
Abstract
Nonacog beta pegol is a 40-kDa polyethylene glycosylated (PEGylated) human recombinant coagulation factor IX, intended for the treatment of hemophilia B. Human coagulation factors are immunogenic in animals; therefore, to evaluate the long-term toxicity of nonacog beta pegol, an immune-deficient, athymic rat (Rowett nude; Crl:NIH-Foxn1rnu) was used. Rats (n = 216) were given intravenous nonacog beta pegol 0, 40, 150, 600, or 1,200 IU/kg every 5th day for 26 weeks. To avoid infections, the animals were housed in a full-barrier environment with sterilized food and bedding. Standard toxicity end points were unaffected by treatment. All treated animals were exposed to nonacog beta pegol throughout the study, and no animals developed antidrug antibodies. Immunohistochemical staining revealed PEG in choroid plexus epithelial cells in a dose-dependent manner. Transmission electron microscopy showed that PEG was distributed in cytoplasmic vesicles of these cells, with no apparent effect on cellular organelle structures. Fourteen (6.5%) animals were euthanized or died prematurely due to nontreatment-related infections in the urogenital system and skin. In conclusion, the athymic rat is a suitable model for testing chronic toxicity of human proteins that are immunogenic in animals. Nonacog beta pegol was generally well tolerated, with no adverse effect of PEG on choroid plexus epithelial cells.
Introduction
Nonacog beta pegol (also referred to as N9-GP) is a 40-kDa glycoPEGylated human recombinant coagulation factor IX (rFIX), intended for the treatment of hemophilia B. The conjugation of a 40-kDa branched polyethylene glycol (PEG) molecule to rFIX increases the half-life and enables once weekly prophylactic dosing instead of twice weekly dosing with unmodified rFIX (Collins et al. 2014). Human coagulation factors are immunogenic in animals, precluding the possibility to conduct chronic toxicity studies (European Medicines Agency 2004b, 2005; Ivens and Arora 2013; Kirschbaum and U.S. Food and Drug Administration 2014). In line with this, the conducted toxicity studies with nonacog beta pegol have been of limited duration, not exceeding 4 weeks, due to the development of antibodies. According to The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guideline for duration of chronic toxicity testing in animals (2009, 2011), the nonclinical safety evaluation of new drugs should include a repeat dose toxicity study of 6 months’ duration in rodents.
In order to overcome the limitation of study duration related to antidrug-antibody development, immune-deficient rat and mouse models were considered in order to enable long-term dosing of nonacog beta pegol, and the Rowett nude rat (Crl: NIH-Foxn1rnu ) was selected. Immune-deficient mice have been used for many years in different areas, especially in cancer research, to gain knowledge about the immune system and in research involving graft tissue (Jin et al. 2010; Shultz, Ishikawa, and Greiner 2007). However, in contrast to rats, mice have been reported to be very sensitive to human rFIX resulting in widespread thrombosis (U.S. Food and Drug Administration 1997) and were therefore not considered a suitable animal model for nonacog beta pegol. The Rowett nude rat is an athymic rat that does not develop mature functional T cells and thereby lacks the ability to elicit a T cell–dependent immune response toward species foreign proteins (Hanes 2006). Due to the vulnerability to infections, the life span under normal conditions is only about 4 months, but when housed in a pathogen-free environment the life span can be up to 2 years (Schuurman 1995).
The purpose of this study was to evaluate the long-term toxicity of nonacog beta pegol in the Rowett nude rat, that is, an animal model that did not develop neutralizing antidrug-antibodies. To assess the feasibility of the model, a preliminary 6-week study was performed. Since no antidrug-antibodies developed and all rats were exposed, the model was found feasible. Thereafter, a 26-week toxicity study in the Rowett nude rat was conducted and included a range of standard toxicological end points (clinical observations, blood sample analysis [clinical chemistry, hematology, and coagulation], toxicokinetic, antibody analysis, macroscopic, and microscopic evaluation of selected tissues). The study included a 26-week treatment-free period to assess recovery from potential findings. In addition, an immunohistochemical (IHC)-staining method specific for PEG was included to assess the microscopic localization of PEG in selected tissues, that is, brain/choroid plexus and spleen. The choroid plexus was selected as primary tissue for this investigation based on the literature indicating the choroid plexus epithelial cells are a potential target organ for PEG distribution (Center for Drug Evaluation and Research 2012; European Medicines Agency 2009, 2012; Ivens et al. 2015; Rudmann et al. 2013). Spleen was selected as positive IHC control tissue, as this tissue was expected to contain PEG-positive macrophages. Furthermore, transmission electron microscopy (TEM) was included to evaluate the ultrastructural distribution of PEG in the choroid plexus cells and any potential effects on cellular organelles.
Materials and Methods
Animals and Experimental Design
A preliminary 6-week study (twice weekly dosing) followed by a 26-week toxicity study (every 5th day dosing) was conducted in Rowett nude rats (Crl:NIH-Foxn1rnu
), in life from June 2012 to August 2014. The studies were conducted at test facilities of Envigo, Huntingdon, United Kingdom. Formulation analysis, pharmacokinetic (PK) evaluation and analysis, antidrug-antibody analysis, IHC, and TEM were performed at Novo Nordisk A/S, Maaloev, Denmark. The 6-week study was conducted to assess tolerability and immunogenicity of nonacog beta pegol in the Rowett nude rat and verify the feasibility of the model for longer-term dosing. Two groups, each comprising 13 male and 13 female rats, received nonacog beta pegol at twice weekly doses of 40 or 1,200 IU/kg (in the 1st week, only 1 dose was given to allow for full PK sampling and profiling for up to 7 days postdose). A control group, comprising 5 males and 5 females, received the vehicle (sodium chloride 2.34 mg/ml,
The 26-week study design and doses are shown in Table 1. Animals were dosed every 5th day for 26 weeks (dosing phase) followed by a 26-week treatment-free period (recovery phase, control, and high dose only). The dose levels were selected to cover a range including a control group (0 IU/kg) and the typical clinical dose; 40 IU/kg and multiples thereof, that is, 150 (×3.8), 600 (×15), and 1,200 (×30) IU/kg.
Design and Doses of 26-Week Study.
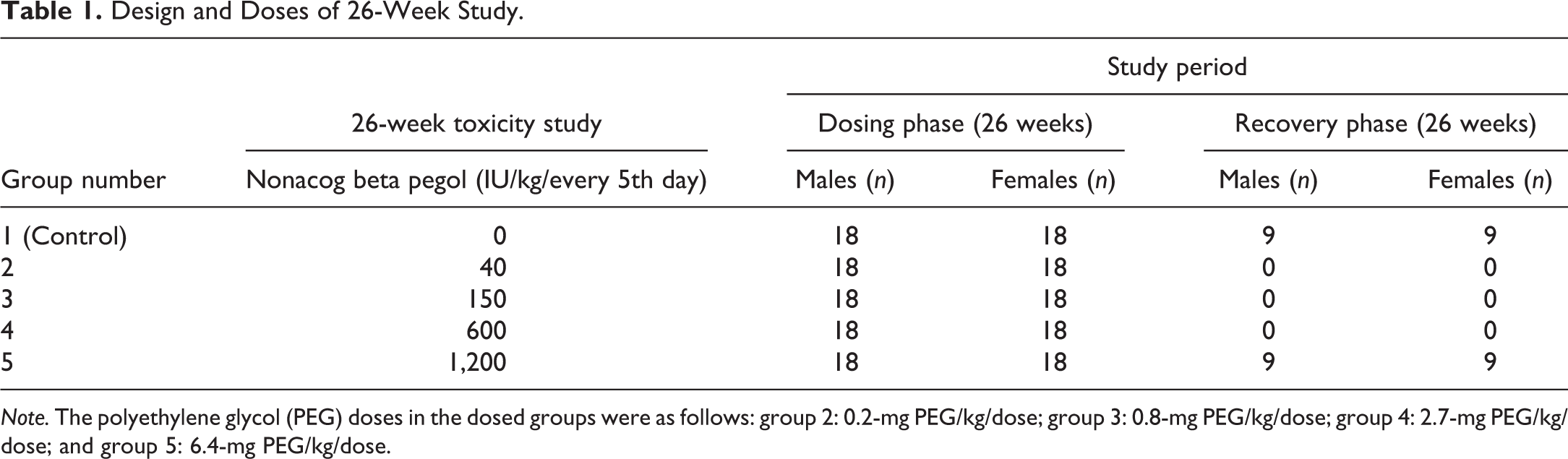
Note. The polyethylene glycol (PEG) doses in the dosed groups were as follows: group 2: 0.2-mg PEG/kg/dose; group 3: 0.8-mg PEG/kg/dose; group 4: 2.7-mg PEG/kg/dose; and group 5: 6.4-mg PEG/kg/dose.
In both studies, the in-life experimental procedures were in accordance with provisions of the United Kingdom Animals (Scientific Procedures) Act 1986 (the Act) under an appropriate project animal license number.
Nonacog Beta Pegol
Human rFIX has been glycoPEGylated with a 40-kDa PEG. Preparation of rFIX and the PEGylation process has been described elsewhere (Ostergaard et al. 2011).
Animals and Housing
The Rowett nude rats (Crl:NIH-Foxn1rnu ) were supplied from Charles River, USA, and Charles River, Germany. At start of treatment, in the 6-week study, the rats were 36 to 57 days of age with males weighing 115 to 243 g and females weighing 90 to 159 g, and, in the 26-week study, the rats were 10 to 14 weeks old with males weighing 204 to 397 g and females weighing 155 to 231 g. Standard principles for randomization and acclimation were applied. Groups of 3, 4, or 5 rats were housed in a Techniplast Sealsafe™ individual ventilated, High-efficiency particulate arrestance [HEPA] filtered, sterilized, polysulfone cage at an approximate size of 480 × 375 × 210 mm height. All animals were kept in a full-barrier rodent facility with filtered air supply, continuous monitoring of temperature (19°C to 23°C) and humidity (40% to 70%), with a 12-hr light:12-hr dark cycle. Sterilized and certified bedding, nonrestricted pellet diet (VRF1 diet, Special Diet Services, Witham, UK), nonrestricted water, and environmental enrichment were provided for the animals and changed at appropriate intervals.
Administration of Test Substance
Test substance or vehicle control formulations were administrated to all animals by intravenous bolus injection into a caudal vein (rotating between right and left caudal vein) using a suitably graduated syringe and a new sterile disposable needle per animal. Volume-doses ranged from 0.8 to 4.8 ml/kg body weight.
In-life Observations
All animals were visually observed for ill-health at least twice daily and physically examined once weekly. Body weight and food consumption were recorded weekly. In both studies, blood was sampled for clinical chemistry, hematology, coagulation, PK, demonstration of continuous exposure, and antidrug-antibody analysis. Ophthalmic examination and urinalysis were conducted in the 26-week study but not in the 6-week study.
Clinical Pathology
In the 6-week study, blood was sampled for activated partial thromboplastin time (aPTT), prothrombin time (PT), and fibrinogen analysis once weekly (samples were taken 3 days after dosing). In the 26-week study, blood was sampled 12-hr postdose for clinical chemistry, hematology, and coagulation in weeks 13 and 26 of the dosing phase and in week 26 of the recovery phase. Urine samples were collected in week 26 of the dosing phase and in week 26 of the recovery phase. The animals were placed in an individual metabolism cage overnight and urine was collected over approximately 16 hr. Clinical chemistry parameters: alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, creatinine kinase, total bilirubin, urea, creatinine, glucose, total cholesterol, triglycerides, sodium (Na), potassium (K), chloride (Cl), calcium, inorganic phosphorus, total protein, and albumin. Hematology parameters: hematocrit, hemoglobin concentration, erythrocyte count, absolute reticulocyte count, mean cell hemoglobin, mean cell hemoglobin concentration, mean cell volume, red cell distribution width, total leucocyte count, differential leucocyte count, and platelet count. Coagulation parameters: were PT and aPTT. All blood samples were drawn from the sublingual vein. Samples for clinical chemistry were collected into tubes containing lithium heparin, and analyses were performed on plasma using a Roche P Modular apparatus (Roche Diagnostics, North America). Samples for hematology were collected in tubes containing ethylenediaminetetraacetic acid [EDTA], and analyses were performed on whole blood using a Bayer Advia 120 apparatus (Siemens Healthcare GmbH, Erlangen, Germany). Samples for coagulation were collected in tubes containing citrate (3.2%), and analyses were performed on whole blood using an ACL9000 (Instrumentation Laboratory, Beckman Coulter, Miami). Urinalysis using manual methods: appearance, volume, pH, specific gravity, and microscopic examination of urine sediment. Urinalysis using dipstick (Multistix 10 SG; Siemens Healthcare GmbH, Erlangen, Germany): glucose, ketones, bilirubin/bile pigments, and blood pigments. Urinalysis using Roche P Modular apparatus (Roche Diagnostics): protein, Na, K, and Cl (not normalized to creatinine). Creatinine in the urine and the urine to serum ratio were not measured.
Antidrug-antibody Analysis
In the 6-week study, blood was sampled for antidrug-antibody analysis 2 weeks after the last dose. In the 26-week study, blood was sampled for antidrug-antibody analysis in week 26 of the dosing phase and in week 2 of the recovery phase. Samples were taken into 3.8% trisodium citrate aliquots.
A validated bridging enzyme-linked immunosorbent (ELISA) assay was used for detecting antibodies against human rFIX and PEGylated human rFIX (nonacog beta pegol) in both studies. Briefly, potential immune complexes were acid dissociated with acetic acid for 1 hr followed by neutralization with a Tris pH = 9.5 high-salt buffer containing biotinylated and horseradish peroxidase labeled human rFIX and PEGylated human rFIX (Novo Nordisk, Denmark). After 1-hr incubation at room temperature, the samples were transferred to a streptavidin plate, incubated for 1 hr, and washed thoroughly, prior to addition of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate. Following a 15-min development with TMB substrate, the reaction was stopped with 4 M sulfuric acid. Samples were read at 450 nm. The sensitivity of the assay was approximately 20 ng/ml anti-rFIX antibody.
Pharmacokinetics/Exposure
In both studies, human rFIX concentration in the blood samples was determined using a luminescent oxygen channeling assay (LOCI) and PK calculations were performed using a noncompartmental analysis in Phoenix™ WinNonlin® version 6.2 (Pharsight®, St. Louis, Missouri). A sparse sampling schedule with 3 animals per time point for both males and females was used for PK evaluation in both studies. Samples were taken into 3.8% trisodium citrate aliquots.
In brief, the LOCI assay is a homogenous assay based on the proximity of 2 beads: the europium-coated acceptor beads and streptavidin-coated donor beads. Specific antibodies raised against human rFIX were coupled to the acceptor beads making them specific reagents, while the donor beads were generic. A biotinylated antibody against human rFIX was added to make up the immunosandwich. Upon laser excitation, a photosensitizer present in the donor beads converts ambient oxygen to a more excited state, which in turn channels into the acceptor beads generating chemiluminescence. This further activates a fluorophore contained in the acceptor beads, which subsequently emits light that is measured. The amount of light is proportional to the amount of rFIX (including nonacog beta pegol) in the sample.
In the 6-week study, the PK evaluation included 9 time points up to 168-hr postdose on day 1 and on the final dose administration (week 6).
In the 26-week study, the PK evaluation included 9 time points up to 96-hr postdose on day 1 and in week 26 of the dosing phase. For demonstration of continuous exposure in the 26-week study, exposure samples were taken monthly from 3 animals from each group 4-hr postdose.
Necropsy and Histology
All animals were terminated by exsanguination using the femoral vein after carbon dioxide asphyxiation. A macroscopic pathological examination of all organs was performed in all animals, and organ weights were obtained in both studies (Table 2). Tissues from each animal were fixed appropriately for a minimum of 24 hr (10% neutral-buffered formalin, except testes [modified Davidsons fluid; Latendresse et al. 2002] and eyes [Davidsons fluid; Latendresse et al. 2002]), processed to paraffin wax and cut into 4 to 5 µm sections, which were stained with hematoxylin and eosin (H&E) for histological evaluation. Brain including choroid plexus, spleen, and skeletal muscle were processed to paraffin wax stage within 48 hr. Histological evaluation was performed by a pathologist at Envigo and peer-reviewed by a pathologist at Novo Nordisk A/S according to international best practice standards.
List of Collected Tissues.
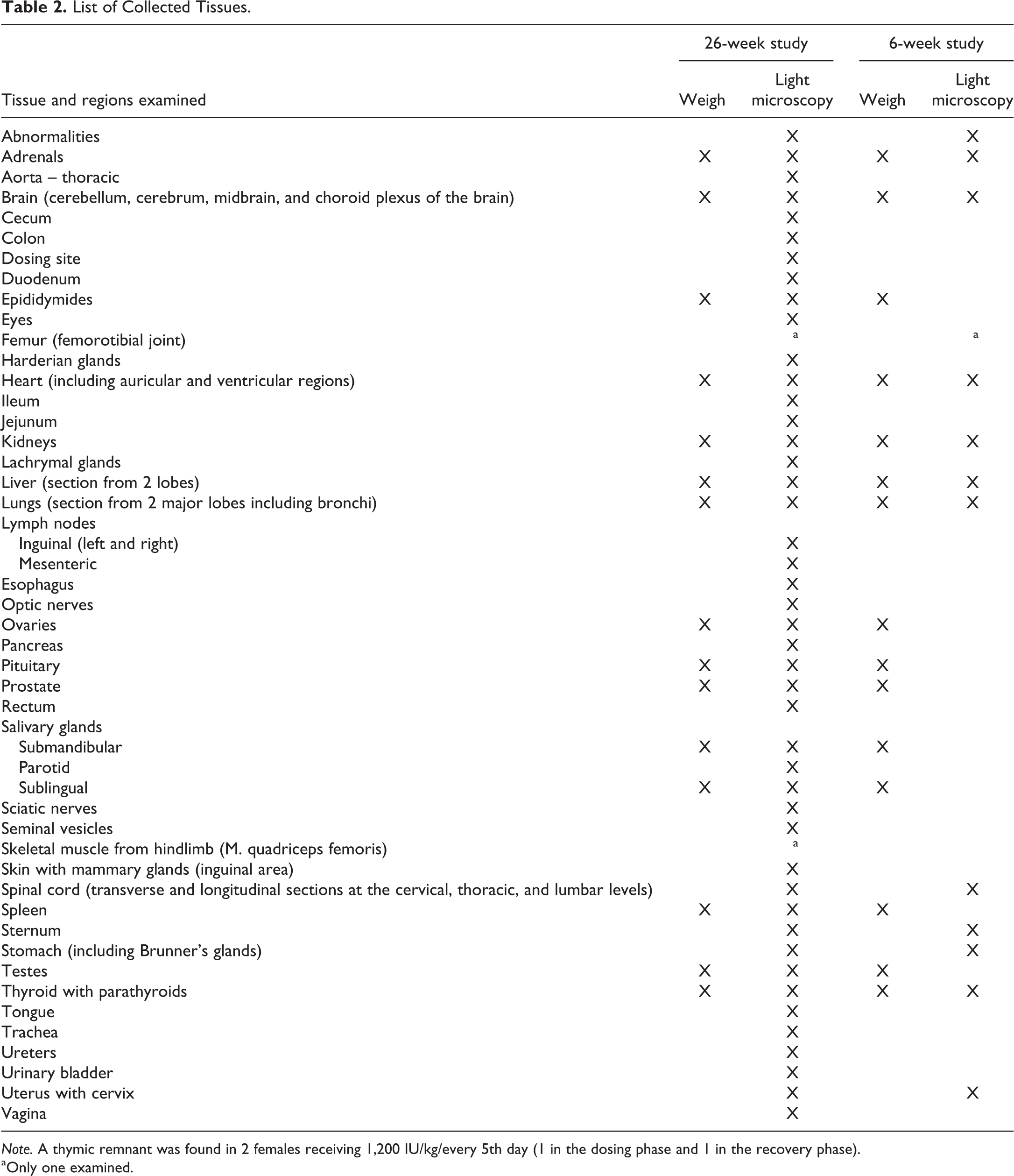
Note. A thymic remnant was found in 2 females receiving 1,200 IU/kg/every 5th day (1 in the dosing phase and 1 in the recovery phase).
aOnly one examined.
In the 6-week study, the scheduled termination was performed 2 weeks after the last dose. In the 26-week study, animals in the dosing phase were terminated 3 or 4 days after the last dose, and animals included in the recovery phase were terminated 26 weeks after the last dose. A full tissue list was histologically evaluated from animals included in the 26-week study (Table 2).
Immunohistochemistry
In both studies, the choroid plexus and spleen were evaluated by IHC for the detection of PEG. Formalin-fixed and paraffin-embedded tissue blocks were sectioned at a nominal thickness of 3 µm, mounted on Superfrost® Plus slides and stained for the detection of PEG using the following IHC protocol: the sections were deparaffinized and rehydrated and washed in Tris-buffered saline (TBS) buffer for 10 min. Nonspecific binding was blocked by incubations with bovine serum albumin. The primary anti-PEG antibody (monoclonal-rabbit anti-PEG, No. PEG-B-47 from Epitomic, California) was diluted (1:1,000) in TBS buffer and incubated at room temperature for 60 min followed by a 30-min incubation using Alexa 488 conjugated Goat antirabbit secondary antibody (No. A11001 from Invitrogen, Thermo Fisher Scientific, Illinois). Nuclei were counterstained with propidium iodide as part of the aqueous mounting medium Vectashield H-1300 (No. H-1300 from Vector Laboratory Inc. California). Positive staining controls included human macrophages cultured for 22 hr in medium added 6.6 µM 40-kDa PEG, muscle tissue injected with 40-kDa PEG, and negative staining included control muscle tissue and muscle tissue injected with rFIX alone. The specificity of the antibody was tested using identical antibody IgG (IgG Rabbit isotype, no. 11000003 from Jackson and IgG1 mouse isotype no. X0931 from Dako) and preabsorption of the primary PEG antibody with 40-kDa PEG before applied on slide as primary antibody. The presence of PEG in choroid plexus and spleen was identified using a fluorescence microscope, with filter cubes U-FUW/U-FBW and U-FGNA (Olympus, DK) and images were taken with a DP72 digital camera (Olympus, DK).
TEM
The electron microscope examinations were in total carried out in 4 nonacog beta pegol treated animals (1,200 IU/kg) and in 4 control animals from the 26-week study. The choroid plexus sampled from the left and right lateral ventricle was, with minor adjustments, fixed and processed for epoxy embedding according to standard procedure for preparative methods for TEM (Spector, Goldman, and Leinwand 1998). The presence of tissue and the tissue quality was verified by light microscopy in approximately 1-µm thick sections of the epoxy blocks stained with toluidine blue (0.1%).
Ultrathin tissue sections at a nominal thickness of 60 nm were collected at copper grids, mesh 200, and contrast stained with aqueous uranyl acetate and lead salt solution preparing the grids for the electron microscopy evaluation. Electron micrographs of the choroid plexus epithelial cells were taken in a Philips Morgagni FP5005 Transmission Electron Microscope, and micrographs at 5,600 to 22,000 magnification were used for the general characterization of organelles in the cytoplasm of the epithelial cells. The vesicles identified in the choroid plexus epithelial cells were graduated and could be categorized into the following 4 distinct types illustrated in Figure 1. Type 1 = rough endoplasmatic reticulum (rER)/Golgi vesicles, mucinous content; type 2 = undefined, considered large mucin droplets; type 3 = vesicles most likely containing PEG as identified by IHC; type 4 = “dense body” lysosome and lysosome-related/lipofucin.
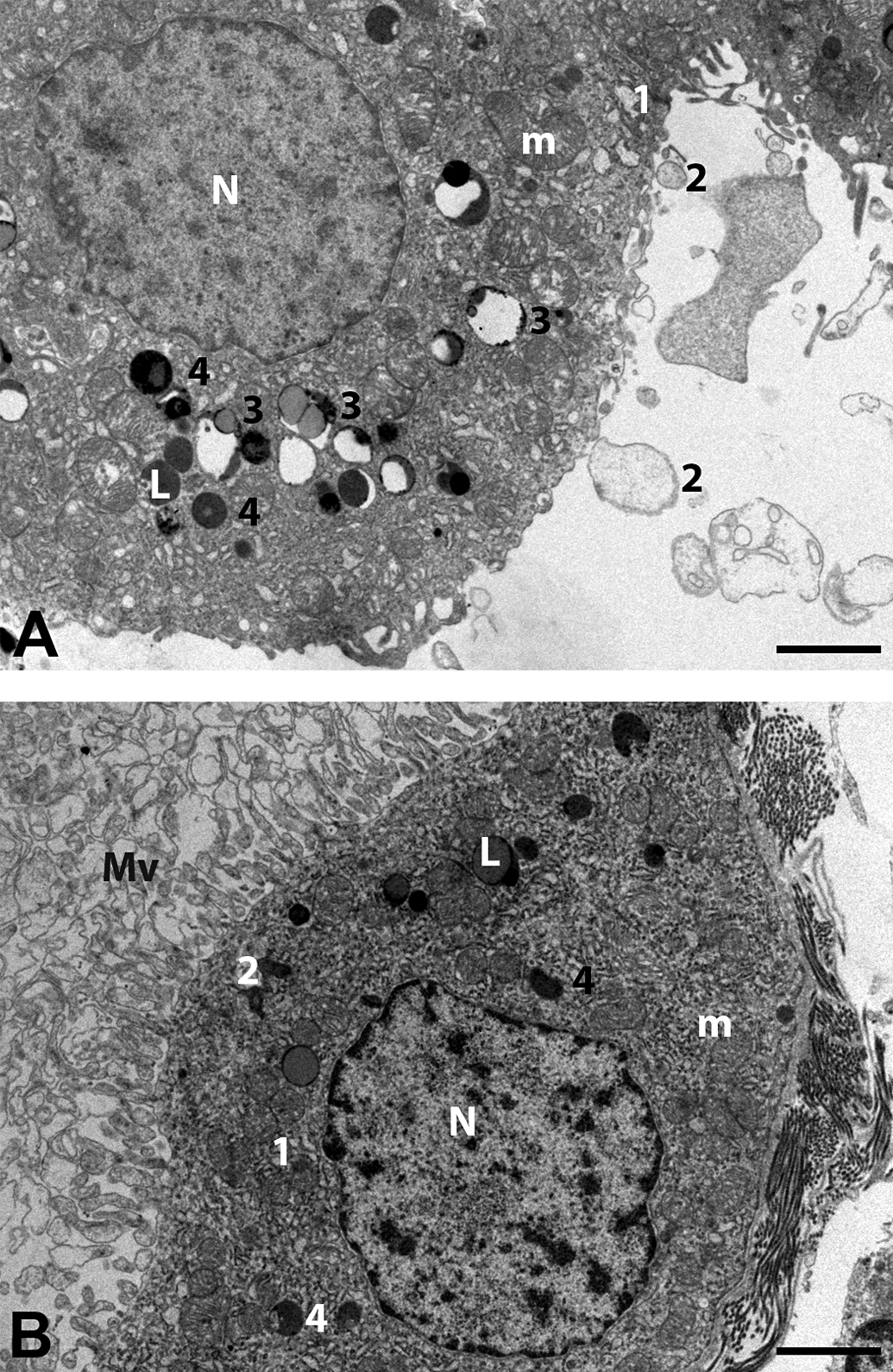
Electron microscopic pictures of choroid plexus epithelial cells from rats in the 26-week study. (A) Animal treated with 1,200 IU/kg nonacog beta pegol for 26 weeks. (B) Animal treated with vehicle for 26 weeks. N = nucleus, m = mitochondria, L = lipid droplet, Mv = microvilli. Definition of the different vesicles types: type 1 = rough endoplasmatic reticulum/Golgi vesicles, mucinous content; type 2 = undefined, but morphologically consistent with large mucin droplets; type 3 = electron-dense droplets most likely containing polyethylene glycol (PEG), as the presence of PEG was identified in these vesicles by immunohistochemistry, type 4 = “dense body” lysosome and lysosome-related/lipofucin. Bar = 2 µm. Letters in the figures are placed on top of the cellular structure they explain. Numbers are located on the midright side of the cellular structure they explain.
Statistical Analysis
In both studies, all statistical analyses were carried out separately for each sex. The analyses were carried out using the individual animal except for food consumption, which was analyzed on a cage basis. Group mean and standard deviation were calculated for the following parameters: body weight, food consumption, clinical pathology parameters, and organ weights. In both studies, the percentage deviation from controls was calculated for body weight and food consumption and additionally for clinical pathology parameters and organ weights in the 26-week study. No further statistical analyses were performed in the 6-week study. In the 26-week study, the following parameters were statistically analyzed at each time point separately, comparing each group to controls: body weight gains and food consumption over appropriate study periods, clinical pathology parameters, and organ weights (absolute and relative to terminal body weight). Data were tested for homogeneity of variance using Bartlett’s (1937) test. If data were parametric (with or without logarithmic or square root transformation), an F1 test was applied to test for monotonicity between means (1% significance level). If the outcome of the F1 test was significant, a Dunnett’s test for multiple comparison (1955, 1964) was applied to the data, and if not, a Williams’ test for monotonic trend (1971, 1972) was applied. If there were only 2 groups, a t-test was applied to these parametric data. If data were nonparametric, a H1 test was applied instead of F1, Steel’s test (1959) instead of Dunnett’s test, Shirley’s test (1977) instead of Williams’ test, and Wilcoxon rank sum tests (1945) instead of the t-test. For some clinical pathology parameters, more than 75% of these data were the same across groups and here Fisher’s exact tests (1973) were used.
Results
In-life Observations
There were no treatment-related clinical signs, effect on body weight, or food consumption in either of the studies. Unscheduled deaths occurred in both studies, but due to the distribution among groups and the histopathological findings/cause of death (Table 3; none of them were considered related to treatment). In the 6-week study, there were 3 unscheduled deaths: 2 females receiving 40 IU/kg and 1 male receiving 1,200 IU/kg. The survival percentage was 95 in the 6-week study. In the 26-week study, there were 14 unscheduled deaths: 2 control male and 4 control females, 2 males receiving 40 IU/kg, 2 males and 1 female receiving 600 IU/kg, and 1 male and 2 females receiving 1,200 IU/kg. The survival percentage was 94 in the 26-week study.
List of Unscheduled Deaths and Contributing Cause.
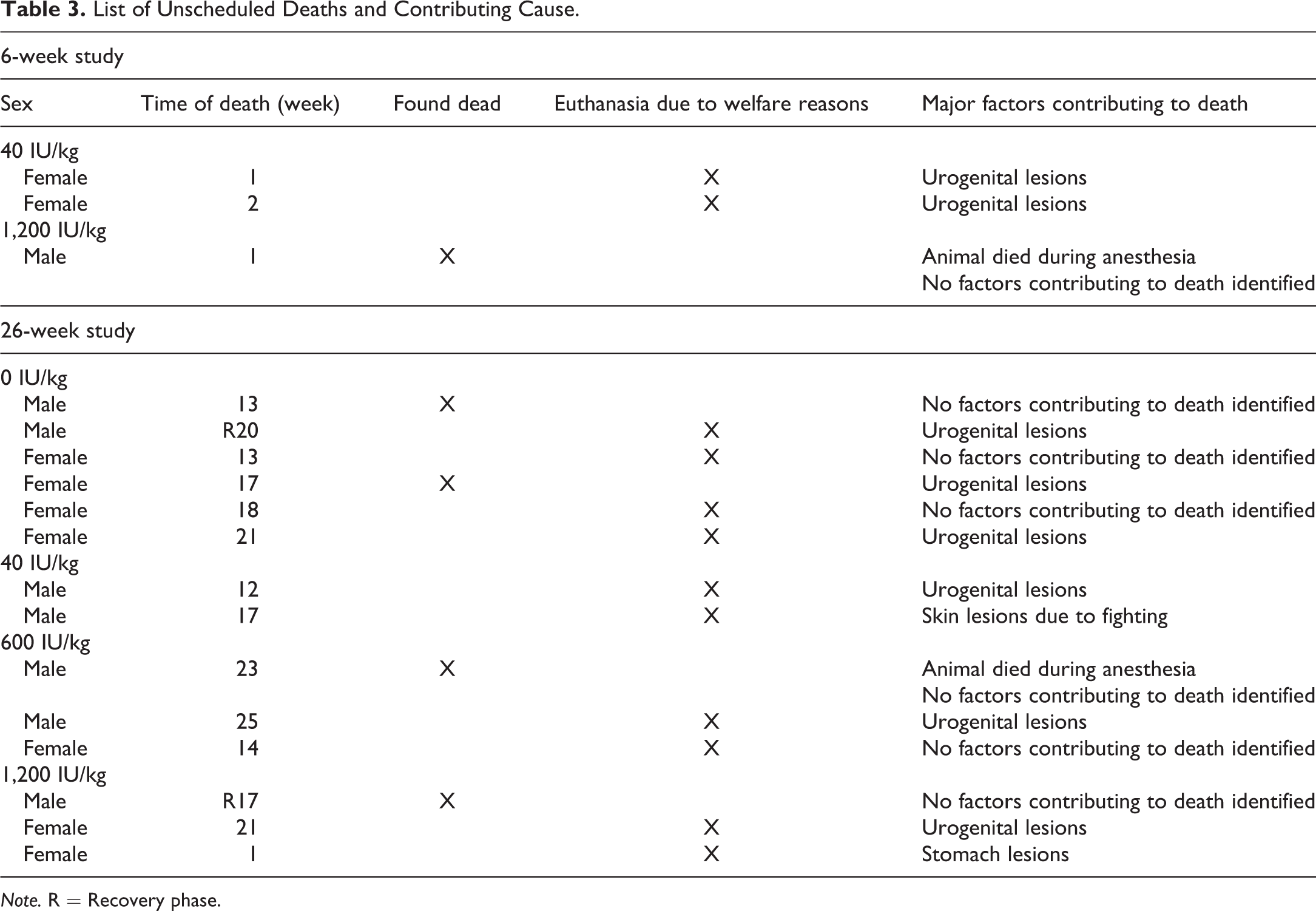
Note. R = Recovery phase.
Clinical Pathology
Differences between groups were only observed in relation to coagulation parameters: at all weekly sampling points in the dosing phase of 6-week study, there was a marked shortening of an aPTT in animals dosed with 1,200 IU/kg (range of means 11.4 to 14.8 sec) compared with controls (range of means 17.7 to 20.9 sec), whereas the shortening was only slight in animals dosed with 40 IU/kg (range of means 14.0 to 18.2 sec). After the 2-week washout period, the aPTT values for animals dosed with 40 and 1,200 IU/kg were similar to controls. There was no treatment-related difference in PT or fibrinogen concentrations.
Twenty-six-week study: the coagulation analysis indicated a dose-related prolongation of PT in animals dosed with 150, 600, and 1,200 IU/kg (Table 4). The PT values for animals receiving 1,200 IU/kg were similar to controls after 26 weeks without treatment. There was no treatment-related difference in aPTT.
Prolongation of Prothrombin Time in 26-Week Study.
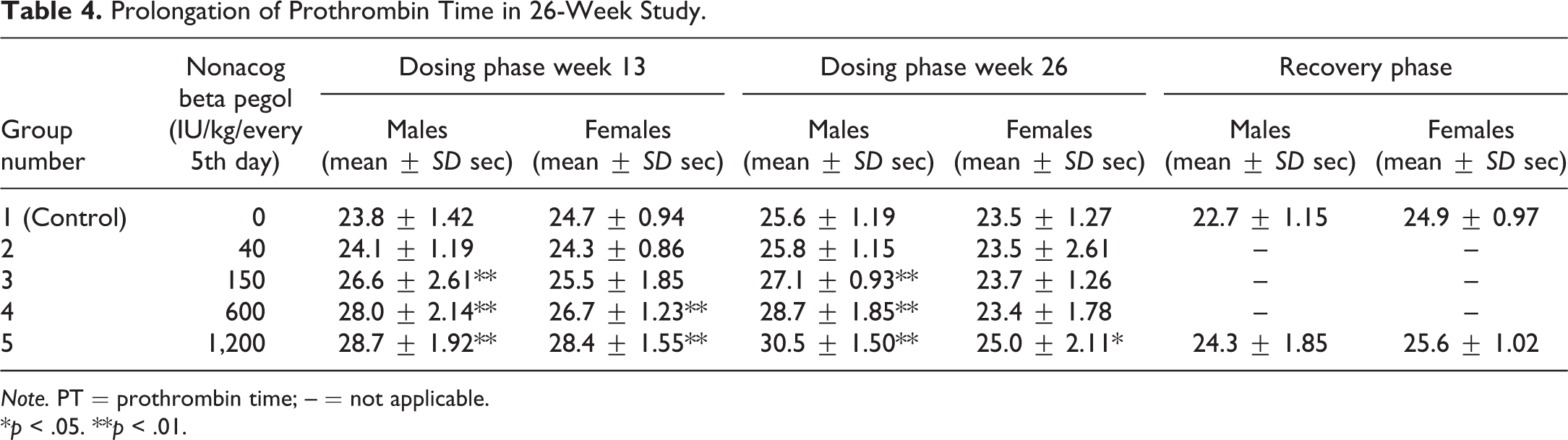
Note. PT = prothrombin time; – = not applicable.
*p < .05. **p < .01.
Antidrug-antibody Analysis
No anti-rFIX-antibodies were detected in either of the studies.
Pharmacokinetics/Exposure
In both studies, the PK investigations confirmed systemic exposure to nonacog beta pegol in the dosed animals. The half-life (t ½) ranged from 12 to 46 hr after repeated dosing in both studies and was generally 26 to 27 hr after a single dose in the 26-week study. The maximum observed plasma concentration (C max) and area under the plasma concentration–time curve (AUC) from dosing to last measurable concentration (AUClast ≡ AUC0–96hr) increased with dose for both sexes, with the increase of C max and AUClast being approximately proportional to dose.
Necropsy and Histology
No findings were considered related to treatment with nonacog beta pegol in animals terminated according to schedule or in the unscheduled death animals. The majority of the findings in the unscheduled death animals in both studies appeared to be related to nonspecific inflammatory processes, seen as urogenital lesions (Table 3) and considered related to the immunodeficiency of the animals and not to the treatment with nonacog beta pegol.
Immunohistochemistry
PEG was detected in small cytoplasmic vesicles in the choroid plexus epithelial cells in both studies in a dose- and time-dependent manner (Table 5). The small cytoplasmic vesicles were not visible on standard H&E staining of the choroid plexus. In addition, PEG was detected weakly in the connective tissue of the choroid plexus and within the blood vessels of the brain and choroid plexus but not in any other brain structures. In the spleen, PEG was detected in macrophages after 26-weeks dosing only. After 26-week recovery phase, no PEG was detected in the spleen, whereas PEG was still detectable in all animals in the choroid plexus epithelial cells but not in the connective tissue in the choroid plexus or in the blood vessels in the brain (only investigated in the 1,200 IU/kg dose group; Figure 2).
Overview of Percentage of Animals Immunohistochemically-positive for Polyethylene Glycol in Either Choroid Plexus or Spleen.

Note. N/A = not analyzed.
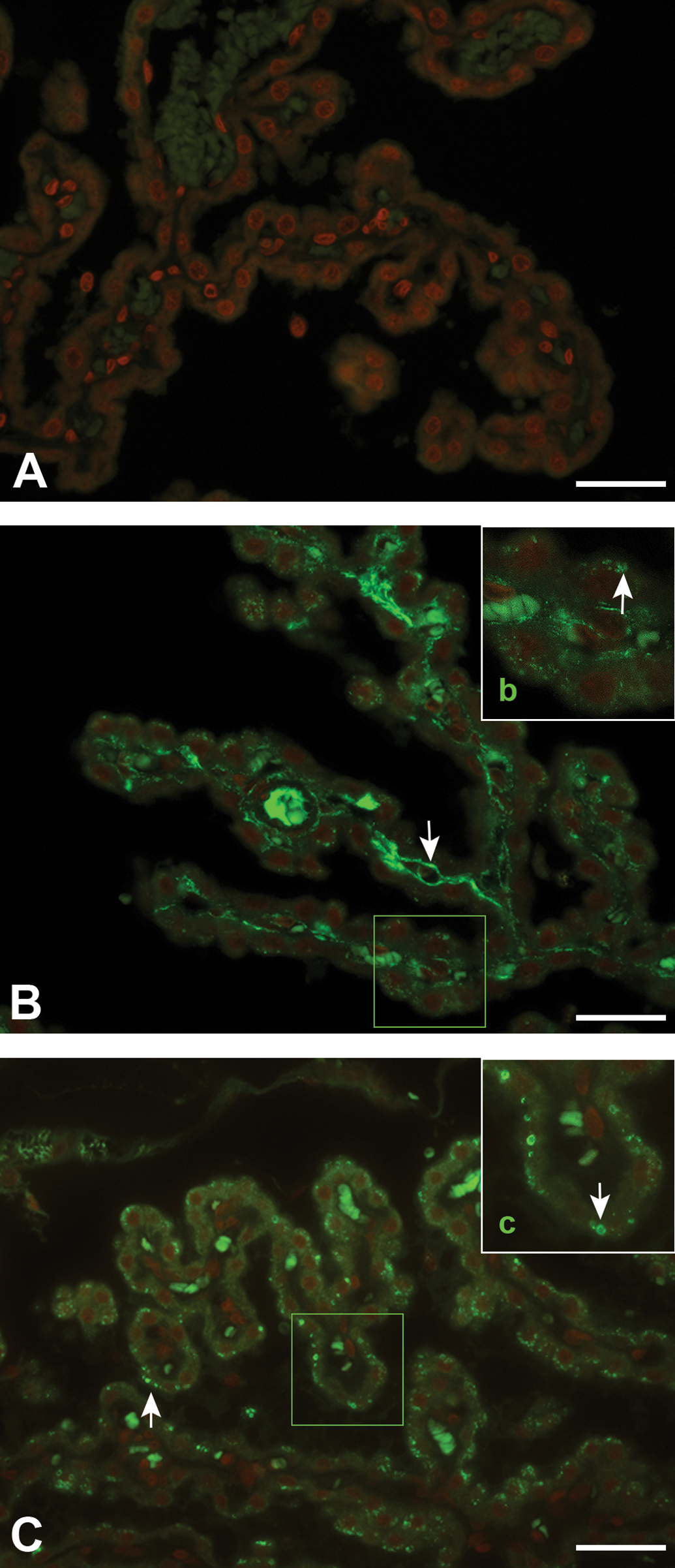
Immunohistochemical detection of polyethylene glycol (PEG) in the choroid plexus epithelial cells from rats in the 26-week study. Color intensity does not represent a quantitative measure of the amount of PEG present in the choroid plexus. Fluorescent green staining of the cores in the choroid plexus, visible in A, B, and C, represents autofluorescence of red blood cells. In addition to autofluorescence of red blood cells in B, PEG represented by fluorescent green is visible in the blood vessels. (A) Animal dosed with 0 IU/kg for 26 weeks. (B) Animal dosed with 1,200 IU/kg for 26 weeks. (C) Animal dosed with 1,200 IU/kg for 26 weeks followed by a 26-week recovery phase. (b) Digital enlargements of encircled area in B. (c) Digital enlargements of boxed area in (C). Bars = 30 µm- PEG fluorescent green indicated by white arrows. Red staining represents nuclei.
TEM
In general, choroid plexus epithelial cells were characterized by having microvilli on the luminal site, plenty of mitochondria, and a large amount of rER and polysomes (Figure 1). Mucinous vesicle defined as rER and Golgi vesicles (type 1) and undefined vesicles, morphologically consistent with large mucin droplets (type 2), were found. Dense bodies defined as lysosomes and lysosome related, for example, autophagic vesicles or lipofucin -containing vesicles were present in the cytoplasm (type 4).
Cells from animals treated with nonacog beta pegol differed from control animals by the presence of small electron-dense droplets (type 3) most likely containing PEG as identified by IHC. Apart from areas in the cytoplasm with vesicles most likely containing PEG, choroid plexus epithelial cells from nonacog beta pegol–dosed animals and control animals were very similar and no differences in cell nuclei, tight junctions, mitochondria, rER, polysomes, and apical cilia board (microvilli) were observed.
Discussion
Evaluation of the Rowett Nude Rat for Toxicity Testing
The immune-deficient Rowett nude rat (Crl:NIH-Foxn1rnu ) proved suitable for a chronic toxicity study of 26-week duration with nonacog beta pegol administered every 5th day. Thus, the rats were exposed during the entire dosing phase without development of antidrug-antibodies and had a high survival rate. The few animals that died prematurely showed signs of infections in the urogenital system and skin consistent with a compromised immune response as expected in this model. Based on the high survival rate, the animal model appears quite robust and not highly susceptible to infections. The nude rat could therefore be considered as an alternative long-term rodent model for testing for other human proteins found to be immunogenic in animals. However, given its immunocompromised status, the Rowett nude rat may not be a suitable model for immune-modulating compounds. Considerations related to the need for sterile conditions and barrier housing need to be taken into account when using this model. For toxicity testing, the nude rat model has a disadvantage in lacking a large amount of background data, meaning that differentiating between pathological findings being related to treatment or to the model could be challenging. However, in this case, the risk of unexplainable toxicity was considered minimal, based on the long-term clinical knowledge of risks related to rFIX (Berntorp et al. 2012; Monahan et al. 2010; Roth et al. 2001). Thus, a case-by-case approach is suggested when considering the nude rat as a chronic toxicity model.
Long-term Toxicity of Nonacog Beta Pegol in the Rowett Nude Rat
Chronic toxicity of nonacog beta pegol was tested in an immune-deficient rat model. Use of this model ensured that exposure was continuous throughout the study and not affected by antidrug-antibody development. In this model, no signs of acute or chronic toxicity were observed following 26 weeks of dosing of up to 1,200 IU/kg every 5th day.
The preliminary 6-week study tested the tolerability of 2 doses (40 IU/kg and 1,200 IU/kg twice weekly) in the Rowett nude rat. Nonacog beta pegol was well tolerated, no antidrug-antibody development was observed, and the animals were exposed throughout the 6 weeks.
In the 26-week study, nonacog beta pegol was generally well tolerated. No antidrug-antibodies developed and animals were exposed throughout the study. A dose-dependent shortening of an aPTT was observed in all 6 weeks of the dosing phase in the 6-week study but not in the 26-week study. A dose-dependent prolongation of PT was observed in the 26-week study and is considered a result of PT assay interference by high rFIX concentrations as described previously by others (Lefkowitz et al. 1993; Osterud et al. 1981; Spitzer et al. 1990). Shortening of aPTT and prolongation of PT is interpreted as an effect of unphysiologically high levels of rFIX on the assays and not a direct effect on the coagulation system. This interpretation is further supported by the macro- and microscopic pathology data, where no findings were identified, which could otherwise indicate in vivo effects of the shortened aPTT or prolonged PT. The inconsistency in occurrence of shortened aPTT and prolonged PT between studies was most likely caused by different postdose sampling time points, but preanalytical errors could not be ruled out.
None of the toxicity end points (clinical observations, clinical chemistry and hematology, macroscopic, and microscopic evaluation of tissues) were affected by treatment and no treatment-related vacuole formation was observed in any tissue covering a range of all major tissues and various cell types examined microscopically. In total, 45 tissues were examined for each animal. Presence of PEG in the choroid plexus was examined by IHC in both studies and a dose- and time-dependent increase in the number of animals with PEG IHC positive choroid plexus epithelial cells was observed. There was an increase in PEG positive animals at the dose levels of 40 and 1,200 IU/kg from 6 to 26 weeks dosing and an increase in PEG positive animals with increasing dose levels in the 26-week study (Table 5). In the 26-week study, a 26-week recovery phase was included. Following this, PEG was still detectable in the choroid plexus epithelial cells in animals previously dosed with 1,200 IU/kg (the only group with recovery). Presence of PEG was not confirmed in any other brain structures. In the spleen, PEG was detected by IHC in macrophages after 26 weeks of treatment, whereas no PEG was observed in macrophages after 26 weeks of recovery.
In contrast to our study, in which no vacuolation was observed in the H&E slides, other PEGylated products have been associated with macrophage and epithelial cell vacuolation (Bendele et al. 1998; Center for Drug Evaluation and Research 2012; European Medicines Agency 2004c, 2006, 2009, 2012; Ivens et al. 2015; Rudmann et al. 2013; Savient Pharmaceuticals 2009), and this has led to regulatory focus on careful assessment of the choroid plexus in the safety evaluation of PEGylated therapeutics (European Medicines Agency 2012). The observed retention of PEG in some tissues may be partly explained based on data describing the distribution and excretion of PEG (Bjoernsdottir et al. 2013). The authors have investigated the fate of a free 40-kDa PEG (1, 12, 100, and 200 mg/kg) and describe a faster initial excretion of PEG (over the first 1 to 2 weeks), followed by a longer elimination phase with a t 1/2 of 25 days to 54 days (shortest for the 2 lower doses). In addition, when looking at the PEG retained in the animals 12 weeks after a single dose, retention in carcass is increased with increasing doses of PEG. The dose-dependent increase in PEG retention in carcasses is in agreement with our studies, where PEG was detected by IHC in small cytoplasmic vesicles in choroid plexus epithelial cells in a dose- and time-dependent manner.
A literature review has identified no evidence that FIX is actively transported into the choroid plexus. Therefore, the observed uptake of PEG in the choroid plexus epithelial cells is most likely a result of unspecific uptake. This is supported by a recent paper published by Rudmann et al. (2013), where it was shown that the induction of treatment-related vacuole formation in various tissues is dependent on the size of the PEG molecule and that vacuole formation is primarily dependent on PEG and less dependent on the protein. However, protein-related uptake of the PEG is likely involved in some cases (European Medicines Agency 2012). The mechanism behind unspecific uptake of PEG in the choroid plexus may be related to cells being exposed to PEG from the fenestrated capillaries and high blood flow in choroid plexus (Redzic and Segal 2004) leading to high concentrations of nonacog beta pegol or PEG in the connective tissue (seen as positive PEG IHC in the connective tissue). Macromolecules, like nonacog beta pegol or PEG, are known to enter the epithelial cells in vesicles derived from the cell membrane, for example, by receptor-mediated endocytosis, fluid-phase endocytosis, or adsorptive endocytosis. However, the exact uptake mechanism is unknown. In the studies conducted in the nude rats, PEG was still present in the choroid plexus epithelial cells after the 26-week recovery phase, which may be explained by a very slow cell turnover time of the epithelial cells in the choroid plexus (Chauhan and Lewis 1979; Liddelow 2015), a very slow degradation of the PEG molecules in the epithelial cells or both (Ivens et al. 2015). This is further substantiated by the finding of recovery from PEG staining in macrophages in the spleen, a cell type with a higher turnover rate (Furth 1989). In some of the published studies, vacuole formation related to PEG retention has been observed by histopathological examination (Bendele et al. 1998; Center for Drug Evaluation and Research 2012; European Medicines Agency 2004c, 2006, 2009; Ivens et al. 2015; Rudmann et al. 2013; Savient Pharmaceuticals 2009), whereas in other studies, administration of PEG did not result in histopathological recognizable treatment-related vacuole formation (Center for Drug Evaluation and Research 2007; European Medicines Agency 2004a; Ivens et al. 2015; Pilaro 2000, 2001; Savient Pharmaceuticals 2009; U.S. Food and Drug Administration 1997; Webster et al. 2007). There was no indication of an inflammatory or degenerative response in relation to the choroid plexus in any of the cases. In the present studies with nonacog beta pegol, PEG retention (seen as positive PEG IHC) was minor and did not lead to treatment-related vacuole formation or pathological changes in the choroid plexus or other tissues. TEM examination of control and high-dose animals from the present 26-week study identified small droplets, most likely containing PEG, in membrane-delimited vesicles in the epithelial cells of the choroid plexus in the treated animals. Judged from the overall cell ultrastructure, including rER and polysomes, there was no indication that the membrane-delimited vesicles, which most likely contained PEG, affected normal epithelial cell function. Taken together, PEG seems, by an unknown mechanism, to be transported into and retained in the choroid plexus epithelial cells without influence on the cell ultrastructure. Based on the absence of neither degenerative nor inflammatory reactions, the retention of PEG in membrane delimited vesicles in the cytoplasm in the choroid plexus epithelial cells is considered a nonadverse adaptive response.
Conclusion
The immune-deficient rat proved suitable for a 26-week chronic toxicity study and could be considered in cases where a model is needed for the chronic toxicity testing of human proteins found to be immunogenic in animals. Based on the absence of treatment-related adverse findings evaluated by standard toxicological end points and complemented by IHC staining for PEG and an ultrastructural investigation, the presence of PEG observed in this study in the choroid plexus epithelial cells is not considered an adverse finding.
Footnotes
Acknowledgments
The authors would like to thank the following personnel in Non-clinical Development at Novo Nordisk A/S for their contribution to this study: Helene Jacobsen from Nonclinical Development Project Management, Anja Kirkeby Goldenbæk from Biopharm Toxicology and Safety Pharmacology, Ingrid Sjögren from Pathology, Joan Hummeluhr Rasmussen from Investigative Pathology, Wendela Kappers from Development DMPK, Mette Høgh Sørensen, Theresa Willer Dallerup, and Pernille Rasmussen from Bioanalysis & ADME, and Anders Holm Millner and Jette Bruun Tvenge from DMPK, Cell and Antibody Analysis.
Authors’ Contribution
Authors contributed to conception or design (CR, JN, JL, AB, AR, and HO); data acquisition, analysis, or interpretation (CR, JN, JL, AB, AR, and HO); and drafting the manuscript (CR, JN, and HO). All authors critically revised the manuscript, gave final approval, and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Authors’ Note
The study has previously been presented as a poster at the 8th Annual Congress of the European Association of Haemophilia and Allied Disorders, Helsinki, Finland 2015.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was sponsored by Novo Nordisk A/S, Maaloev, Denmark. Caroline E. Rasmussen, Jette Nowak, Julie M Larsen, and Hanne Offenberg are employees of Novo Nordisk A/S. Anna Bottomley and Alison Rowles are employees of Envigo where the study was conducted.