
Abstract
This review illustrates common lesions of peripheral nerve myelinated fibers that occur in toxic neuropathy. These distinctive structural changes help to define the site of toxicant activity and thus predict the course of neurotoxic disease and recovery. Neuronopathy is the condition where the primary injury is directed to the neuronal cell body giving rise to a peripheral nerve axon. Axonopathy occurs when the axon is the primary target, and myelinopathy develops where the Schwann cell and/or myelin sheath is the primary target; these conditions can be discriminated early during the course of nerve fiber degeneration, but reciprocal influences between axon and myelin result in degeneration of both structures late in the disease.
This review article is derived from a talk on common toxicant-induced structural lesions of peripheral nerves that was given at a ½-day continuing education course on toxicologic pathology of the peripheral nervous system (PNS) that was presented at the 37th annual meeting of the Society of Toxicologic Pathologists, June 2018 (Pardo et al. 2018). There are several components to this article. First is a brief review of the organization and histologic features of the PNS. Next, are examples of classic experimentally induced lesions of peripheral nerve myelinated fibers representing three classic categories of toxic neuropathy: neuronopathy, axonopathy, and demyelination. To reflect contemporary practice of peripheral nerve pathology (Schröder 2001), most of the light microscopic material presented in this review is from toluidine blue–stained, semithin sections of aldehyde-fixed, osmium tetroxide postfixed, epoxy resin–embedded nerve samples. Some aspects of the present article are drawn from a previously published work (Jortner 2011).
Organization and Components of the PNS
The PNS is defined as those portions of the motor, primary sensory, and autonomic neurons that extend outside the central nervous system (CNS; Schaumburg, Berger, and Thomas 1983). Elements of the PNS thus include dorsal and ventral spinal nerve roots, spinal and cranial nerves (except for the first and second cranial nerves, which represent extensions of the CNS), dorsal root and other sensory ganglia, sensory and motor terminals, and the bulk of the autonomic nervous system. Supporting glia in the PNS include Schwann cells and ganglionic satellite cells. The concept of separate PNS and CNS is artificial, since cell bodies of motor neurons with peripherally directed axons lie within the CNS and some peripheral sensory neurons have extensive central projections.
A summary of the components and organization of the PNS is demonstrated in Figure 1. This diagram demonstrates both the somatic and autonomic elements (Schaumburg, Berger, and Thomas 1983). As noted above, this review article is focused on the somatic myelinated components of the PNS, including peripheral motor and sensory fibers, along with neuronal cell bodies that reside in ganglia. The organization of a prototypical peripheral nerve as seen in cross section is demonstrated in Figure 2 (as a diagram) and Figure 3 (as a photomicrograph), which show its components including the connective tissue sheaths, the outer epineurium, and the specialized perineurial barrier. The latter is a multilayered structure with tight junctions between cells, forming a diffusion barrier isolating the interior (endoneurium) of the fascicles. Within this protected endoneurial space lie fascicles of myelinated and unmyelinated nerve fibers each encompassed by a sheath formed by a linear series of Schwann cells. In myelinated fibers, individual Schwann cells form the internodal segments of myelin, with each cell separated from its neighbors at the nodes of Ranvier (Figures 1 and 2). Multiple unmyelinated fibers are enfolded by the cytoplasm of a single Schwann cell (Figure 2). An example of a normal ganglion, in this case a dorsal root (or spinal) ganglion, is provided in Figure 4.
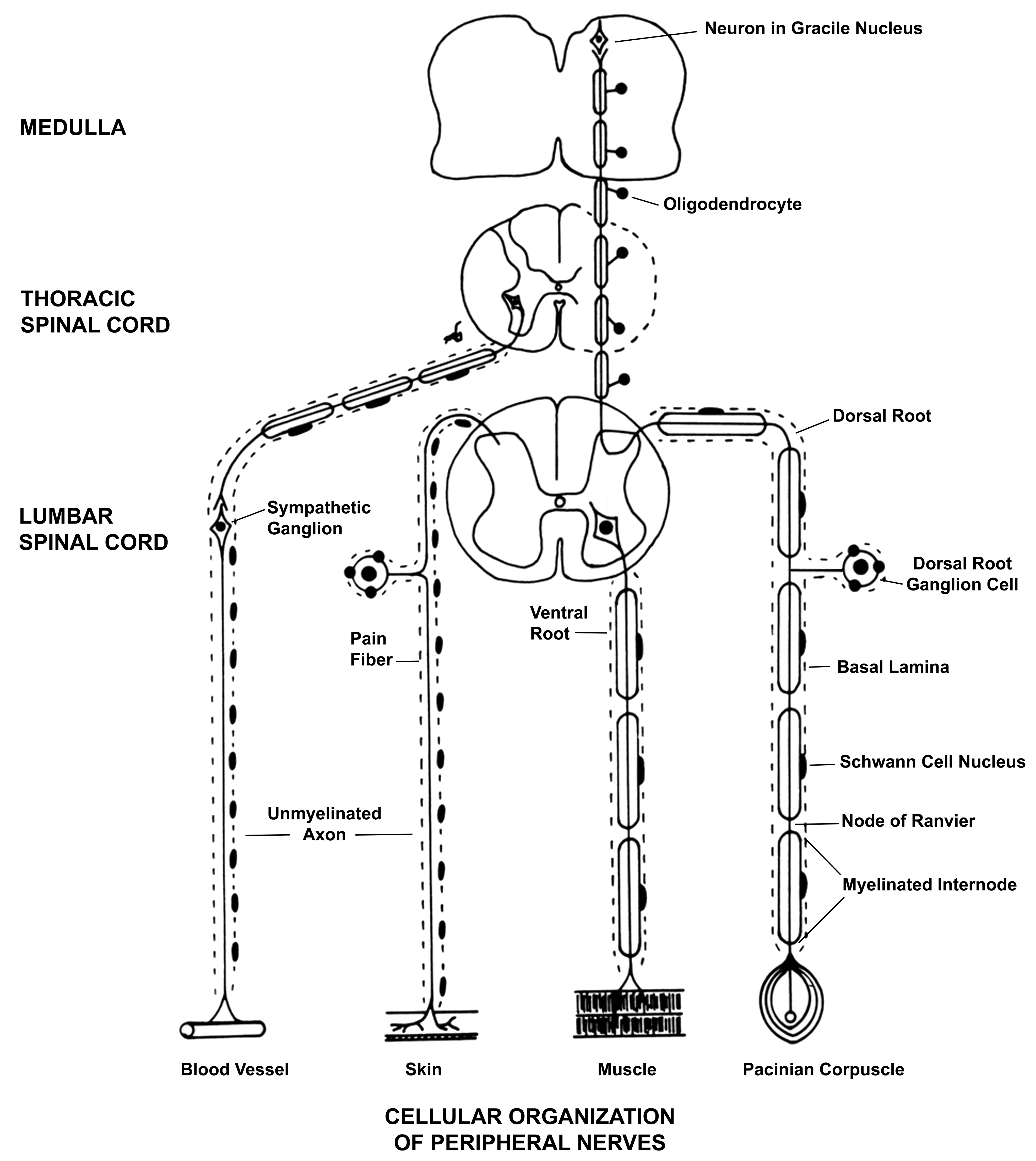
The principal components of the peripheral nervous system and the cellular organization of peripheral nerves. The image shows examples of myelinated and unmyelinated components of the somatic nerves and the sympathetic component of the autonomic system. As noted in the text, the focus in this article is on lesions in myelinated fibers, illustrated by the two fibers to the right of the image. From Schaumburg, Berger, and Thomas (1983), by permission of Oxford University Press, USA.
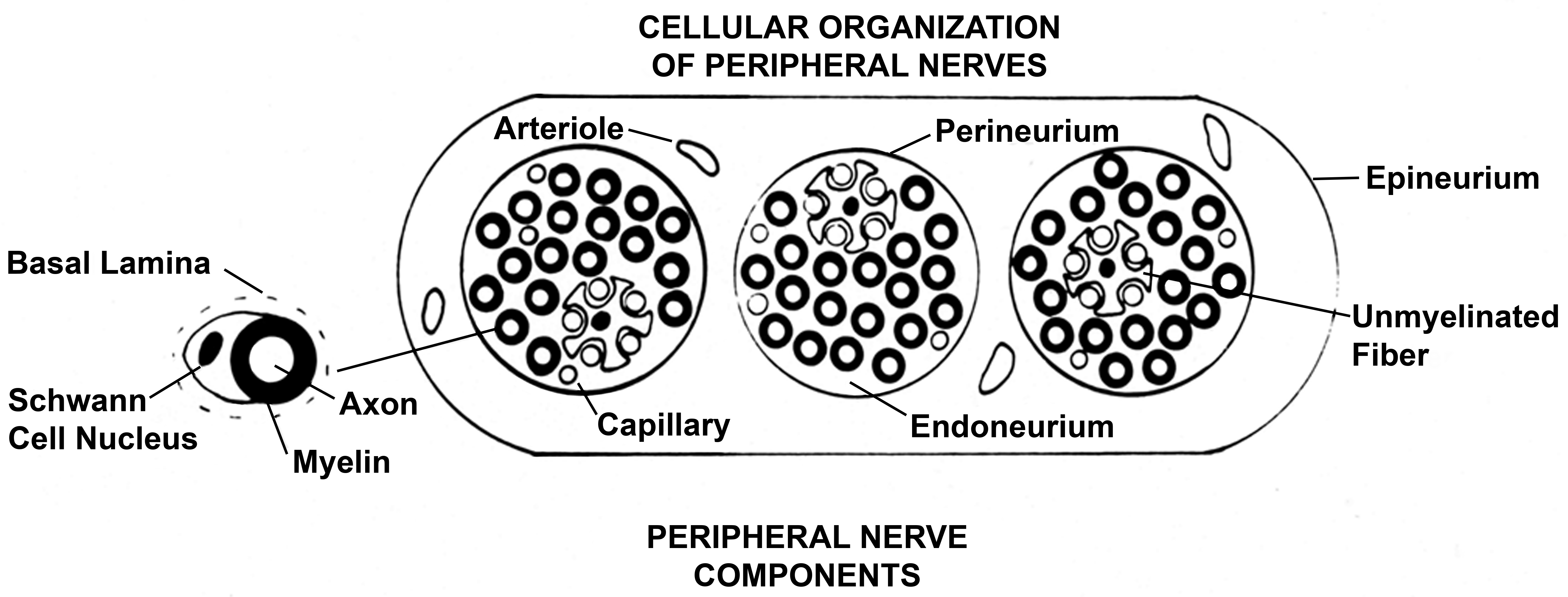
Diagram of peripheral nerve in cross section, showing three fascicles of myelinated and unmyelinated nerve fibers, each surrounded by a perineurial sheath. A connective and adipose tissue covering, the epineurium, enwraps this sheath. The left side of the image shows the relationship of Schwann cell, the myelin sheath it generates, associated axon and basal lamina around the Schwann cell. From Schaumburg, Berger, and Thomas (1983), by permission of Oxford University Press, USA.
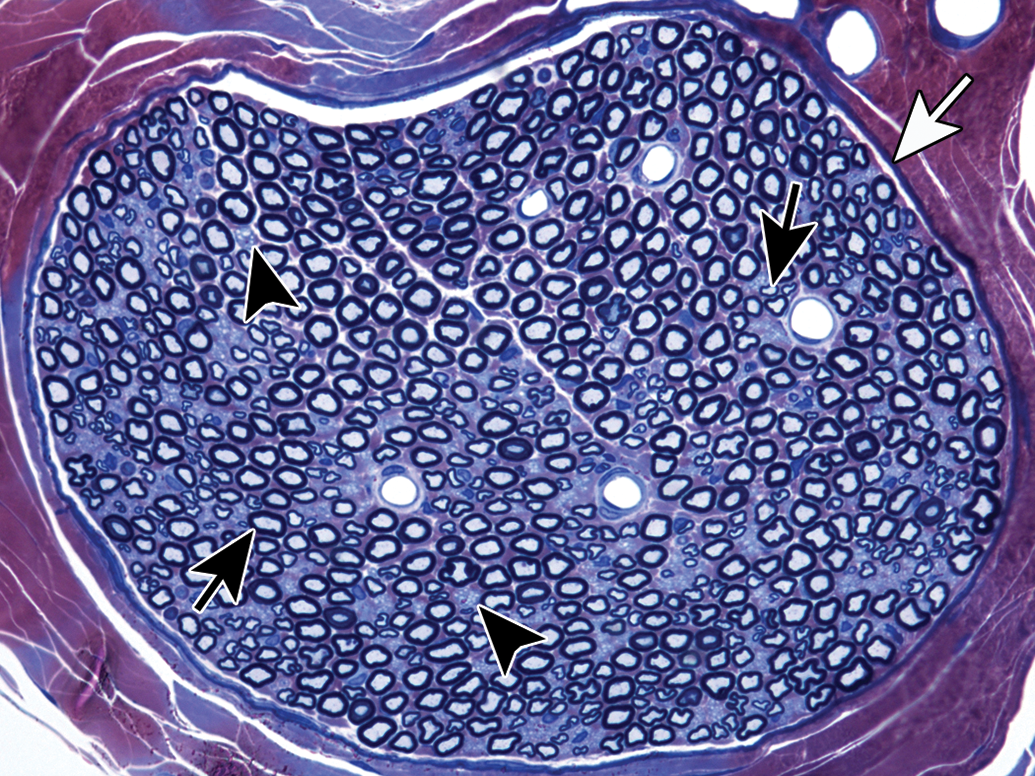
Cross section of a rat sural nerve, with the perineurium (white arrow) surrounding the endoneurium, which is filled with varying sized myelinated fibers (black arrows) and small pale-stained regions containing clusters of unmyelinated fibers (arrowheads). Blood vessels are distended due to perfusion fixation of the nerve. Toluidine blue and safranin stain. From Jortner (2011), used with permission of SAGE Publications, Inc.
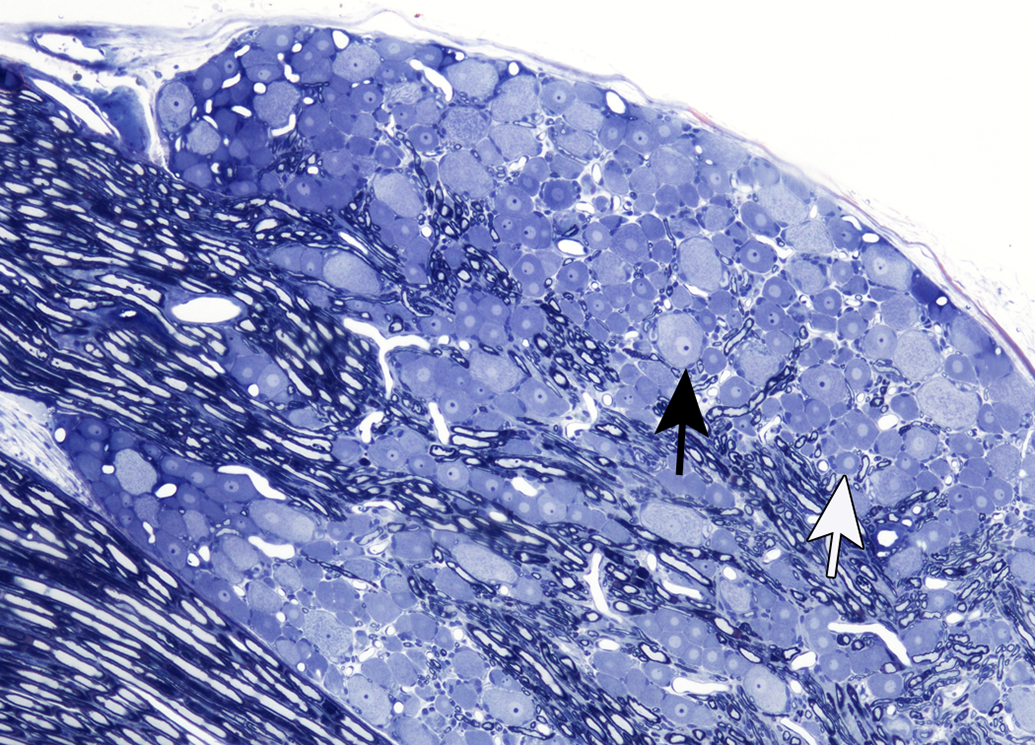
Longitudinal section of a rat dorsal root ganglion with associated nerve roots. Note the presence of varying sized neuronal cell bodies, mainly large light (black arrow) and small dark cytons (white arrow). Dorsal root fibers course through the ganglion, and the ventral root (lower left corner of image) lies adjacent to it. Toluidine blue and safranin stain. From Jortner (2011), used with permission of SAGE Publications, Inc.
Pathology of Toxic Peripheral Neuropathy
As shown in Figure 5, there are three basic pathologic reactions of peripheral nerves induced by toxic agents: neuronopathy, in which the injury is concentrated on the neuronal cell body; axonopathy, in which the axon is the major site of injury; and myelinopathy (demyelination), in which the myelin sheath or Schwann cell are involved. Selected examples of these lesion categories follow.
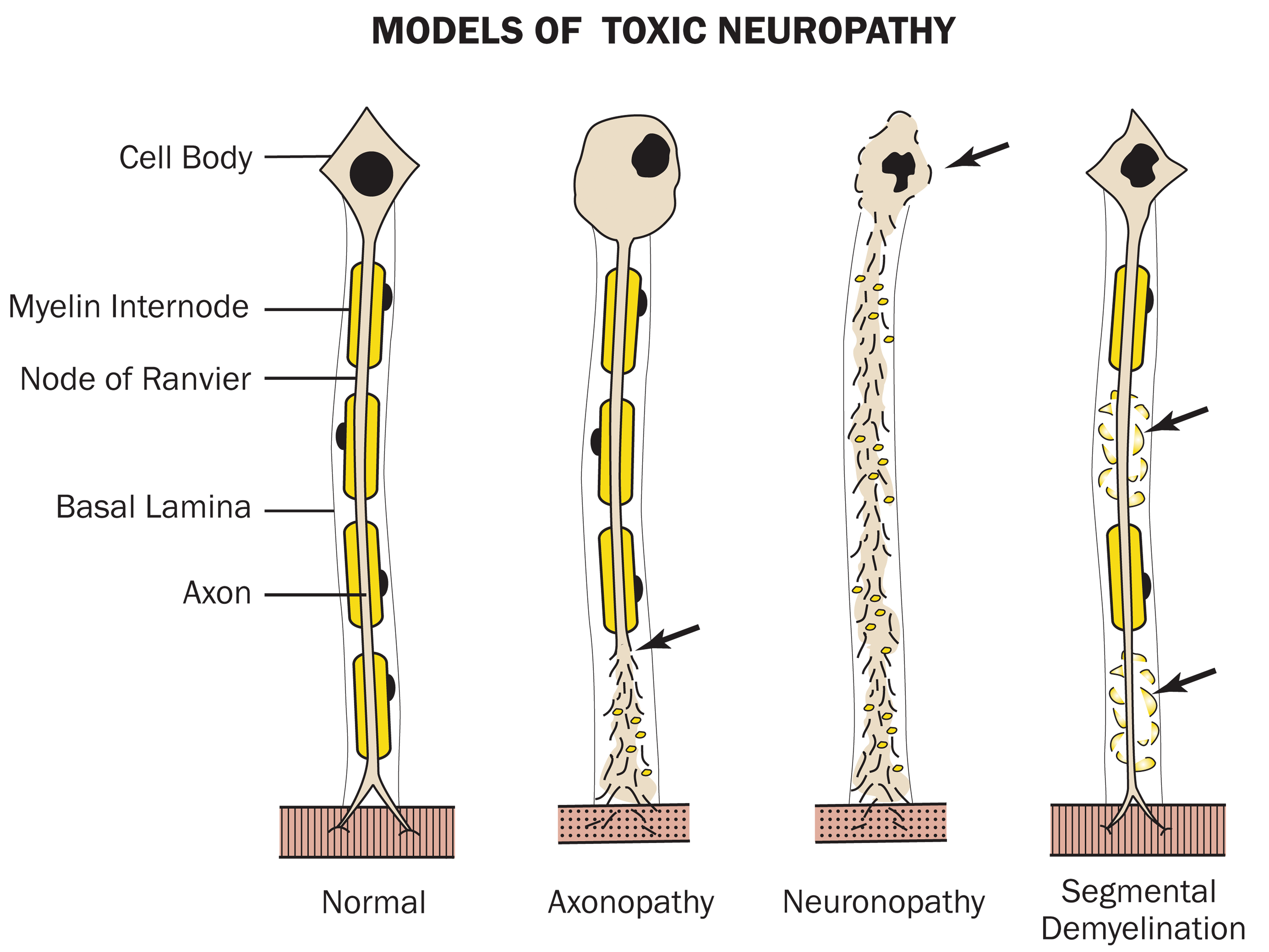
Compared to the normal condition (left), injury (shown by arrows) leads to different patterns of toxicant-induced injury to myelinated peripheral nerve fibers depending on the target. Primary damage to the axon (axonopathy) leads to loss of the distal axon and secondary disintegration of its myelin sheath sometimes with swelling and chromatolysis of the neuronal cell body (i.e., the “axonal reaction”) to support axon regrowth. Primary injury to the cell body (neuronopathy) can result in destruction of the neuron cell body and irreparable secondary loss of its axon and associated degeneration of the entire myelin sheath. Primary damage to the myelin sheath or Schwann cell leads to segmental demyelination in association with retained axons early (as shown), although in later stage the axon also may undergo secondary degeneration due to the loss of trophic factors produced by Schwann cells. Rectangular structures at end of axons are representative of portions of innervated skeletal muscle fibers. Reproduced from Pardo et al. (2018), by permission of SAGE Publications, Inc.
The classic example for neuronopathy (Figure 5) is toxic exposure to vitamin B6 (pyridoxine). Megadoses of this vitamin can injure PNS neurons such as dorsal root (spinal) ganglia by some as yet unknown mechanism (Levine and Saltzman 2004). Toxic exposure of this population of neurons is related to the diminished blood-nerve-barrier in ganglia, in particular in their cell body-rich central region, making it accessible to a variety of low and high molecular weight toxic agents (Jimenez-Andrade et al. 2008). This includes the markedly elevated blood levels of pyridoxine. Such toxic neuronal exposure leads to sensory neuropathy. This has been seen with daily oral exposures of 7 to 10 g for two months in man (Schaumburg 2000) or 100 to 300 mg/kg for 12 weeks or 600 to 1,200 mg/kg intraperitoneally for 6 to 10 days in rats (Xu et al. 1989). As noted above, the primary insult in such toxic exposures is to the neuronal cell body (cyton) of ganglionic neurons (Krinke and Fitzgerald 1988; Windebank et al. 1985), and its severity depends on dosage and rate of exposure (Figures 6 and 7). In addition to the primary cell body effect, there is associated failure of these affected cells to maintain their axons, leading to breakdown of the myelinated nerve fibers in the PNS. Larger neurons are more susceptible. The sensory nature of the neuropathy can best be appreciated in the spinal nerve roots, where the dorsal (sensory) one is affected and the ventral (motor) one is spared (Figure 8). In addition, more distal levels of peripheral nerve are affected. The features of the peripheral lesion include axonal degeneration progressing to fiber breakdown, eventually being represented by myelin ovoids within Schwann cells (Figure 9). Axonal atrophy may occasionally be noted, possibly a reflection of sublethal injury to the dorsal root ganglionic neurons with resulting impaired export of neurofilaments from the cyton to the axon (Krinke and Fitzgerald 1988). This entity is an example of a neuropathy involving both peripheral and central regions of the nervous system, since degenerating axons originating from dorsal root ganglion neurons extend for a distance within the spinal cord dorsal funiculus in the gracile and cuneate fascicles. Recovery following toxicant exposure relates to the degree of neuronal injury. If sublethal, the cell body may recover and thus support peripheral nerve axonal regeneration. If neuronal necrosis occurs, clearly such regeneration will not be seen (Figure 10).
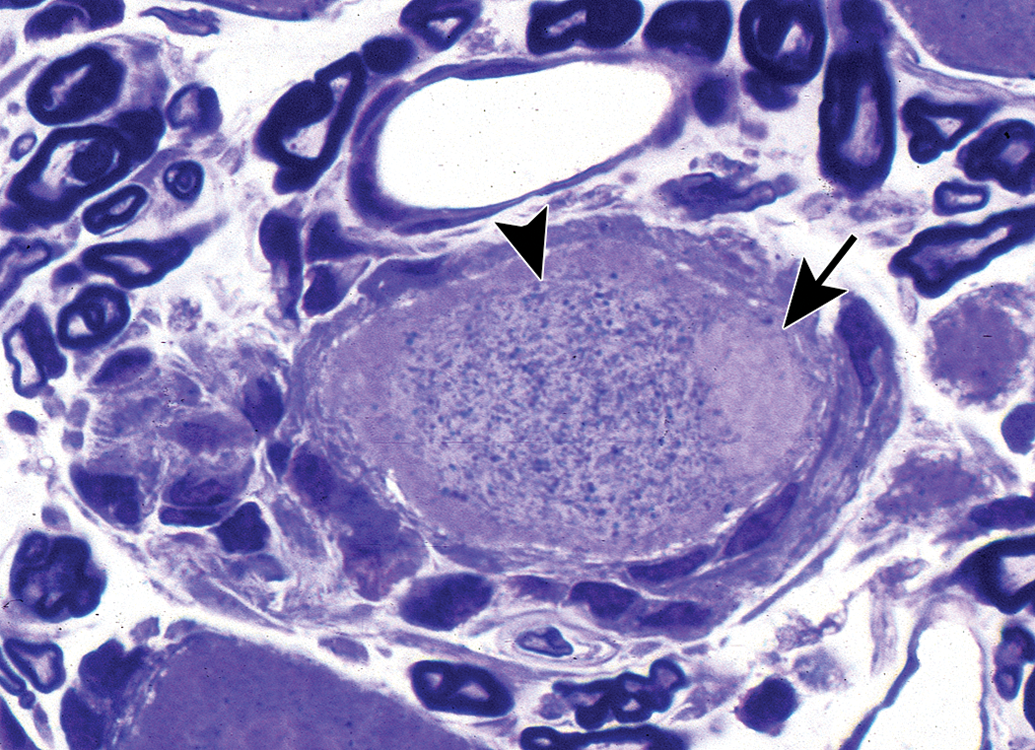
Neuronopathy presents initially as an injured (degenerating) dorsal root ganglion neuron from a rat exposed to 600 mg/kg of pyridoxine twice a day for four days, with a subsequent two-day recovery period. There is a pale-stained eccentric nucleus (arrow) and cytoplasmic masses of blue-stained granular material (arrowhead), which likely consist of autophagosomes, dense bodies, and altered mitochondria. Toluidine blue and safranin stain (from Jortner, Toxicologic Pathology 2011, used with permission of SAGE Publications, Inc.).
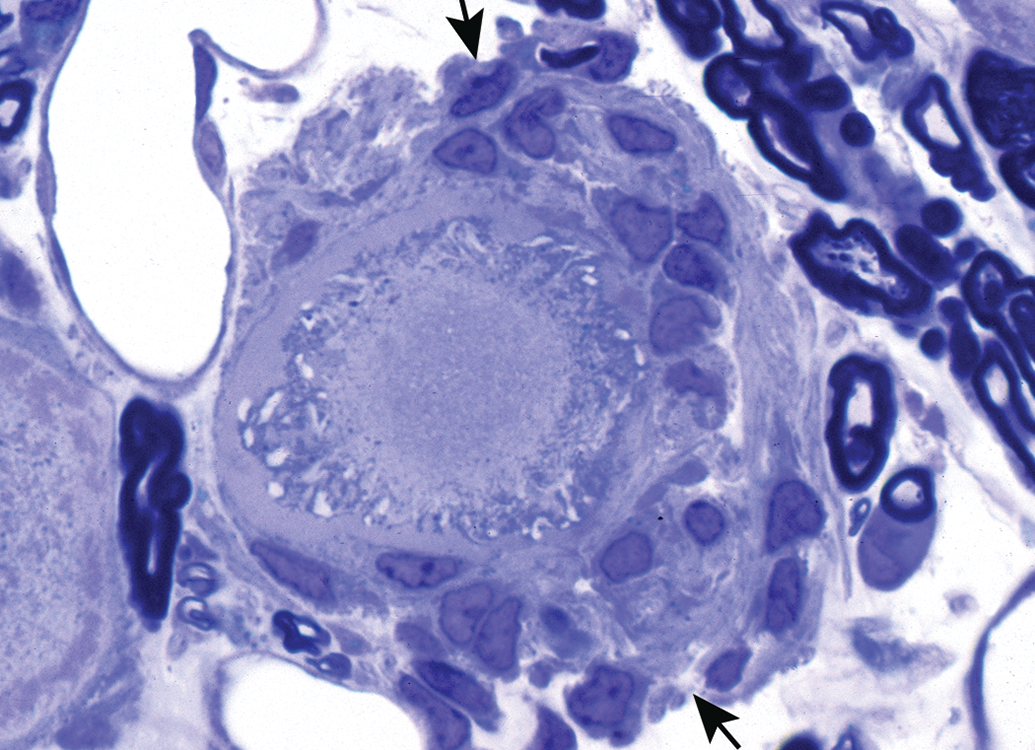
Neuronopathy that has produced irreversible injury leads to neuronal necrosis, as seen here in a dorsal root ganglion from the animal illustrated in Figure 6. The neuronal cell body is being phagocytized by numerous reactive satellite cells (arrows). Toluidine blue and safranin stain (from Jortner, Toxicologic Pathology 2011, used with permission of SAGE Publications, Inc.).
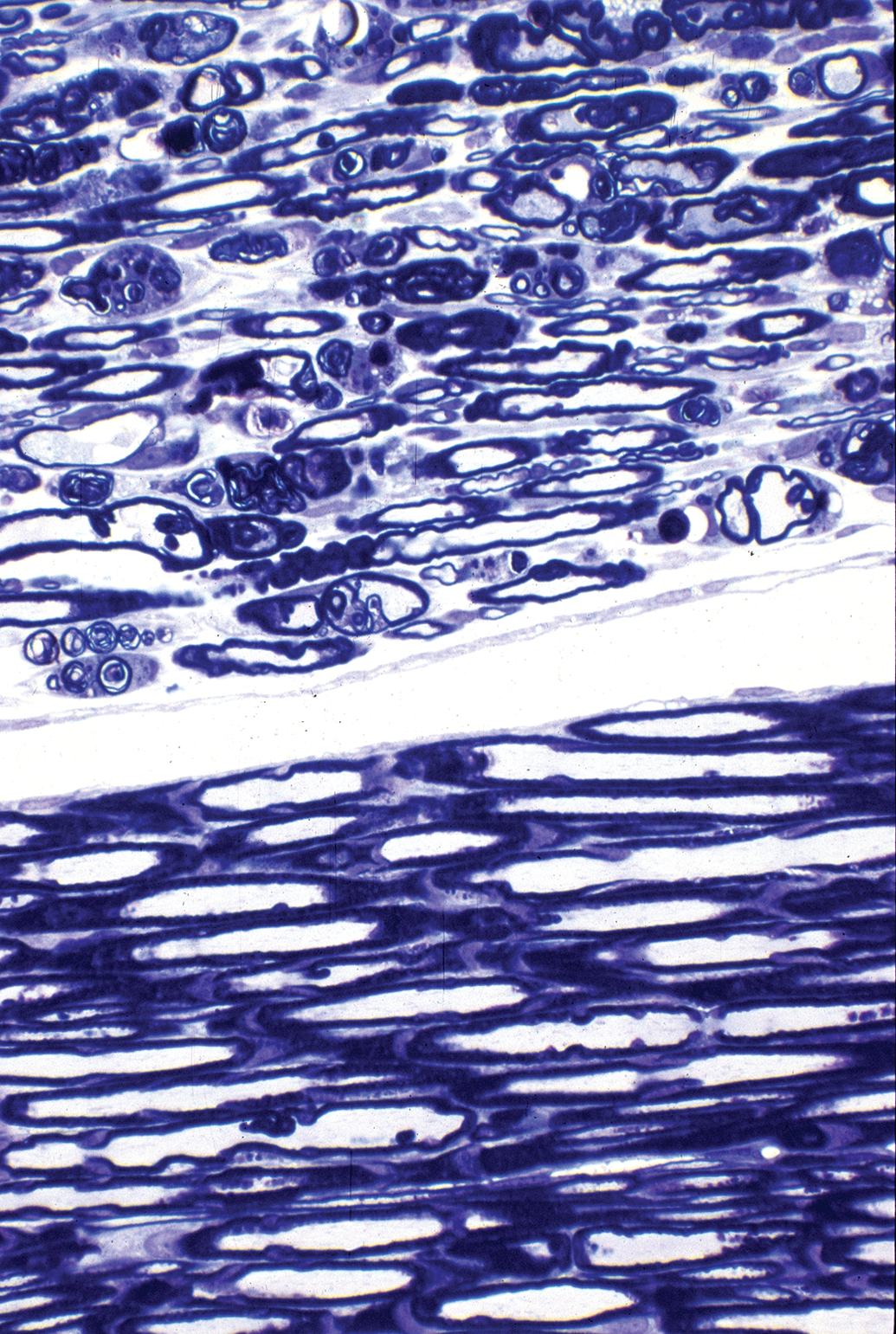
Secondary nerve fiber degeneration related to primary neuronopathy is seen in the spinal nerve roots from the pyridoxine-treated rat. Injury to the dorsal root ganglion neurons in such an animal is similar to those shown in Figures 6 and 7. There is marked disruption of myelinated nerve fibers in the dorsal root (upper part of image) compared to the ventral root (lower part of image), which is spared. This demonstrates the effect of toxicant-related injury to dorsal root neurons with sparing of neurons of spinal cord ventral horns. Toluidine blue and safranin stain (from Jortner, Toxicologic Pathology 2011, used with permission of SAGE Publications, Inc.).
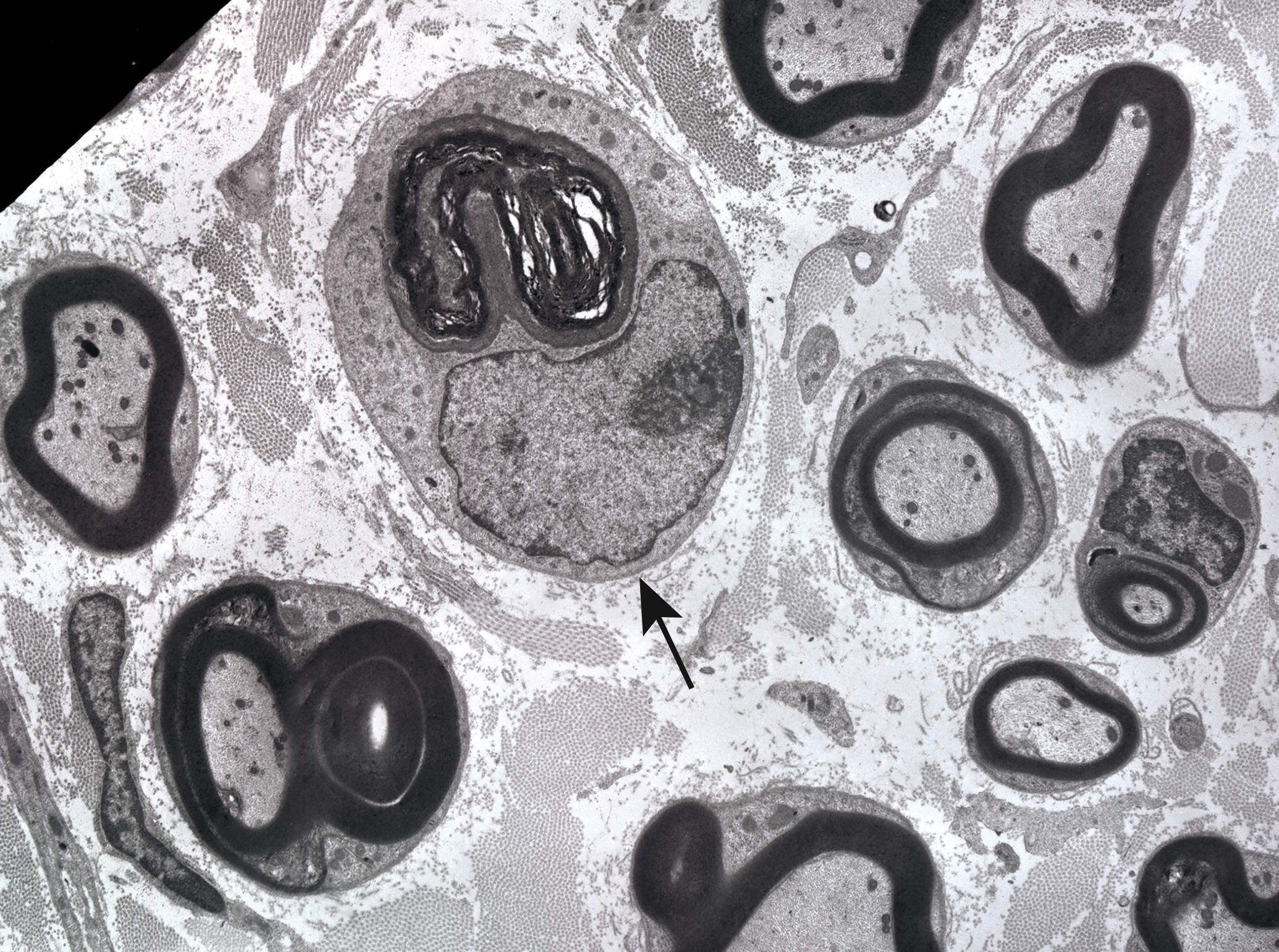
Advanced degeneration of a myelinated nerve fiber with myelin-rich debris (horseshoe-shaped, lamellar material inside a membrane-bound cytoplasmic vacuole) in a phagocytic cell (arrow), likely a Schwann cell. In a rat exposed to 600 mg/kg pyridoxine twice a day for four days, with a two-day recovery period, to induce a sensory neuronopathy. Whisker follicle sensory mystacial nerve in vibrissal whisker capsule (sometimes referred to as vibrissal nerve), a branch of infraorbital nerve arising from trigeminal ganglion). Osmium postfixation, lead citrate, and uranyl acetate staining.
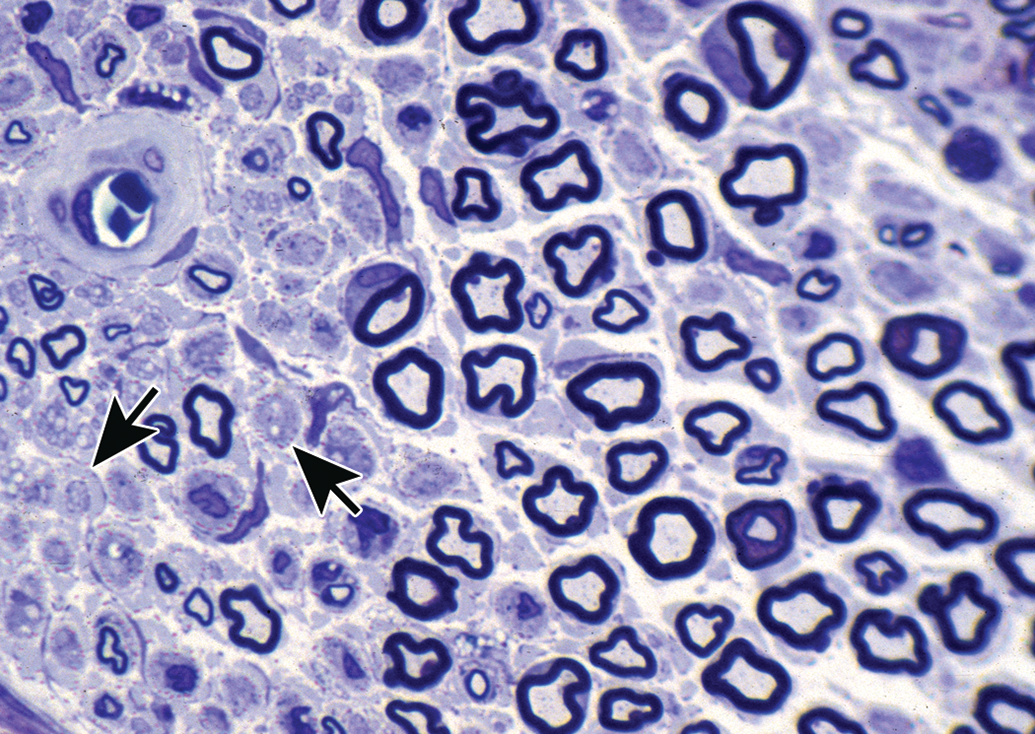
Cross section of sural nerve from a pyridoxine-exposed rat (600 mg/kg pyridoxine twice a day for four days), with a 40-day recovery period. Neuronopathy is reflected here as multiple denervated bands of Büngner (arrows), which are devoid of axons and therefore indicative of failed axonal regeneration due to necrosis of dorsal root ganglion neurons. Toluidine blue and safranin stain (from Jortner, Toxicologic Pathology 2011, used with permission of SAGE Publications, Inc.).
An example of toxic axonopathy is the classic entity of organophosphorus ester–induced delayed neurotoxicity (OPIDN). The modifier “delayed” indicates that clinical signs of neuropathy are not manifest until one week or more postexposure. This neuropathy has been known for over 100 years and has been the cause of a number of outbreaks of human disease. The lesions of OPIDN consist of axonal degeneration, most prominent in distal regions of the peripheral nerve and certain tracts of the CNS. A number of organophosphorus compounds elicit this lesion. These include pro-toxicants (needing prior activation by liver microsomes to produce a toxic metabolite) such as leptophos and tri-ortho-tolyl phosphate (TOTP) and direct toxicants such as diisopropyl phosphorofluoridate, mipafox, and tolyl saligenin phosphate. Chemical features of these neurotoxicants include phosphorus in the pentavalent state, phosphorus with an oxygen attached by a coordinate covalent bond, and at least one oxygen or amine bridge linking an R group to phosphorus (Ehrich and Jortner 2010).
These neurotoxicants inhibit esterases by phosphorylation of their active site. A number of molecular events occur during the development of OPIDN, although the definitive neuropathic temporal sequence leading to axonopathy has not been identified (Ehrich and Jortner). An important feature is the toxicant-associated inhibition of the nervous system enzyme neurotoxic esterase (NTE). As a precursor to the neuropathy, the toxicant must strongly bind to and inhibit NTE to a significant degree and produce a molecular rearrangement resulting in a negative charge (which has been termed “aging”) on the enzyme. The mechanistic effect of such inhibition/aging of NTE on the subsequent neuropathy is unknown. It is thought to be more of a biomarker of impending OPIDN rather than as a specific initiator of the neuropathy (Ehrich and Jortner). However, the association of NTE inhibition and developing OPIDN is strong enough that regulatory agencies require assessment of enzymatic inhibition in safety testing of candidate organophosphorus compounds. These compounds are usually tested in hens (domestic chicken), as this species is particularly sensitive. Using that standard experimental model, a single neurotoxic dose in the domestic chicken, there is NTE inhibition/aging of 70% or more 1 to 2 days postdosing, with the appearance of axonal lesions within 2 to 3 weeks (Ehrich and Jortner 2010).
The lesions of OPIDN in the single neurotoxic dose chicken model mentioned above consist of bilateral axonal degeneration of distal regions of larger diameter, longer myelinated fibers in peripheral nerves and selected CNS tracts. The primary lesion manifests as axonal swelling, with either pallor or increased intensity of staining, progressing to axonal collapse with associated secondary breakdown of the myelin sheath resulting in development of myelin ovoids (Figures 11 –13). Given the qualitative resemblance of affected fibers to that seen in Wallerian degeneration (which is a fiber lesion induced distal to the site of crushing or cutting a nerve), this toxic process has been termed “Wallerian-type” degeneration. Lesions of OPIDN are more severe in more distal fiber regions. With appropriate dosing schemes, a progressive spread toward the cell body (“dying-back”) may be seen. At the ultrastructural level, affected axons exhibit axonal swelling as well as granular degeneration of the cytoskeleton, disorganized structures composed of normal and altered mitochondria, cytoskeletal components, lysosome-like dense bodies, or membranous components (Figure 13). These changes are thought to be related to toxicant-associated excess in intra-axonal calcium stores leading to activated proteases and/or hyperphosphorylation of axonal cytoskeletal elements mediated by calcium–calmodulin-dependent protein kinase (El-Fawal et al. 1989; Lapadula et al. 1992).
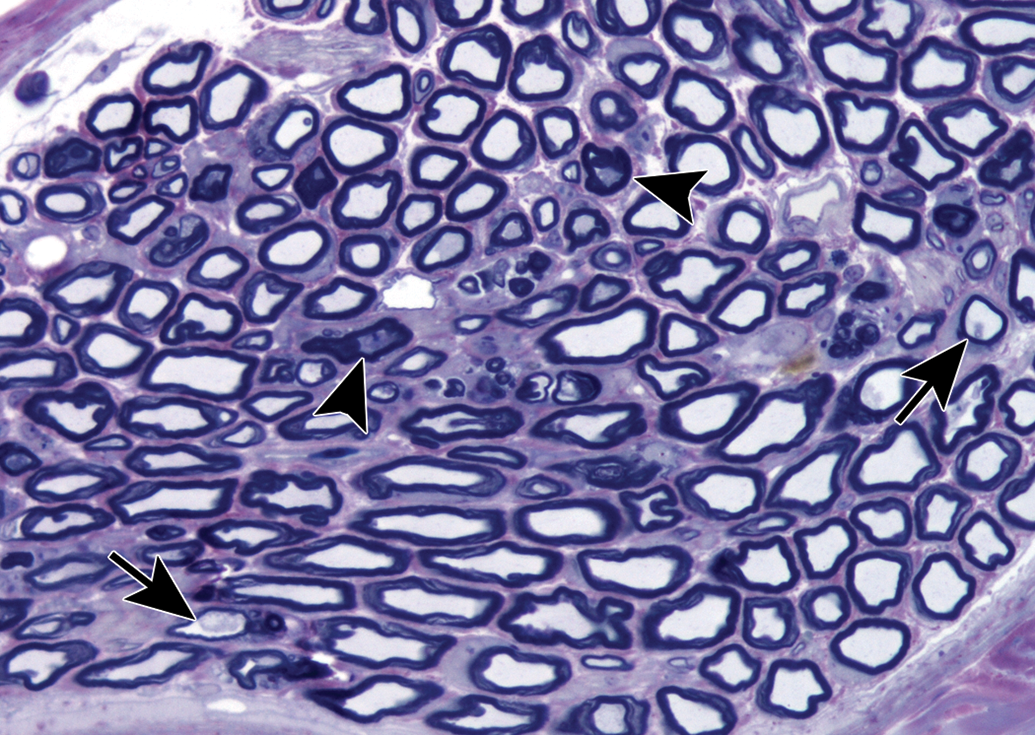
Axonopathy of organophosphorus ester–induced delayed neurotoxicity is apparent in a branch of the tibial nerve that supplies the lateral head of the gastrocnemius muscle from a hen dosed with 30 mg/kg of Mipafox 21 days earlier. There are both dark-stained collapsing axons (arrowheads) as well as swollen ones (arrows). Toluidine blue and safranin stain.
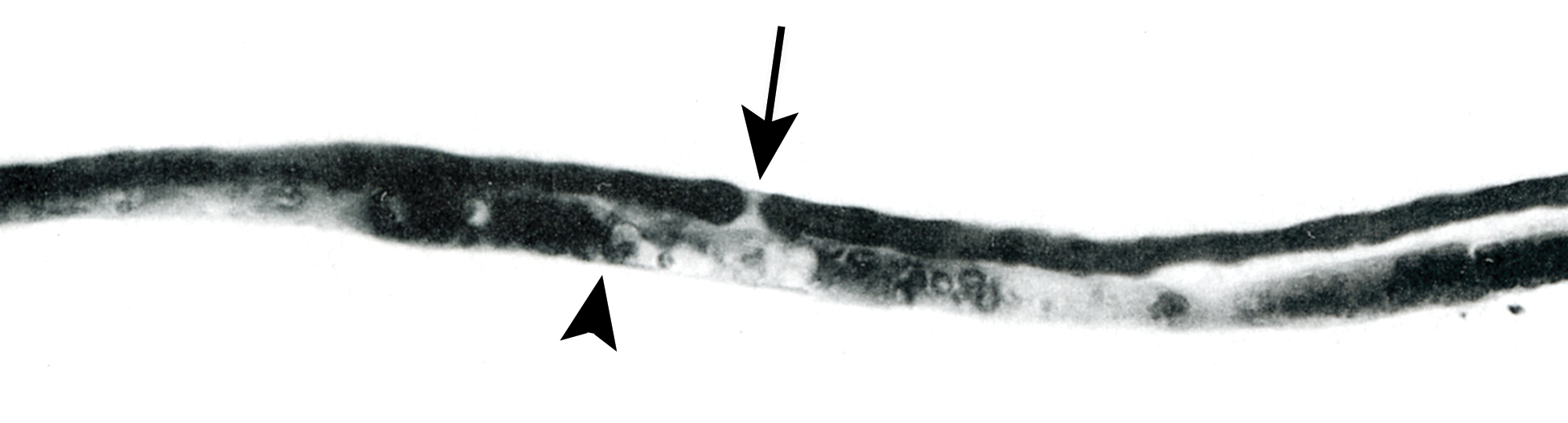
Teased nerve fiber preparation demonstrating axonal degeneration in a tibial nerve branch to the gastrocnemius muscle of a hen with organophosphorus ester–induced delayed neurotoxicity 14 days after exposure to phenyl saligenin phosphate. One nerve fiber is undergoing Wallerian-type degeneration (arrowhead), while the adjacent fiber is intact as indicated by as indicated by a uniformly dark myelin sheath and a distinct node of Ranvier (arrow). Osmium tetroxide stain (from Jortner, Toxicologic Pathology 2011, used with permission of SAGE Publications, Inc.).
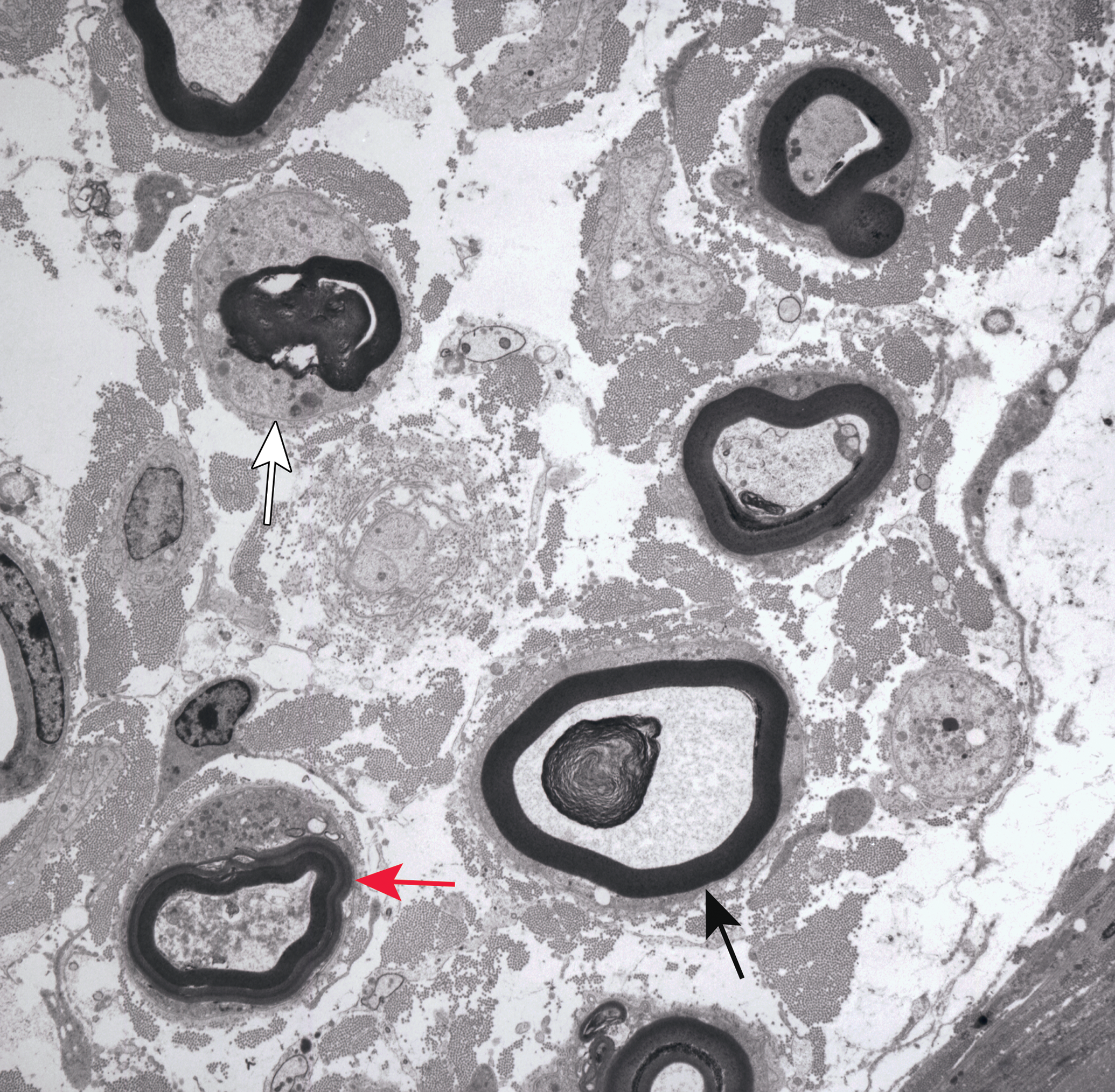
Electron micrograph demonstrating axonal degeneration associated with organophosphorus ester–induced delayed neurotoxocity of the dorsal metatarsal nerve in a hen 14 days after dosing with 1 mg/kg diisopropyl phosphorofluoridate. The cross-sectioned nerve shows several stages of myelinated fiber axonal degeneration including axonal swelling with granular degeneration of the cytoskeleton and a large membranous body (black arrow), collapsing axon with osmiophilic debris (red arrow), and a fully degenerated fiber with myelin ovoid inside a Schwann cell (white arrow). The increased separation of the neuritic elements indicates endoneurial edema. Osmium tetroxide postfixation, lead citrate, and uranyl acetate staining.
The phagocytized elements of the injured peripheral nerve fibers are degraded in Schwann cells or macrophages, forming what are sometimes referred to as “digestion chambers.” This clearance is more rapid in the PNS than in the CNS and is largely accomplished within four weeks following a single neurotoxic dose of TOTP in the domestic chicken (Jortner et al. 1989). With advanced breakdown of the fiber debris comes proliferation of Schwann cells within the intact basal lamina tube, forming a solid mass of cells termed the “band of Büngner.” This column of proliferating Schwann cells extends from the intact proximal part of a nerve fiber and serves as a guide through which regenerating axon can sprout (King 1999). Such regenerating PNS axons can extend distally into the band of Büngner and will myelinate over time (Figure 14). This process is robust in the avian single toxic dose model of OPIDN, with significant reconstitution of the peripheral nerve by seven weeks postdosing (Jortner et al. 1989). As is typical in other forms of axonal degeneration, such repair does not occur in the CNS.
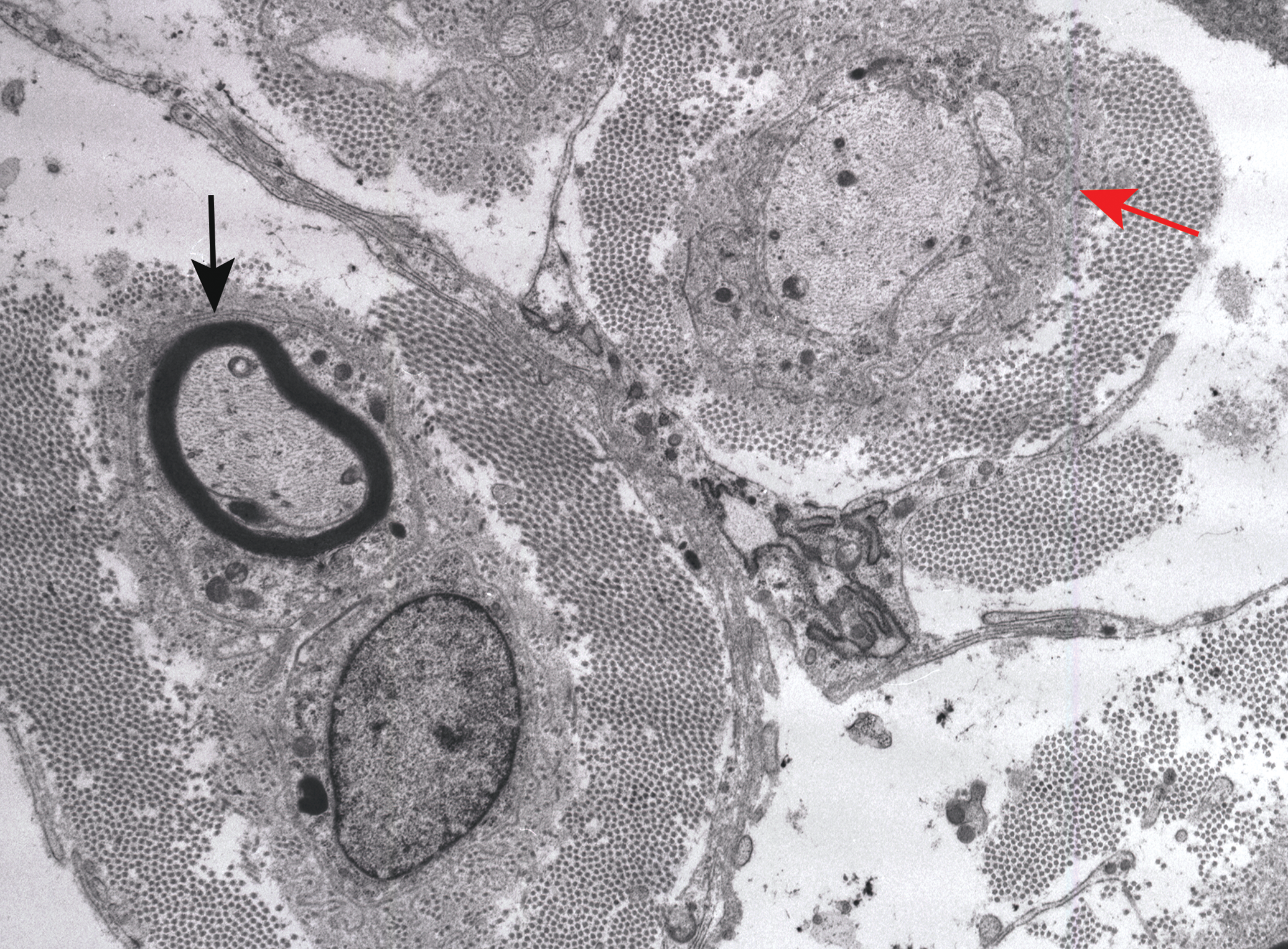
Electron micrograph showing early axonal regeneration associated with organophosphorus ester–induced delayed neurotoxocity in cross-sectioned nerve to the biventer muscle of a chicken 28 days after exposure to 360 mg/kg of tri-ortho-tolyl phosphate. There is a thinly myelinated regenerating nerve fiber in a band of Büngner (black arrow). An adjacent band of Büngner contains several small, oval regenerating axonal sprouts (red arrow). Endoneurial collagen is prominent. Osmium tetroxide postfixation, lead citrate, and uranyl acetate staining.
The last type of neuropathy to be considered is myelinopathy, which presents as demyelination. The example described here is the dietary riboflavin deficiency model in rapidly growing chickens (Jortner et al. 1987), and though this is not a toxic entity such as diphtheria, lead poisoning, or tellurium toxicity, it is still a useful model for demonstrating the key features of myelin-focused injury along with its repair. Riboflavin is an important component of flavin adenine dinucleotide and flavin mononucleotide, which are part of pathways involved in carbohydrate and lipid metabolism as well as energy production via the respiratory chain. Because of the complexity of myelin biosynthesis, compaction, and maintenance, riboflavin deficiency-related alterations that affect Schwann cell energy supply or lipid metabolism may lead to demyelination in rapidly growing nerves (Jortner et al. 1987). For instance, Ross-Arbor Acres cross meat-type chickens are genetically programmed for a 16-fold increase in body weight by 23 days of age, although growth is somewhat slower in riboflavin deficient chicks (Jortner et al. 1987). Since chickens are precocial animals, peripheral nerve (and other) myelin is well-developed at hatching. However, dietary riboflavin deficiency from day 1 of age in such rapidly growing broiler chicks leads to lower tissue levels of the vitamin resulting in injury to Schwann cells manifested by hypertrophy, regions of cytoplasmic lysis, and the presence of lipid droplets (Figure 15). These lesions are indicators of reduced Schwann cell functions with a resulting failure to meet the demands required for growth-related enlargement of myelin internodes and to maintain already formed myelin. This presents morphologically as projections of and/or sequestration of myelin fragments in Schwann cell cytoplasm with subsequent segmental demyelination (Figures 15 and 16). Additional lesions that can be seen include endoneurial edema, which can be marked (Figure 15), and small numbers of degenerated axons. In the riboflavin deficiency chicken model, demyelinating lesions are seen after 10 days on the deficient diet and were marked by day 21 (Jortner et al. 1987). From 14 days onward, small numbers of remyelinating internodes may be seen, a process that is enhanced if supplemental riboflavin is supplied (Figures 16 and 17).
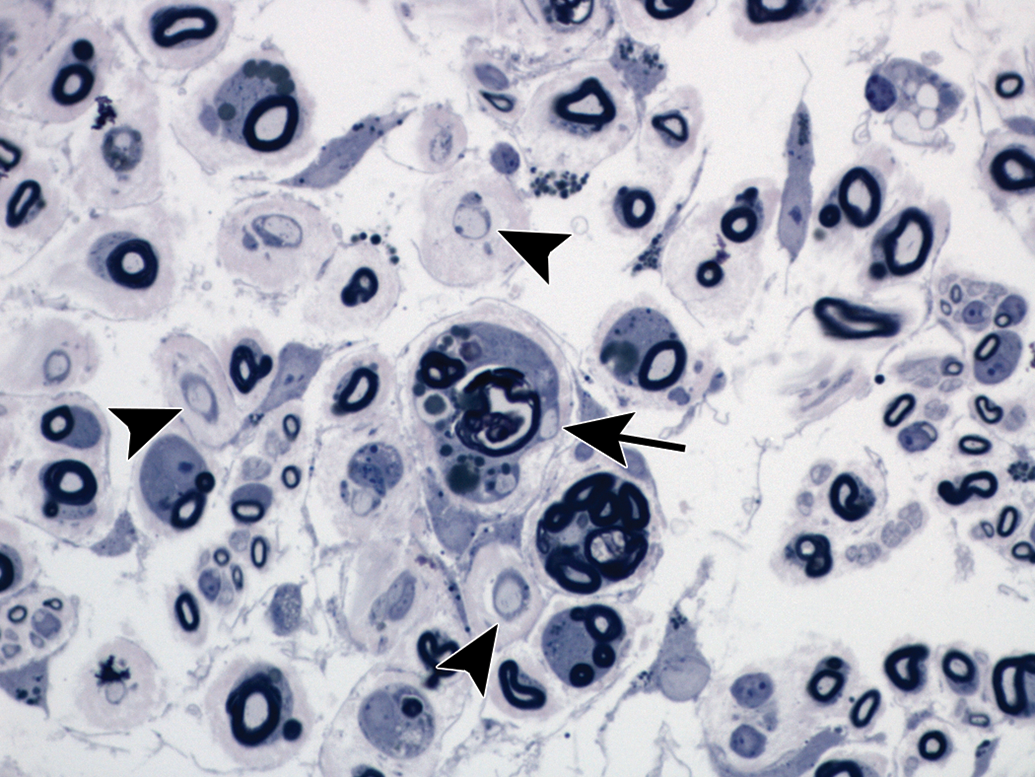
Primary myelinopathy leading to demyelination of the sciatic nerve from a 14-day-old riboflavin-deficient chick. Schwann cells are enlarged (hypertrophied). Features of demyelination include intracytoplasmic fragments of blue-stained degraded myelin and associated demyelinated axon profiles within Schwann cells (arrow). Axon profiles showing more advanced stages of demyelination also are present (arrowheads). Separation of nerve fibers indicates endoneurial edema. Toluidine blue and safranin stain.
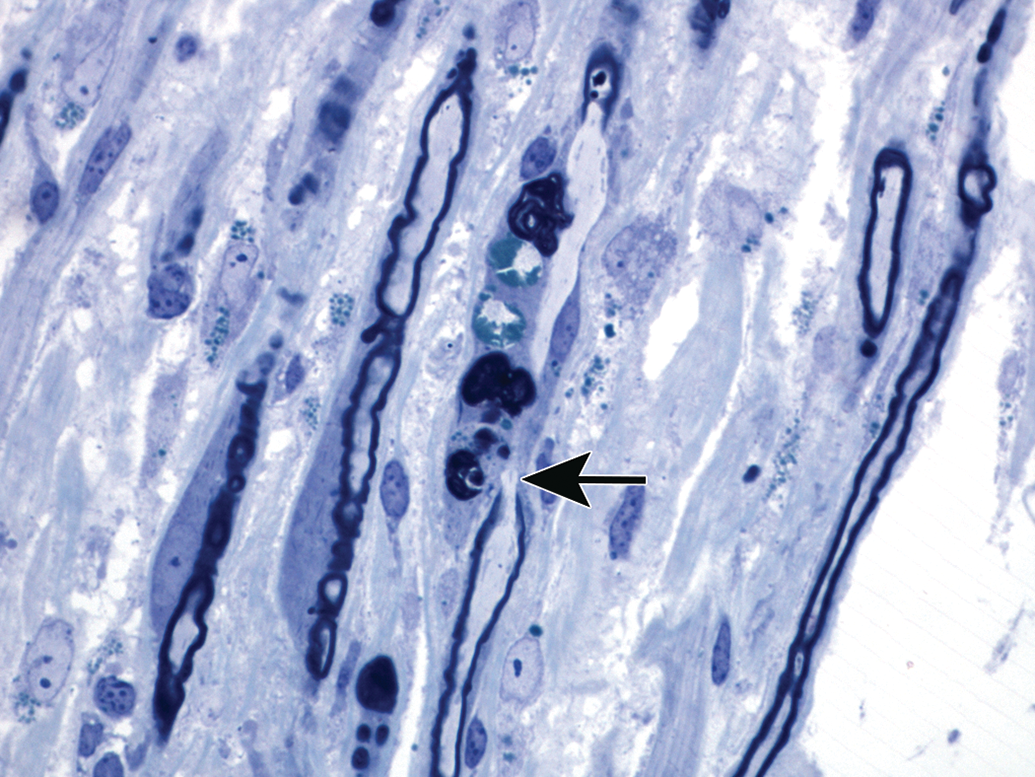
Primary segmental myelinopathy with associated demyelination as seen in a longitudinal section of sciatic nerve in a riboflavin-deficient chick. The black arrow indicates a node of Ranvier. The internode located above the node is demyelinated as indicated by the absence of a dark myelin sheath and the presence of myelin debris (dark-stained, irregular masses) segregated within the Schwann cell. The internode below the node is thinly myelinated, which likely is an indication of remyelination. Toluidine blue and safranin stain.
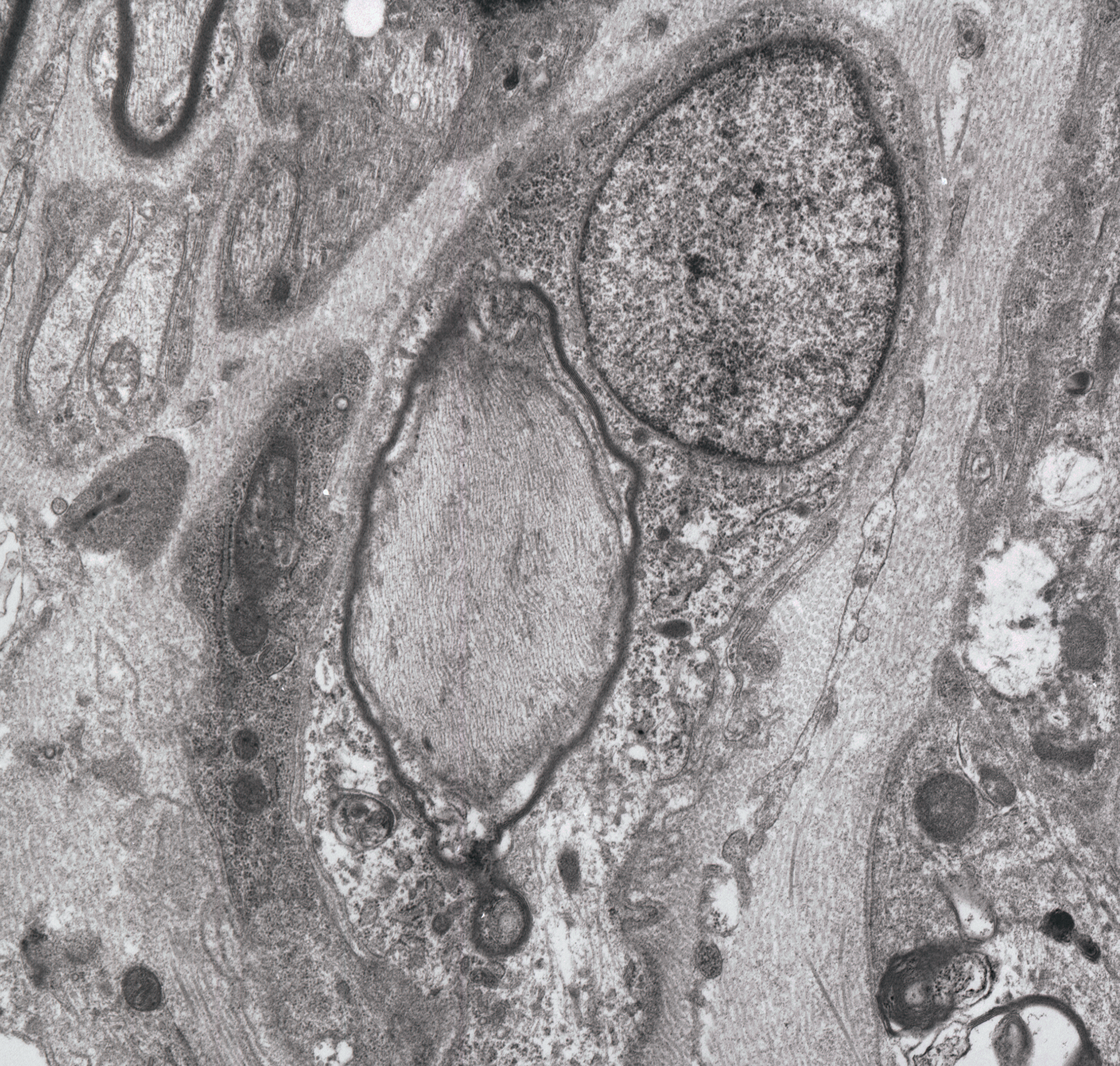
Remyelination occurring in primary myelinopathy of a chicken on a riboflavin-deficient diet for 18 days followed by one day of supplemental riboflavin. Early restoration of myelin is indicated by a thin myelin sheath (dark line that surrounds the pale axon in the center of the image); the nucleus of the Schwann cell generating the new myelin is located just above and to the right of the affected axon. This remyelination process likely began prior to the riboflavin supplementation. Osmium tetroxide postfixation, lead citrate, and uranyl acetate staining.
Summary
This overview of toxic injury to the PNS began with a brief summary of the elements and organization of the PNS, followed by examples of experimental models of specific types of such injury to this system. As elsewhere, myelinated fibers in peripheral nerve consist of axons with supporting Schwann cells which produce successive segments of myelin. There are several responses by which such fibers react to injurious agents such as toxicants. One model is seen when the toxicity primarily affects the neuronal cell body giving rise to the axon (neuronopathy). Since the axon depends on the cell body for maintenance, injury to the neuronal cell body can also result in axonal degeneration extending over the entire length of the fiber. The neuronopathy example described in this review was pyridoxine (vitamin B6) toxicity, which as noted above primarily involves neurons of peripheral ganglia, such as dorsal root and trigeminal ganglia. A second pattern of PNS injury described in this review was primary axonal injury (axonopathy), which initially develops in distal axonal regions, although a “dying-back” pattern subsequently involving more proximal regions of axon can also occur with appropriate dosing schemes. Since the neuron supporting the injured axon generally survives, postintoxication regeneration is possible. The axonopathy example described in this article was organophosphorus ester–induced delayed neuropathy. The last form of peripheral neuropathy described in this review was injury to the myelin sheath and/or supporting Schwann cell was (myelinopathy) with the riboflavin deficiency model in rapidly growing meat-type young chickens provided an example. With this type of injury, remyelination can occur if the axon survives.
Footnotes
Author’s Note
Much of the material included in this review was derived from work conducted in the Virginia Tech Laboratory for Neurotoxicity Studies.
Acknowledgments
I would like to recognize the contributions of colleagues Sandra Hancock and Marion Ehrich to these studies. I appreciate Dr. Brad Bolon critically reviewing the article, but any errors are my responsibility.
Author Contribution
All authors (BJ) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The studies on organophosphorus ester-induced delayed neurotoxicity were supported in part by USEPA 68D80098.