mechanical Injury to Peripheral Nerve
As noted above, in contrast to the central nervous system, injured peripheral nerves have a robust potential for axonal regeneration, a subject later dealt within the context of toxic neuropathy. To provide background for the latter, what follows is a brief review of repair after nerve trauma, a much better studied event. Such nerve injury has been classified by the extent of damage, to include focal segmental demyelination (grade 1), axon damage with intact endoneurium (including Schwann cell basal lamina; grade 2), axon and endoneurial damage with intact perineurium (grade 3), axon, endoneurium, and perineurium damage with intact epineurium (grade 4), complete nerve transection (grade 5). 3 As might be expected, repair is move efficient with less intense injury, and repair of them in man may be protracted and incomplete. 2 There has been much interest in events following traumatic nerve injury, beginning with the work of Waller, 4 hence the term Wallerian degeneration applied to this process. In the classic model, there is interruption of a peripheral nerve. The nerve fiber segment distal to the site of injury undergoes Wallerian degeneration, resulting in its fragmentation and disintegration of the axon and its myelin sheath. This occurs fairly rapidly and in a proximodistal fashion. 5 There is sealing of the severed ends of the axons, loss of the axonal cytoskeleton, swelling of the periaxonal space, and failure of axonal transport, with associated pooling and degeneration of axonal organelles at nodal constrictions and within myelin ovoids. 2,6 Degradative events are likely initiated by early elevation of intra-axonal Ca++ leading to activation of phospholipases and calpains. The blood–nerve barrier is altered, allowing large molecules to enter the endoneurial space. 7 The degrading distal axonal segment is unable to conduct an action potential upon external stimulation. There is subsequent removal of axon and myelin debris by macrophages and Schwann cells. 8 -11 Since myelin debris contains inhibitors of axonal regeneration, such as myelin-associated glycoprotein, effective removal is important. 12 Relative to the central nervous system, this is a rapid process. Proximal to the site of injury, breakdown in the nerve fiber segment is limited to the region distal to the first node of Ranvier, although if injury occurs near the cell body neuronal apoptosis may occur. This may involve loss of some primary sensory neurons (within dorsal root ganglia), and less often motor neurons, following nerve section, related to loss of target-derived neurotrophic factors. 13
As removal of fiber debris proceeds in the distal segment, there is an initiation of events that would lead to regeneration of damaged axonal segment. Schwann cells convert to a nonmyelinating (regenerative) phenotype, within the persisting basal cell tube of the degenerated axonal segment, forming the band of Büngner, under influence of enhanced c-Jun production. In this process, there is downregulation of structural proteins P0, myelin basic protein, and myelin-associated glycoprotein and upregulation of cell adhesion molecules (CAM) such as L1-CAM, neural CAM, glial fibrillary acidic protein, a number of growth factors (nerve growth factor glial-derived neurotrophic factor, basic fibroblast growth factor, insulin-like growth factors), and neurotrophin 3. 13 Schwann cells in the band of Büngner generate extracellular matrix components such as fibronectin and laminin that allow adhesion of growth cones to the matrix. In combination with debris removal by action of macrophages and Schwann cells, these form a permissive environment rich in trophic factors enabling guided axonal regeneration. 14 Thus, the bands of Büngner provide a trophic site for regenerating axons, which initially arise as mobile growth cones from the distal region of the proximal segment. The growth cones contain tyrosine kinase receptors for neurotrophins generated by Schwann cells in the band of Büngner, along with extracellular matrix components such as fibronectin and laminin that allow adhesion of growth cones to the matrix in the band of Büngner.
There are concordant changes in the proximal segment and attached neuronal cell body. Following axotomy, neurons with axons extending into the peripheral nervous system upregulate numerous regeneration-associated genes. These include c-Jun, activating transcription factor-3, SRY-box containing gene 11, small proline-repeat protein 1A, and growth-associated protein-43 (GAP-43). 1 After such an injury, one or more sprouts arise from the terminal swelling or the first rostral node of Ranvier, under stimuli provided by the cell body. 12,15 Faroni et al 14 describe effects on involved neurons. An initial antidromic burst of action potentials is seen, which open calcium channels and initiate Jun-kinase cascades that influence transcription. There also is withdrawal of target-derived neurotrophic support, leading to gene and protein expression. The balance of these determines whether the neuron undergoes apoptosis or survives and attempts regeneration. The latter is manifest by upregulation of proteins associated with growth (GAP-43, tubulin, actin) and downregulation of functional proteins (neurotransmitters, neurofilaments). Chromatolysis of the neuronal cell body is a morphologic manifestation of these events. There is production of growth cones and axonal sprouts from the distal level of the proximal axon segment, as noted above.
The degree of injury affects the efficacy of regeneration. As reviewed by Griffin et al, 12 compared to nerves subjected to transection and reconnection, nerves that have undergone less severe injury, such as focal crush, regenerate much faster and more completely. Because the latter leave the Schwan cell basal lamina intact, there is a higher probability of the sprouts growing within the Schwann cell bands of the original fibers. Such lesions thus optimize the opportunities for axons to grow down their original paths. Not only does this arrangement enhance guidance back to original targets, but it also allows fibers the benefits of “their” kind of daughter Schwann cells and growth factors. Denervated motor Schwann cells differ from denervated sensory Schwann cells in their pattern of growth factor expression as well as in aspects of their extracellular matrix. This is shown by the better growth of motor nerve fibers in motor pathways. Sunderland 3 cites studies indicating 1 to 5 mm/(day). Griffin et al 12 note that successful regenerating fibers can grow fairly rapidly in the mouse, ≥4 mm/d. In uncomplicated regeneration, the connectivity with the end organ is attained. Axonal sprouts increase in diameter and will become myelinated by Schwann cells upon reaching a certain size, in centrifugal fashion. 3,15 A frequent feature of regenerating sprouts is their arrangement in small bundles. 15 A potential complication of incomplete healing after peripheral nerve trauma, especially if the proximal and distal stumps of the nerve are not approximated, is the traumatic neuroma. These are often painful, disorganized non-neoplastic masses of undirected axonal sprouts, Schwann cells, fibroblasts, and collagen.
Regeneration Following Toxic Peripheral Neuropathy
There are many toxins with the potential to injure the peripheral nervous system and eliciting peripheral neuropathy, 16 often with associated with CNS damage. In many of these conditions, axonal damage is a primary focus (axonopathy). 17 As noted above, in contrast to the central nervous system, peripheral nerve has substantial capability to regenerate following injury. That process as seen after traumatic injury and Wallerian degeneration has been noted above. Here some examples of such repair following chemical damage are reviewed. There have been a number of studies combining toxic effects and regeneration in which the experimental paradigm is to study the effect of the test article on regeneration following traumatic injury to the nerve. 18 These are not included in this article, since the combination of toxin plus local trauma complicated the data. Instead the following discussion is restricted to states in which regeneration in peripheral nerve is seen following systemic administration of toxicants, analogous to what might be seen in standard drug/chemical safety assessment. Under optimal conditions, such a chemically mediated nerve fiber injury leaves the Schwann cell basal lamina tube and endoneurial vascular and connective tissue support intact, allowing ready formation of the band of Büngner in the affected distal segment of the fiber, which enhances orderly axonal regeneration. This mimics the Sunderland 3 grade II traumatic injury, a condition that enhances axonal regeneration following Wallerian degeneration. Of course other support for regeneration, such as the overall state of health of the individual and of the neuronal cell body and adjacent proximal axonal stump, also can affect the repair process. To illustrate peripheral nerve fiber regeneration in this setting, 3 experimental animal models using a variety of toxicants have been chosen. These are organophosphorus ester-induced delayed neurotoxicity (OPIDN) in domestic chickens, neuropathy following exposure of rats to carbon disulfide vapor, and oral dosing of antituberculosis antibiotic isoniazid in rats. In all of these, the test article is administered systemically and results in peripheral nervous system degeneration and subsequent regeneration. The major focus of the following is on neuropathologic changes seen in the studies.
From the standpoint of pathology, the best described study is that of Jortner and colleagues 19 using the hen OPIDN model. This is a well-known entity in which exposure to the toxicant elicits axonal degeneration, most prominent in distal regions of the peripheral nerve and certain tracts of the CNS. 20 Jortner et al 19 used a single toxic gavage dose of 360 mg/kg tri-ortho-tolyl phosphate in adult hens (the standard test animal for OPIDN) and evaluated clinical signs and lesions over the next 64 days. This model has value, since the clinical effect of OPIDN is easily recognized and quantitated during the evolution of the neuropathy. Peripheral nerve lesions were studied in the tibial nerve branch to the lateral head of the gastrocnemius muscle or in the nerve of the biventer cervicis muscle, both distal, not terminal, nerves susceptible to injury in the hen model of OPIDN. 21,22 As typical of OPIDN in hens, Jortner et al 19 noted bilateral lesions of axonopathy progressing to myelinated fiber Wallerian type degeneration were prominent on days 14 (the first day of such sampling) and 16, with larger myelinated fibers preferentially affected (Figures 1 and 2). As fiber debris was diminished, Schwann cells subsequently proliferated within the basal lamina tubes of affected fibers, forming the band of Büngner (Figure 3). As noted above, these hyperplastic Schwann cells enhanced fiber regeneration and, with time, were noted to contained one or more axonal sprouts. In this model, it was common to see a single regenerating axonal sprout dominate a band of Büngner (Figure 4). By day 23 to 28, many of these neurites became thinly myelinated by Schwann cells of the band of Büngner (Figures 5 and 6). It was not unusual to observe multiple stages of degeneration and regeneration at this interval (Figure 7), although regeneration became more advanced as the postdosing period lengthened. Myelin thickness progressively increased by day 42, in the reinnervated bands of Büngner. By day 64, the nature and concentration of the fibers in the regenerated nerves was indistinguishable from the controls (Figure 8). In contrast to this ready repair in peripheral nerve, lesions in central nervous system, as evaluated in distal (cervical) levels of the spinocerebellar tracts, evolved more slowly. 19 Swollen, debris-laden large myelinated fibers undergoing Wallerian-type degeneration were noted from postdosing day 14 and remained present, although at reduced incidence later in the course, through day 64. There was associated fiber loss, gliosis, and perivascular fibrosis, but no axonal regeneration was seen throughout the period of observation.
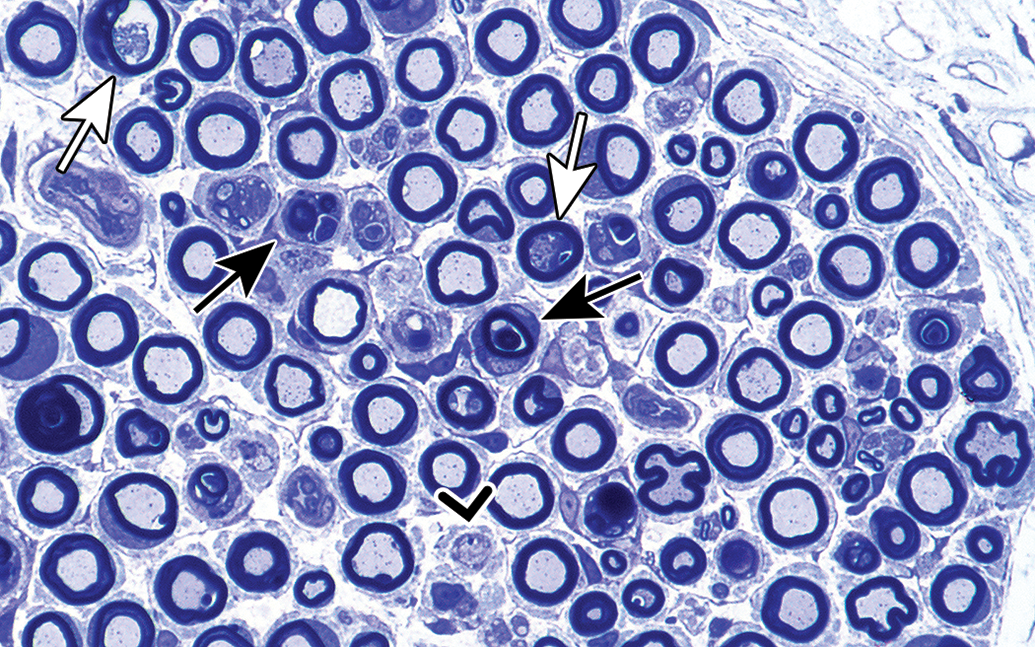
Myelinated fiber degeneration 14 days after dosing with 360 mg/kg tri-ortho-tolyl phosphate. There are profiles of axonal degeneration (white arrows), which progress to fiber breakdown manifest by myelin-rich debris within phagocytes, likely Schwann cells (black arrows). Rare bands of Büngner were recognized in this section (caret). Transverse section, tibial nerve branch to lateral head of gastrocnemius muscle, toluidine blue stain.
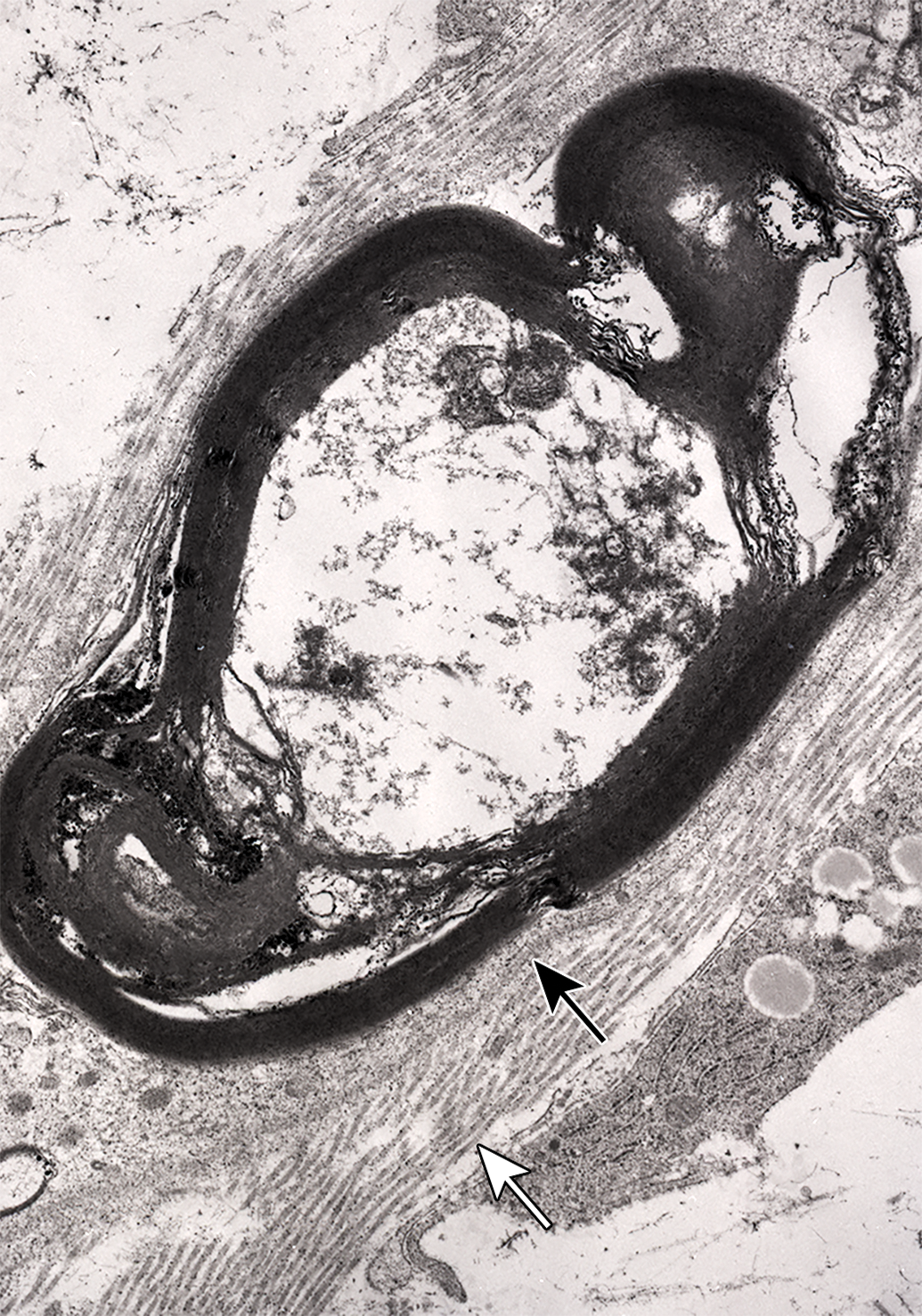
Advanced stage of myelinated fiber degeneration, with electron-dense myelin-rich debris in Schwann cell. Note the basal lamina of the Schwann cell (black arrow) and adjacent collagen fibrils (white arrow). Tibial nerve branch to the lateral head of the gastrocnemius muscle, 16 days following administration of tri-ortho-tolyl phosphate.
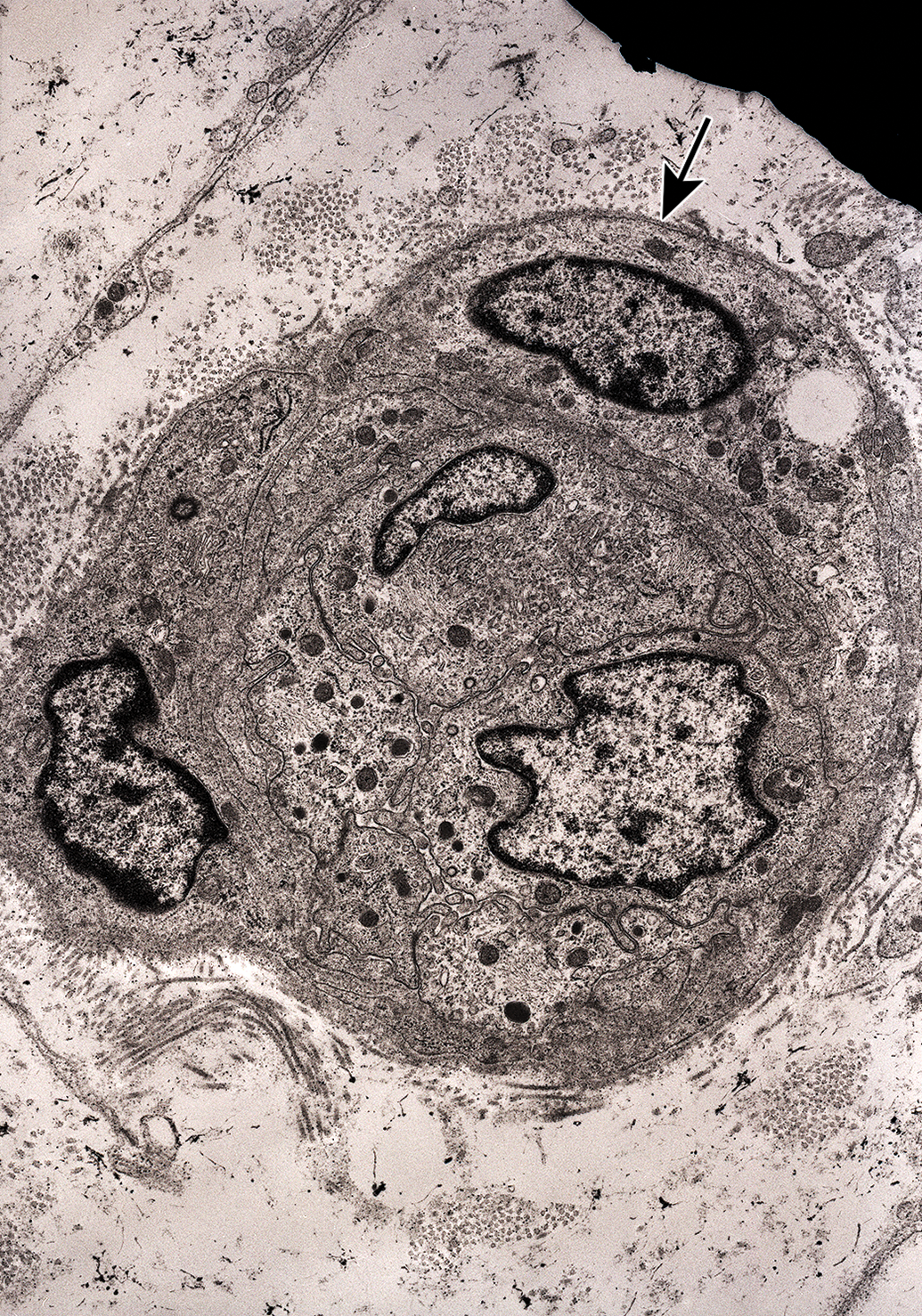
Transverse section of a band of Büngner with multiple Schwann cell profiles within a tube outlined by basal lamina (arrow). Nerve to biventer cervicis muscle. Sixteen days after tri-ortho-tolyl phosphate administration.
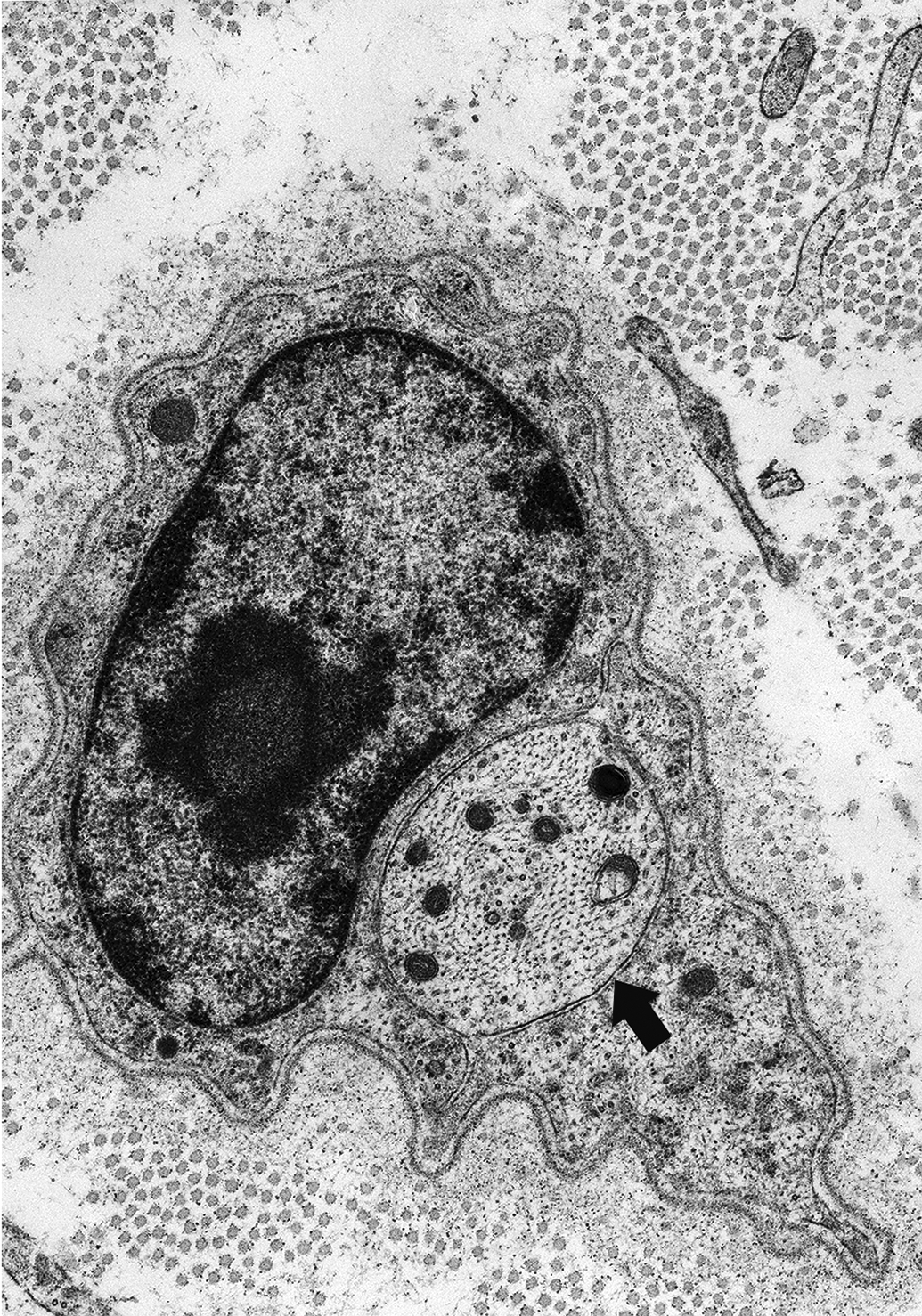
Premyelinated regenerating axon (arrow) in band of Büngner, adjacent to a Schwann cell nucleus. Tibial nerve branch to the lateral head of the gastrocnemius muscle, 23 days following administration of tri-ortho-tolyl phosphate.
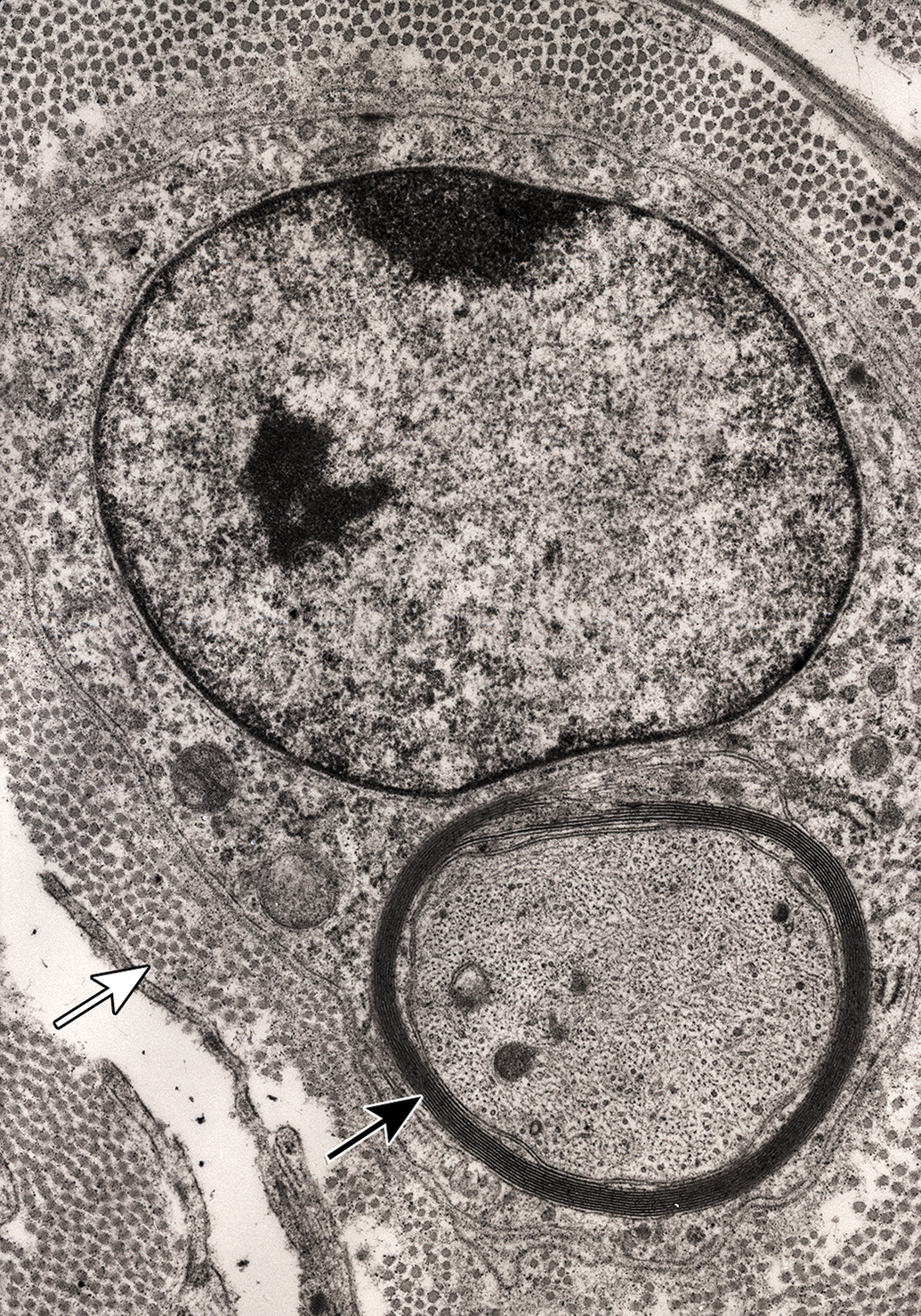
Juxtanuclear thinly myelinated regenerating axon (black arrow) in band of Büngner and adjacent collagen fibrils (white arrow). Tibial nerve branch to the lateral head of the gastrocnemius muscle, 28 days following administration of tri-ortho-tolyl phosphate.
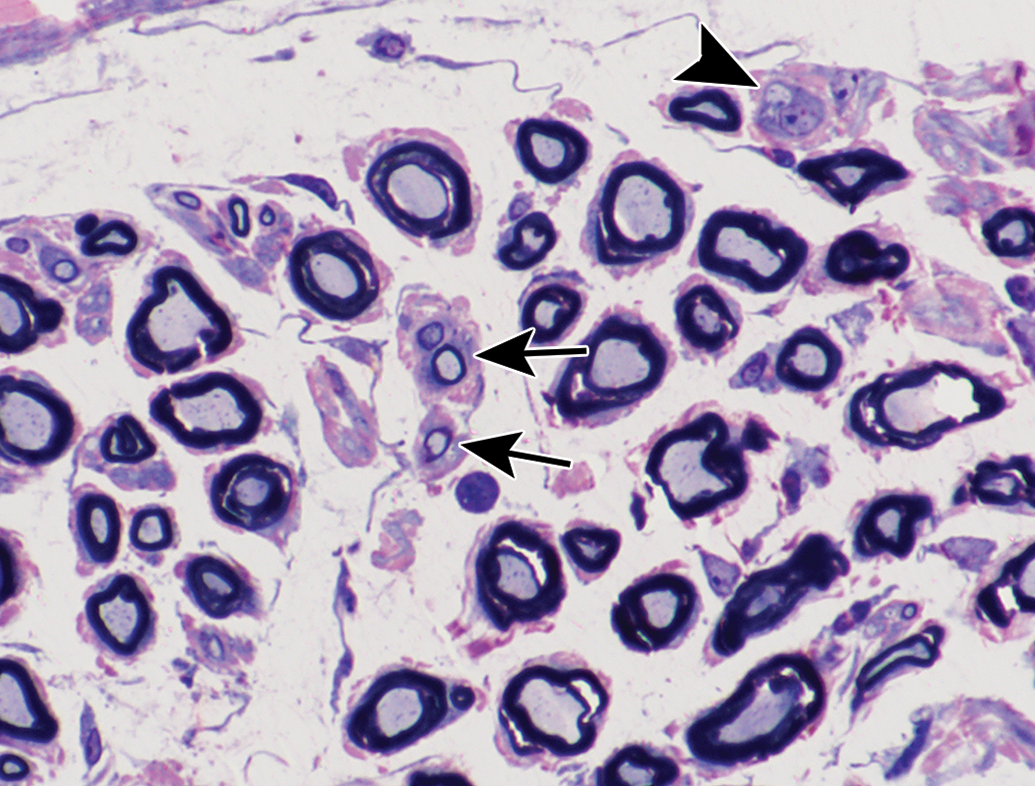
Small thinly myelinated regenerating fibers (arrows) and a single premyelinated axon (arrowheads) are present in this transverse section of the tibial nerve branch to lateral head of gastrocnemius muscle 28 days following administration of tri-ortho-tolyl phosphate, toluidine blue, and safranin stain.
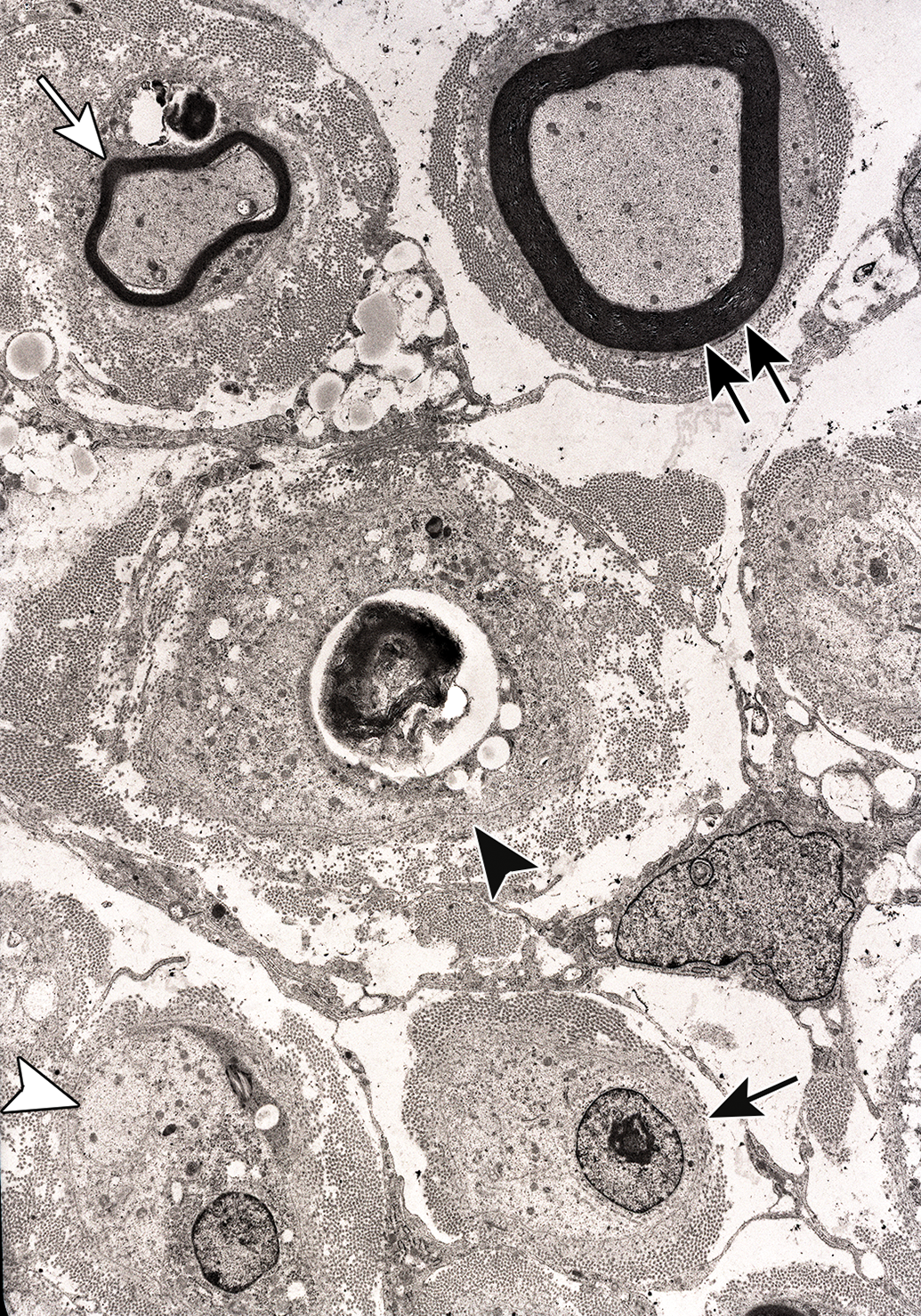
Survey of endoneurial region seen 23 days after tri-ortho-tolyl phosphate administration. There are transverse sections of a debris-laden Schwann cell/band of Büngner (black arrowhead), a noninnervated band of Büngner (black arrow), a premyelinated regenerating axon in a band of Büngner (white caret), a thinly myelinated regenerating axon (white arrow), and a more advanced myelinated regenerating fiber (double arrows) each in a band of Büngner. Tibial nerve branch to the lateral head of the gastrocnemius muscle.
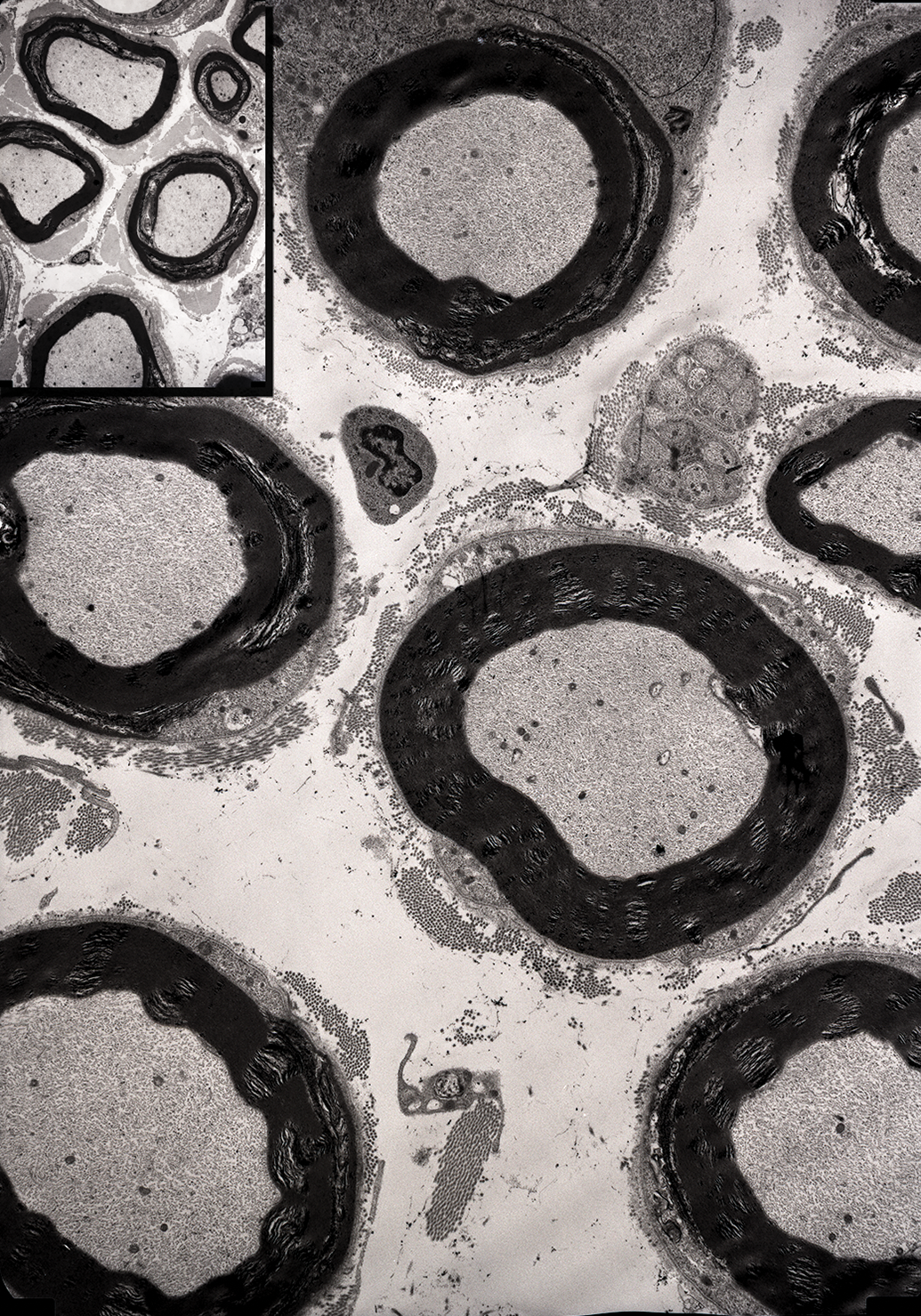
Advanced regeneration of myelinated fibers 64 days after tri-ortho-tolyl phosphate administration. These resemble fibers seen in control hen (inset). Tibial nerve branch to the lateral head of the gastrocnemius muscle.
In the above-noted study, Jortner et al 19 focused on axonal regeneration into distal, nonterminal levels of peripheral nerve following chemical injury. The regenerative fibers were not traced to their end organs, admittedly an important issue in restoring function. 2 Despite this, there is clinical evidence that this has occurred in affected peripheral nerve in this study of OPIDN in hens. 19 Clinical deficits of OPIDN such as varying intensities of ataxia and paresis are readily measured in that animal model and are scored using a semiquantitive 1 to 4 scale system. 20 These were measured in the 14-to 64-day postdosing period and were most profound on day 14. 19 There was progressive improvement in clinical performance through day 49, when the deficits plateaued around a score of 1. The authors felt this reflected regeneration of peripheral nerve that, although not demonstrated, likely extended to the appropriate terminals. The low level of residual deficit may reflect the ongoing lack of central nervous system regeneration. In summary, this study reflects toxic effects that make regeneration of peripheral nerve fiber more likely. That is a single toxic exposure that elicits a distal axonopathy, no disruption of the basal lamina of affected fibers or of the endoneurium, and the general toxic effect is transitory, with neuronal cell bodies being able to support regeneration.
There are other examples of peripheral nerve fiber regeneration seen after toxic exposures producing axonal degeneration. One of these, carbon disulfide, is an industrial chemical and known human neurotoxicant. Appropriate exposure elicits massive aggregates of neurofilaments with a predilection for larger longer axons in the peripheral and central nervous system. 23 These are related to dose-dependent covalent cross-linking in neurofilament proteins by the toxicant prior to the onset of lesions, contributing to the development of the malaligned neurofilamentous axonal swellings characteristic of carbon disulfide neurotoxicity. 23 Such lesions may progress to fiber degeneration. Colombi and colleagues 24 exposed rats to 700 parts per million of carbon disulfide vapor for 12 weeks, followed by a 4-month recovery period. This exposure produced axonal degeneration in the common peroneal nerve, characterized by massive aggregates of neurofilaments leading to axonal swelling proximal to nodes of Ranvier at 10 weeks’ exposure. By the third week of the recovery period, these lesions progressed to breakdown of affected fibers (Wallerian-type degeneration). In samples examined at the eighth week of recovery, regenerating nerve fibers with early, and a few with more advanced, myelination were noted. Clinical effects paralleled these morphologic findings. Sciatic nerve conduction velocity progressively decreased during the 12 weeks of carbon disulfide exposure and the first 3 weeks of recovery and then slowly improved. There also was progressive impairment of hind limb function beginning with weakness following initial exposure to carbon disulfide and progressing to reduction in spontaneous movements and muscle atrophy by week 3 of recovery. This improved after that time, and function was near normal by the end of the 4-month recovery period. 24 This carbon disulfide study shows peripheral nerve myelinated fiber degeneration featuring axonal lesions progressing to fiber breakdown during and shortly after toxicant exposure, with regeneration seen following a period of recovery. In a similar study, Jirmanová and Lukáš 25 exposed adult Wistar rats to carbon disulfide vapor at 2.4 mg/L of air for 5 d/wk, 6 h/d for 6 months, and saw similar axonal lesions characterized by giant axonal swellings related to disorganized masses of neurofilaments. Some affected fibers underwent Wallerian-type degeneration, but at the same time, there was some fiber regeneration, despite ongoing exposure to the carbon disulfide vapor.
Another example of regeneration after toxic neuropathy is reported in rats given a single 1500 mg/kg gavage dose of the antituberculosis antibiotic isoniazid, 26 a drug which may elicit peripheral neuropathy by interfering with pyridoxine species and related reactions. In the model of Chua et al, 26 there was an axonopathy progressing to fiber myelinated degeneration involving peripheral nerves and the gracile fasciculus, more prominent distally. This was noted as early as post-dosing day 2 in peroneal and distal sural nerves and was prominent by day 14 in the sural nerve and ventral spinal nerve root. Larger myelinated fibers were preferentially affected. Neuronal cell bodies giving rise to affected nerves, such as in spinal ganglion of lumbar level 6 and ventral horn cells of the lumbar spinal cord, appeared intact. Regeneration was seen in spinal nerve roots as early as day 7 and extended into the 14 to 30 days postexposure period. This extended in a proximal–distal sequence during the recovery and was manifest by axonal sprouts and clusters of thinly myelinated fibers. In a related study, Jacobs et al 27 elicited prominent axonopathy progressing to fiber degeneration in adult male Wistar rats administered isoniazid as a single large (up to 2000 mg/kg) gavage dose or at 1.33 mg/mlL in the drinking water for up to 105 days. As in the Chua’s 26 study, this was manifest by axonopathy progressing to Wallerian-type degeneration in peripheral nerve fibers, more prominent distally. There was greater involvement of motor relative to sensory fibers. Interestingly, there also was fiber regeneration even in the face of ongoing drug administration. Nerve fiber sprouting also was noted in an earlier isoniazid study. Cavanagh 28 exposed adult rats to 250 mg/kg/d isoniazid in the diet. This elicited Wallerian-type degeneration of peripheral nerve, primarily motor, fibers, most severe below the knee (distal). A striking finding was prominent regional regenerative collateral axonal sprouting from surviving nerve fibers, even in face of ongoing exposure to the drug. This was seen in rats on the experimental diet for 18 to 89 days and was enhanced when exposure to INH was stopped and rats placed on a normal diet for up to 45 days.
Summary
A consideration of the potential and effects of axonal regeneration should part of a neurotoxicologic investigation. This is a process that is markedly restricted in the central nervous system, but available in the peripheral nerves. The regenerative capacity of peripheral nerve following mechanical injury producing Wallerian degeneration is well known. A special issue of Toxicologic Pathology devoted to toxic neuropathy seemed a good place for a paper drawing attention to such a reparative potential following chemical injury. This work is an attempt to do so. There are 2 major sections to the paper. First is a review of the extensive literature on regeneration of mechanically injured peripheral nerve fibers undergoing Wallerian degeneration, to consider repair mechanisms that may be relevant following toxic injury. This is followed by some examples of regeneration in toxic neuropathy. Given that the literature on the latter is much less extensive, 3 toxicants eliciting peripheral (and central) nerve degeneration were selected for detailed discussion. These agents, the organophosphate tri-ortho-tolyl phosphate, the industrial chemical carbon disulfide, and the antituberculosis drug isoniazid, were selected to illustrate systemic exposure to the toxicant, development of the toxic axonopathy, and a subsequent recovery period, to optimize the potential for regeneration. Toxicologic pathologists, and other workers, should be aware of the repair potential of peripheral nerve in assessing effects of toxicants.
Footnotes
Acknowledgments
I wish to indicate my appreciation for my long-term association with colleagues Sandra Hancock and Marion Ehrich of the Virginia Tech Laboratory for Neurotoxicity Studies and recognize their contributions to material included in this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.