
Abstract
Cardiotoxicity was an unanticipated side effect elicited by the clinical use of imatinib (Imb). This toxicity has been examined in only a limited number of experimental studies. The present study sought, by a variety of approaches, to identify important characteristics of Imb-induced cardiac alterations. Male spontaneously hypertensive rats (SHRs) received oral doses of 10, 30, or 50 mg/kg Imb or water daily for 10 d. Cardiac lesions, detected at all doses, were characterized by cytoplasmic vacuolization and myofibrillar loss. In a second experiment, cardiac lesions were found in Sprague Dawley (SD) and SHR rats given 50 or 100 mg/kg Imb for 14 d. Mean cardiac lesion scores and serum levels of cardiac troponin I were higher in SHRs than in SD rats. Imb induced myocyte death by necrosis, autophagy, and apoptosis. Dose-related increases in cardiac expression were observed for several genes associated with endoplasmic reticulum stress response, protein folding, and vascular development and remodeling. Imb caused alterations in isolated myocytes (myofibrillar loss, highly disrupted and disorganized sarcomeric α-actinin, apoptosis, and increased lactate dehydrogenase release) at low concentrations (5 mM). The authors conclude that Imb exerts cardiotoxic effects that are manifest through a complex pattern of cellular alterations, the severity of which can be influenced by arterial blood pressure.
Keywords
Introduction
Tyrosine kinase inhibitors (TKI) are a relatively new class of anticancer drugs that target cellular pathways overexpressed in certain types of tumors (Sawyers 2003). Interest in these drugs arose from studies showing that imatinib (Imb) markedly inhibited human BCR-ABL1–positive tumor cells (le Coutre et al. 1999). Such studies ultimately led to adding Imb and other similarly targeted drugs to the treatment of neoplasms arising from abnormal tyrosine kinase (TK) regulation. These drugs were active against neoplastic disease such as chronic myelogenous leukemia, the pathogenesis of which has been attributed to the activation of BCR-ABL1 TK (Deininger et al. 2003; Nakajima and Togo 2003; O’Brien et al. 2003; Krause and Van Etten 2005). Drugs targeting tumor-specific pathways are believed to be more effective than conventional chemotherapeutic drugs, which tend to affect rapidly dividing cells in both normal and cancerous tissues.
Current TKI drugs interfere primarily with pathways mediating the mitotic growth of tumor cells (Krause and van Etten 2005). These same pathways have been identified as also being important for maintaining the viability of cells in normal tissues, such as the heart. It is this off-target action that is responsible for the initiation of alterations in normal tissues. Indications of off-target toxicity were observed in clinical trials with Imb (Force, Kraus, and Van Etten 2007; Kerkela et al. 2006). These trials revealed that Imb not only slowed the progression of chronic myologenous leukemia but also caused unanticipated cardiotoxicity. Kerkela et al. (2006) reported a decline in left ventricular ejection fraction and congestive heart failure in patients treated for chronic myeloid leukemia. In that study, biopsy samples were obtained from two patients who had experienced congestive heart failure while taking Imb. Electron micrographs of both samples revealed mitochondrial abnormalities that were associated with intracellular membrane whorls. Similar declines in cardiac function and alterations in myocyte morphology have been reported in patients treated with a second, multitargeted TKI (sunitinib; Chu et al. 2007; Telli et al. 2008; Kerkela et al. 2009).
Imb-related cardiotoxicity was also demonstrated in both in vitro and in vivo experiments (Kerkela et al. 2006). For example, mice treated with Imb for 3 to 6 wk developed left ventricular contractile dysfunction and ultrastructural myocyte alterations (Kerkela et al. 2006). These investigators suggest, as indicated above, that the pathogenic mechanism of Imb-related toxicity may involve interfering with one or more of the antitumor targets (Abelson proto-oncogene) also present in the heart. However, other studies in mice did not confirm the cardiotoxic actions of Imb reported by Kerkela et al. (2006). Wolf et al. (2010) found that cardiac alterations (necrosis), in rats, occurred only at high non–clinically relevant doses.
Studies reporting Imb-induced cardiac lesions describe various types of changes in myocyte morphology. Kerkela et al. (2006) reported mitochondrial abnormalities and accumulation of membrane whorls. Wolf et al. (2010) found that Imb was associated with myocyte necrosis in rats. Recent in vitro studies showed that autophagy was associated with sunitinib (a related TKI)–mediated cell death in H9c2 cardiac muscle cells (Zhao et al. 2010). Although changes in myocyte morphology have been identified following exposure to Imb, a consistent pattern of these alterations has not been reproduced in all studies. In addition, the clinical relevance of the doses used to alter the myocardium in experimental studies has also been questioned (Wolf et al. 2010). It is apparent that many important issues regarding the potential adverse effects of Imb on the heart need further clarification.
In the present study, we investigated a number of factors related to Imb-induced cardiotoxicity in both in vitro and in vivo experiments. The in vitro experiments were designed to determine the properties of lesions and the concentrations of Imb that were capable of causing such alterations in isolated myocytes. The in vivo experiments were designed to characterize the changes in cardiac morphology and gene regulation caused by different doses of Imb in rats. Rat models have been used extensively to explore various aspects of doxorubicin-induced cardiotoxicity (Herman and Ferrans 1998). These studies revealed that hypertensive rats are more sensitive than normotensive rats to the cardiotoxic effects of doxorubicin (Herman et al. 1985). Thus, we also sought to determine whether hypertension could also be a risk factor for Imb-related toxicity and whether hypertensive or normotensive rats would be a more appropriate model to explore the pathogenesis and characteristics of TKI-induced cardiotoxicity.
Methods and Materials
All procedures involving animals were approved by the Institutional Animal Care and Use Committee, Center for Drug Evaluation and Research, Food and Drug Administration, and complied with the guidelines for the 1996 Institute of Laboratory Animals Resources Guide for the Care and Use of Laboratory Animals (National Research Council 1996).
In Vivo Experiments
Forty-five adult male spontaneously hypertensive rats (SHRs) and 25 adult male Sprague Dawley (SD) rats, 10 to 12 wk of age, were obtained from Harlan Laboratories (Indianapolis, IN). Animals were housed individually and given rodent chow (Lab Diet 5002) and water ad libitum. Experiments began after a week of acclimation.
In Vivo Experimental Procedures
The study included two experiments. The first determined whether 10 to 50 mg/kg Imb given daily for 10 d could cause myocardial lesions in SHRs. The second compared the incidence and severity of myocardial alterations induced by doses of 50 or 100 mg/kg Imb given daily for 14 d in SHR and SD rats. Imb doses selected were intended to be in the range of those used in clinical treatment regimens (400-800 mg/d or 228-456 mg/m2 based on a weight of 70 kg). The highest dose of Imb used in the present study was 100 mg/kg, which is equivalent to 590 mg/m2.
Experiment 1.
Imb methanesulfonate salt was obtained from LC Laboratories (Woburn, MA). Twenty SHRs were divided into four experimental groups of five animals each. Three of these groups received 10 mg/kg, 30 mg/kg, and 50 mg/kg Imb, respectively, and the fourth or control group received distilled water at a volume equivalent to the 50-mg/kg per dose (0.5 mL/100 g body weight).
Imb was dissolved in distilled water (10.5 mg/mL) and administered by gavage in dose volumes of 0.1 to 0.5 mL/100 g body weight daily for 10 d. Each animal was observed for overt signs of toxicity and weighed on days 0, 3, 7, and 10.
Experiment 2.
In this experiment, twenty-five male SHR and twenty-five male SD rats were divided into a high-dose treatment group of ten rats, a low-dose treatment group of five rats, and a control group of ten rats. Imb was dissolved in distilled water to a concentration of 15.5 mg/mL. The ten SHR and ten SD rats in each of the two high-dose groups received 100 mg/kg Imb (0.63 mL/100 g body weight). The five rats in each low-dose group received 50 mg/kg Imb (0.33 mL/100 g body weight). The ten rats in each control group received distilled water at a dose volume of 0.63 mL/100 g body weight. All animals were dosed by oral gavage daily for 14 d. Each animal was observed daily for overt toxicity and weighed on days 0, 3, 7, 10, and 14.
Study Termination Procedures
Animals in both experiments were euthanized with isoflurane 24 h after the final dose was given. The inferior vena cava was exposed through a midline abdominal incision. Terminal blood samples (whole, plasma, and serum) for hematological, clinical chemistry, and cardiac troponin I analysis were collected through a 19-G Venocath inserted into the vena cava. Blood samples collected for plasma and serum were immediately centrifuged at 3000 × g for 10 min and frozen at –80°C until analyzed.
Pathologic Evaluation
The heart, kidneys, and portions of the liver and small intestine were excised and fixed in 10% neutral buffered formalin. In experiment 1, hearts were weighed prior to fixation. Hearts were embedded in glycol methacrylate plastic resin. Sections (1-µm thick) of the plastic embedded left ventricular heart tissue were stained with alkaline toluidine blue. The noncardiac tissues and some cardiac tissues were embedded in paraffin and stained with hematoxylin-eosin.
All toluidine blue–stained plastic sections of cardiac tissues (two to four sections/heart) were examined by a research pathologist (J.Z). The evaluation was blinded to treatment and any related biomarker data. The severity of myocardial lesions was scored on a semiquantitative scale of 0 to 3 under light microscopy (Billingham 1991), but necrosis of myocytes was also added to the scale. Thus, the scoring system used to access cardiac lesion severity in the present study was based on the proportion of muscle cells showing myofibrillar loss, cytoplasmic vacuolization, and necrosis: 0, none or a negligible percentage; 1, less than 5%; 1.5, between 6% and 15%; 2, between 16% and 24%; 2.5, between 26% and 35%; and 3, greater than 35%.
Immunohistochemical Studies
The terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and indirect immunoperoxidase staining for nitrotyrosine (an indicator for detecting the generation of reactive nitrogen oxidants such as peroxynitrite) and LAMP-2 (lysosome-associated membrane glycoprotein-1, an indicator for detection of autophagosome fusion with lysosomes) were applied to the sections of formalin-fixed, paraffin-embedded cardiac tissues. The procedures for detecting cardiac apoptosis are described elsewhere (Zhang et al. 2008). The CardioTACS in situ apoptosis detection kit was purchased from Trevigen, Inc. (Gaithersburg, MD).
To detect immunoreactivity of nitrotyrosine and LAMP-2, immunostaining was performed on the Discovery XT Autostainer (Ventana Medical System Inc., Tuscon, AZ) using the Ventana DAB MAPTM kit. In brief, sections were incubated 1 h with polyclonal goat anti-LAMP-2 antibody (Invitrogen, Carlbad, CA) and monoclonal antibody (Cell Science, Norwood, CA). After the sections were incubated for an additional 30 min with a biotinylated second antibody, the peroxidase reaction was initiated with diaminobenzidine. Each section was then counterstained with hematoxylin. Negative controls were stained manually with isotype mouse IgG2b as described elsewhere (Haskins et al. 2011). LAMP-2 primary antibodies were omitted from the incubation step and replaced by the isotype IgG at the same dilution of 1:100.
Sample Analysis
Terminal serum clinical chemistry analysis was performed on the VetScan model #200-1000 using the Comprehensive Diagnostic and Large Animal rotors (Abaxis Inc., Union City, CA). Blood cell counts and other hematological values were determined with the VetScan HMT (Abaxis).
Terminal serum levels of cardiac troponin I were measured with the ultra-sensitive Erenna Immunoassay System (Singulex, Alameda, CA).
Statistical Methods for Analyte and Histopathology Analysis
The Kruskal-Wallis test for nonnormally distributed data was used to assess differences in the myocardial lesion scores between the various treatment and control groups. The Tukey-Kramer multiple comparisons test was used to assess group-related differences in hematologic values, clinical chemistry values, and cardiac troponin concentrations. All data met the assumptions of the tests used to analyze them. Alpha was set at .05, and all tests were two-tailed. The InStat (GraphPad Software Inc., San Diego, CA) statistical software package was used for all analyses.
Microarray Analysis
Gene expression was profiled on a subset of heart samples from SHRs in experiment 2 that possessed a similar severity of myocardial lesions. We analyzed samples from five of the ten rats in the control group, the five in the low-dose group (50 mg/kg Imb) with lesion scores of 1.5, and four in the high-dose group (100 mg/kg Imb) with lesion scores of 2 to 2.5.
A transverse section of heart tissue was taken from the midpoint of the left ventricle and stabilized in RNA later. RNA was isolated using the manufacturer’s recommended modification of the Qiagen RNeasy kit protocol for RNA isolation from heart tissue. Total RNA was reverse transcribed and labeled for hybridization to Affymetrix RAE230A arrays (Santa Clara, CA) using the Affymetrix IVT kit and the manufacturer’s recommended methods. Microarray data were normalized using the Robust MultiChip Average algorithm and analyzed using BRB-ArrayTools developed by Dr. Richard Simon and the BRB-Array Tools Development Team. Alpha was set at .001 for the univariate F-test with a random variance model. Functional analysis on significantly changed genes was performed using Ingenuity Pathway software and the Database for Annotation, Visualization, and Integrated Discovery version 6.7 (DAVID; http://david.abcc.ncifcrf.gov).
In Vitro Experiments (Isolated Myocyte Experiments)
Materials
Imb mesylate (Toronto Research Chemicals, Toronto, Canada, or Advanced Technology & Industrial, Hong Kong, China) stock solutions for myocyte treatments were prepared in sterile water. Trypsin, collagenase, and deoxyribonuclease were from Worthington (Freehold, NJ). Unless specified, other reagents were obtained from Sigma (Oakville, Canada). DF-15 medium (with 7.5% (v/v) fetal bovine serum and 7.5% (v/v) horse serum), fetal bovine and horse serum, penicillin, streptomycin, and fungizone were obtained from Invitrogen (Burlington, Canada). Dexrazoxane hydrochloride was a gift from Adria Laboratories (Columbus, OH). For the in vitro studies, the errors shown are standard errors. Where significance is indicated (p < .05), an unpaired t-test was used (SigmaPlot, Systat, San Jose, CA).
Myocyte Isolation, Culture, and Epifluorescence Microscopy
This study was carried out with the approval of the University of Manitoba Animal Care Committee. Ventricular myocytes were isolated from two- to three-day-old SD rats as described (Hasinoff 2010). Briefly, minced ventricles were serially digested with collagenase and trypsin in PBS/1% (wt/v) glucose at 37°C in the presence of deoxyribonuclease and preplated in large petri dishes to deplete fibroblasts. The preparation, which was typically greater than 90% viable by trypan blue exclusion, yielded an almost confluent layer of uniformly beating cardiac myocytes by day 2. For the lactate dehydrogenase (LDH) release experiments, the myocyte-rich supernatant was plated on day 0 in 24-well plastic culture dishes (5 × 105 myocytes/well, 750 µL/well) in DF-15. On days 2 and 3, the medium was replaced with 750 µL of fresh DF-10 containing 10% (v/v) fetal bovine serum (FBS). To lower the background LDH levels, on day 4, 24 h before the drug treatments, the medium was changed to DF-2 and again on day 5, just before the drugs were added.
The anti-α-actinin/Hoechst 33258 staining and imaging by epifluorescence microscopy of fixed myocytes to score for both myofibrillar disruption and for apoptotic nuclei was carried out as previously described (Hasinoff, Patel, and O’Hara 2008; Hasinoff, Patel, and Wu 2007). Typically, twenty microscope fields containing on average fifty-five myocytes each were analyzed for apoptosis, and sixteen on average were analyzed for myofibrillar damage. Only those myocytes that also stained positive for α-actinin were counted as apoptosis positive.
Drug Treatments and LDH Measurements
Myocytes were treated with Imb for 24, 48, or 72 h. Starting on day 5 after plating, samples (80 µL) of the myocyte supernatant were collected every 24 h for up to 3 d after exposure to Imb. The samples were frozen at –80°C and analyzed within 1 wk. After the last supernatant sample was taken, the myocytes were lysed with 250 µL of 1% (v/v) Triton X-100/2 mM EDTA/1 mM dithiothreitol/0.1 M phosphate buffer (pH 7.8) for 20 min at room temperature. The total cellular LDH activity, from which the percentage of LDH release was calculated, was determined from the activity of the lysate plus the activity of the three 80-µL samples previously taken. The LDH activity was determined in quadruplicate in a spectrophotometric kinetic assay in a 96-well plate in a Molecular Devices (Menlo Park, CA) plate reader as previously described (Hasinoff 2010; Hasinoff, Patel, and O’Hara 2008; Hasinoff, Patel, and Wu 2007). In other experiments, myocytes were pretreated with 100 µM dexrazoxane for 3 h to determine whether this cardioprotectant could reduce Imb-induced myocyte LDH release.
Effect of Imb on Myocyte Oxidation of Intracellular 2′,7′-dichlorofluorescin
The effect of Imb on the oxidation of 2′,7′-dichlorofluorescin was determined essentially as described (Hasinoff, Patel, and Wu 2007). In brief, 2′,7′-dichlorofluorescin diacetate was loaded into attached myocytes 5 d after plating in 96-well plates (125,000 myocytes/well, 200 µL medium/well). Myocytes were incubated with 50 µM 2′,7′-dichlorofluorescin diacetate (the cell permanent ester of 2′,7′-dichlorofluorescin) for 20 min at 37°C in Hanks’ balanced salt solution (pH 7.4) with added Ca2+ (1.2 mM), Mg2+ (0.4 mM), and NaHCO3 (4 mM), followed by two changes of medium at room temperature to remove the extracellular ester.
The kinetic fluorescence measurements were made on a BMG (Durham, NC) Fluostar Galaxy fluorescence plate reader (excitation wavelength of 485 nm, emission wavelength of 520 nm, 37°C) equipped with excitation and emission probes directed to the bottom of the plate. After initial baseline fluorescence intensity data were collected, Imb or H2O2 was added to the attached myocytes and gently mixed with a pipette, and the increase in fluorescence was recorded for 60 min. The change in the rate of fluorescence increase was computed from data 3 min directly after addition of Imb. H2O2, which rapidly enters myocytes and oxidizes 2′,7′-dichlorofluorescin, was used a control.
Results
In Vivo Results
General Toxicity and Body Weight Gain
Experiment 1.
Rats at all doses gained between 32 and 40 g in body weight (Table 1). The mean weight gain did not differ between the control rats and the various treatment groups.
Characteristics of 70 Spontaneously Hypertensive and Sprague Dawley Rats after 10 or 14 Days of Daily Treatment with Imatinib, by Dose
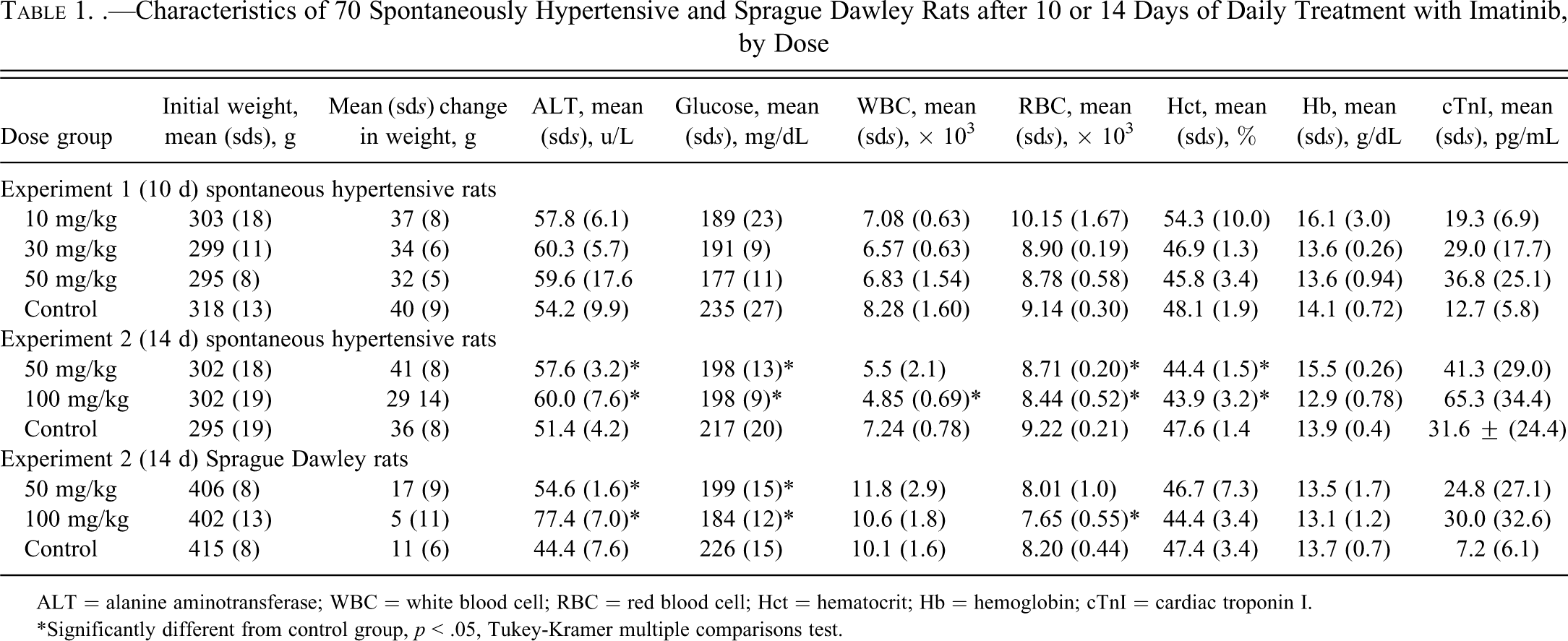
ALT = alanine aminotransferase; WBC = white blood cell; RBC = red blood cell; Hct = hematocrit; Hb = hemoglobin; cTnI = cardiac troponin I.
*Significantly different from control group, p < .05, Tukey-Kramer multiple comparisons test.
Experiment 2.
Three SHRs (one from the high-dose group and two controls) died after the third or fourth day of dosing. In each case, death occurred shortly after oral gavage and thus was considered to be caused by a dosing error. Data related to these animals were not included in subsequent analysis. SHRs gained an average of 29 g (100 mg/kg), 41 g (50 mg/kg), and 36 g (control) in body weight (Table 1). Initial mean body weight of SD rats was approximately 100 g more than that of the SHRs. All treated and control SD rats gained weight over the 14-d experiment, but the mean increases were less (5 to 17 g) than those in the SHRs (Table 1). Treatment-related differences in weight gain did not differ significantly in either SHR or SD rats.
Gross Anatomic Changes
At necropsy, no obvious tissue abnormalities were noted in the heart, liver, kidney, spleen, or small intestine of any treated animal in either the 10- or 14-d experiments.
Myocardial Histopathology
Experiment 1.
During necropsy, no macroscopic abnormalities were noted in the hearts from control or Imb-treated rats. The heart weight/body weight ratios determined in experiment 1 were 3.816 ± 0.288, 4.216 ± 0.582, 4.210 ± 0.251, and 4.200 ± 0.172 for the control and 10-, 30-, and 50-mg/kg Imb groups, respectively. The ratios in treated animals were not significantly different from the control or from each other.
Light microscopic evaluation of hearts from control rats showed cardiac myocytes with regular arrangements of thick and thin myofilaments (Figures 1A and 1B). In contrast, the cardiac tissues from rats treated with 10 mg/kg Imb showed cytoplasmic vacuolization and myofibrillar loss in the myocardium (Figures 1C and 1D). Myocyte necrosis was detected in the cardiac tissues from SHRs treated with 30 mg/kg Imb (Figures 1E and 1F). A few cytoplasmic vacuoles were noted within the affected necrotic cells. At the 50-mg/kg dose, foci of myocyte necrosis and fragmentation of necrotic cells were more frequent (Figures 1G and 1H). Focal inflammation (lymphocytes, macrophages, and a few neutrophils and fibroblasts) was occasionally observed in affected areas (figure not shown). The degree of myofibrillar loss, cytoplasmic vacuolization, and necrosis increased with the dose of Imb (Table 2). Of five hearts from SHRs dosed for 10 d with 10 mg/kg Imb, two were normal (lesion score = 0), whereas the other three had lesion scores of 1, 1, and 1.5 (Table 2). Lesions of mild severity (1.5) were observed in the hearts from four of five and from five of five SHRs dosed with 30 or 50 mg/kg Imb, respectively. The lesions in these two dose groups were significantly more frequent and severe than those in the 10-mg/kg and control groups (p < .001).
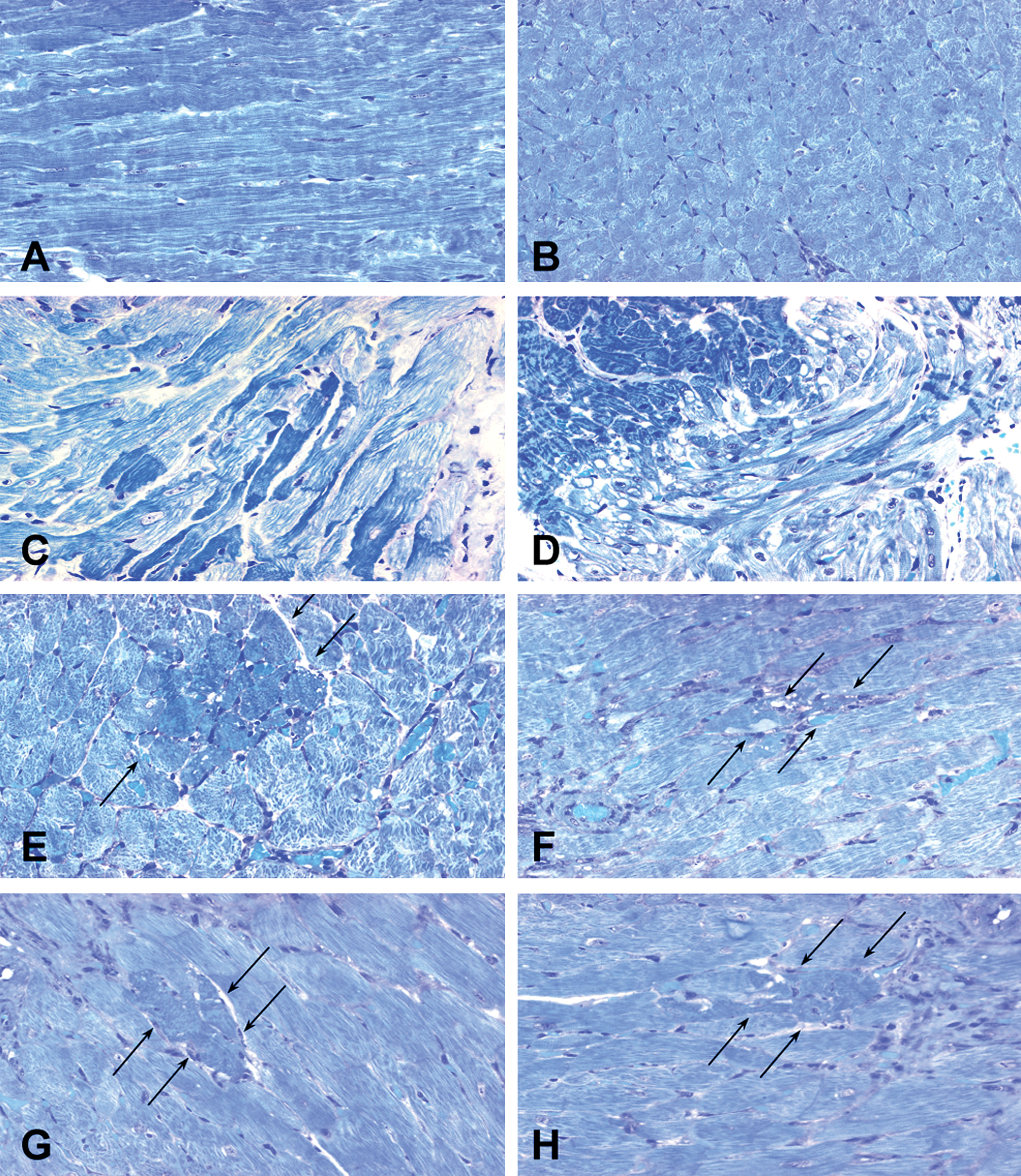
Light micrographs showing cardiac alterations in left ventricle from spontaneously hypertension rats (SHRs) given saline (A, B) and imatinib (Imb) at doses of 10 mg/kg (C, D), 30 mg/kg (E, F), and 50 mg/kg (G, H) for 10 d. (A, B) The cardiac myofibrils from saline-treated rats exhibited regular arrangement of thick and thin myofilaments. (C, D) Focal myofibrillar loss (particularly thin filaments) and formation of variable-sized cytoplasmic vacuoles in the myocardium of SHRs treated with the 10-mg/kg dose of Imb. (E, F) Intact and fragmented necrotic cardiac myocytes (arrows) in association with cytoplasmic vacuoles within the cytoplasm in the myocardium from SHRs treated with the 30-mg/kg dose of Imb. (G, H) Necrosis of cardiac myocytes and fragmentation of necrotic myocytes (arrows) in the myocardium of SHRs treated with the 50-mg/kg dose of Imb. (A-H). Glycol methacrylate–embedded, alkaline toluidine blue–stained, 1-μm-thick plastic sections. Original magnification 250×.
Severity of Cardiomyopathy in 70 Spontaneously Hypertensive and Sprague Dawley Rats Treated Daily with Imatinib for 10 or 14 d
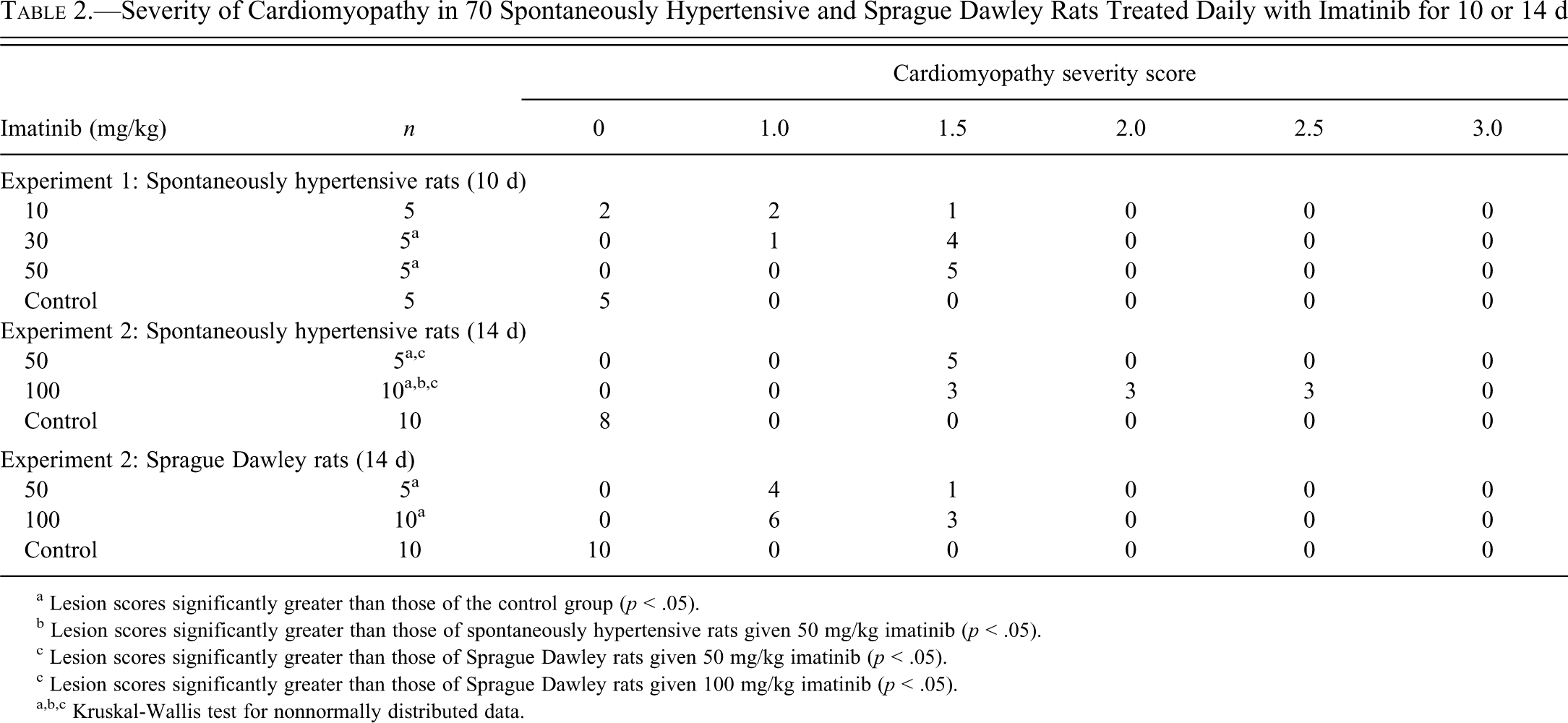
a Lesion scores significantly greater than those of the control group (p < .05).
b Lesion scores significantly greater than those of spontaneously hypertensive rats given 50 mg/kg imatinib (p < .05).
c Lesion scores significantly greater than those of Sprague Dawley rats given 50 mg/kg imatinib (p < .05).
c Lesion scores significantly greater than those of Sprague Dawley rats given 100 mg/kg imatinib (p < .05).
a,b,c Kruskal-Wallis test for nonnormally distributed data.
Experiment 2.
Hearts from all control rats given saline or Imb for 14 d, examined grossly and microscopically, appeared to be normal. Light microscopy revealed that all SHR and SD rats treated with 50 or 100 mg/kg Imb had myocardial lesions characterized by cytoplasmic vacuolization and myofibrillar loss (Figures 2A-H). The myocyte alterations were less severe in rats treated with 50 mg/kg (Figures 2A, B, E, and F) than they were in both rat strains given 100 mg/kg Imb (Figures 2C, 2D, 2G, and 2H; Table 2). The severity of cardiomyopathy scores were significantly different between the SHR and SD rats (p < .001). Of nine SHRs given 100 mg/kg Imb, six had moderate lesions (lesion score = 2 or 2.5). In contrast, of 10 SD rats given 100 mg/kg Imb, six had minimal lesions (lesion score = 1), three had mild lesions (lesion score = 1.5), and only one had moderate lesions (lesion score = 2). Animals receiving 50 mg/kg Imb had similar results. All five SHR had lesions scores of 1.5, whereas only one of five SD rats had a lesion score of 1.5. Again, the lesions were more severe in SHR than in SD rats (p < .02).
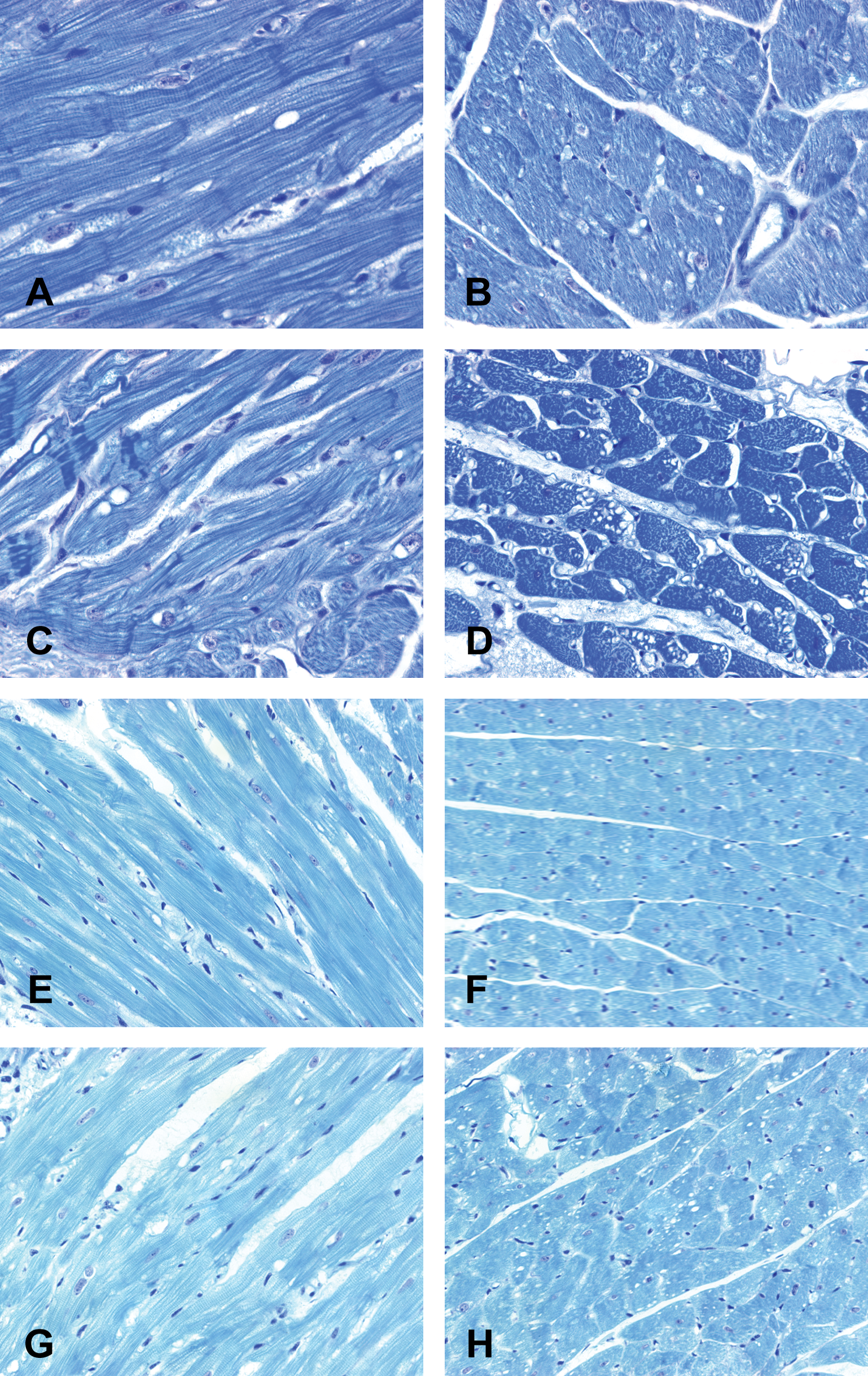
Light micrographs showing cardiac lesions in the left ventricular myocardium of spontaneously hypertension rats (SHRs) given imatinib (Imb) at doses of 50 mg/kg (A, B) or 100 mg/kg (C, D) and SD rats given doses of 50 mg/kg (E, F) or 100 mg/kg for 14 d (G, H). Treatment of SHRs with 50 mg/kg of Imb induced cytoplasmic vacuoles, myofibrillar loss, and disarrangement of myofibrillar filaments (A, B), which were milder than similar lesions noted in SHRs treated with 100 mg/kg of Imb (C, D). Treatment of SD rats caused dose-dependent cytoplasmic vacuolization and myofibrillar loss (E-G and E, F). Both doses of Imb caused lesions that were less severe in Sprague Dawley rats than in SHRs. Compare (A-D) with (E-H). (A-H) Alkaline toluidine blue–stained 1-μm-thick plastic sections. Original magnification 250×.
Pathologic Findings in Noncardiac Tissue
Experiment 1
No treatment-related alterations were observed in the spleen or small intestine from SHRs dosed for 10 d with 10, 30, or 50 mg/kg Imb. However, tubular dilatation and luminal proteins in the kidneys and occasional small foci of hepatic inflammation were observed in SHRs given 50 mg/kg Imb.
Experiment 2
Minor renal tissue changes were noted in three SHRs given 100 mg/kg Imb (pyelectasis) and in one SHR given 50 mg/kg Imb (pyelonephritis) for 14 d.
Myocardial Immunohistochemistry.
Immunostaining detected little or no positive nitrotyrosine reaction in the ventricular myocardium, the ventricular septum, the papillary muscle, or the tendinous cords in control SHRs (Figure 3A). In contrast, hearts from SHRs that had been treated with 100 mg/kg Imb showed positive staining for nitrotyrosine in the cytoplasm of damaged ventricular myocytes (Figure 3B), papillary muscle, and tendinous cords. As an indication of severe necrosis in these hearts, strong nitrotyrosine staining was occasionally observed in individual myocytes (Figure 3C). Additional sites of nitrotyrosine staining in Imb-treated SHRs included endocardial endothelium, vascular endothelial cells, and inflammatory cells.
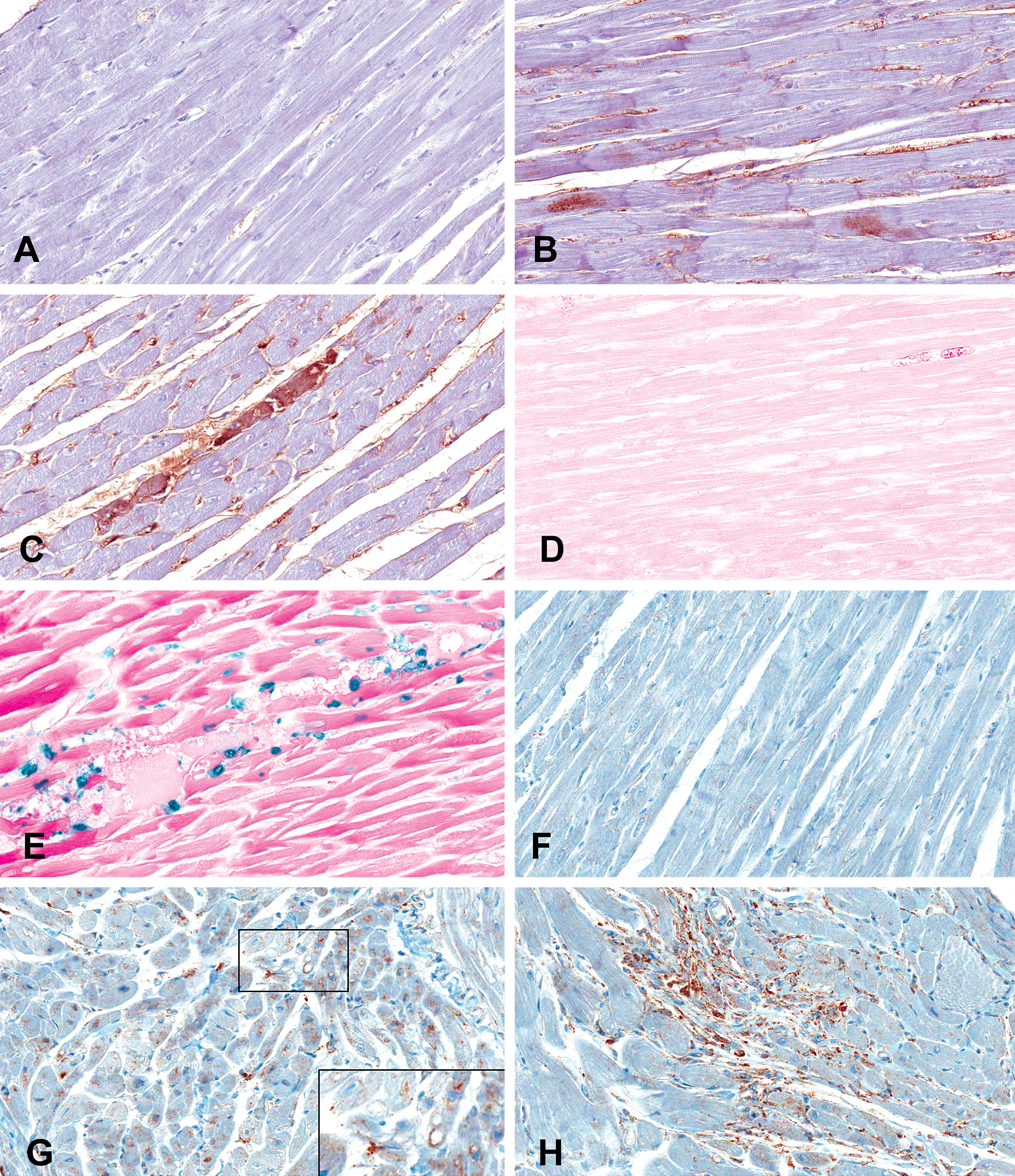
Light micrographs showing nitrotyrosine and lysosome-associated membrane glycoprotein-2 (LAMP-2) immunoreactivity and induction of apoptosis (terminal deoxynucleotidyl transferase dUTP nick end labeling assay) in the hearts from saline- and 100 mg/kg imatinib (Imb)–treated spontaneously hypertension rats (SHR). (A) Little or no positive staining for nitrotyrosine in control SHRs. (B) Positive staining for nitrotyrosine in the cytoplasm of mildly damaged myocytes and endothelial cells of capillaries in Imb-treated SHRs. (C) Strong positive staining for nitrotyrosine in the cytoplasm of necrotic myocytes in Imb-treated SHRs. (D) Absence of nuclear reaction for myocyte apoptosis in the heart from control SHRs. (E) Strong positive staining for myocyte apoptosis in the hearts from Imb-treated SHRs. Note: apoptotic bodies with or without nuclear fragmentation in the interstitium. (F) Little or no positive staining for LAMP-2 in control SHRs. (G) Strong positive staining for LAMP-2 in the cytoplasm of myocytes from Imb-treated SHRs (inset in G). Positive LAMP-2 appearing crescent shaped within cytoplasmic vacuoles (possibly indicative of autolysosomal formation via fusion of lysosomes with vacuoles). Note: also aggregated lysosomes and vacuoles without fusion with lysosomes in the cytoplasm. (H) Positive LAMP-2 within severe atrophic myocytes in the heart of SHRs treated with Imb. (A-H) Original magnification 400×.
TUNEL assay staining was negative in hearts from control SHRs (Figure 3D), but many apoptotic nuclei were noted in the ventricular myocardium (Figure 3E) and papillary muscle from Imb-treated SHRs. Numerous interstitial apoptotic bodies (fragmented cytoplasm with or without nuclear fragmentation) were observed in hearts from Imb-treated SHRs.
Myocytes from control SHRs (Figure 3F) did not stain positive for LAMP-2. However, atrophic myocytes from Imb-treated SHRs showed strong positive staining for LAMP-2 (Figure 3G and 3H). In these hearts, some cytoplasmic vacuoles were fused with positively stained lysosomes (identified by LAMP-2; Figure 3G, inset). This morphology is suggestive of autolysosomal formation. Aggregated lysosomes and nonfusion vacuoles were detected in the cytoplasm of some myocytes. Imb treatment was also associated with positive staining for LAMP-2 in the endocardium and papillary muscles.
Hematology and Clinical Chemistry Measurements
Experiment 1.
Doses of 10, 30, or 50 mg/kg Imb given for 10 d had minimal effects on hematological or clinical chemistry characteristics (Table 2). Mean (sds) blood glucose concentrations in Imb-treated SHRs (189 [23], 191 [9], and 177 [11] mg/dL, respectively) were lower than those in control rats (235 [27]; p < .05] but did not differ among the treatment groups.
Experiment 2.
White blood cells, red blood cells, and hematocrit were modestly but significantly decreased in SHRs treated with Imb (Table 1; p < .05 from the control). The values in the Imb-treated groups did not differ significantly from each other.
After 14 d, SHRs and SDs treated with Imb had mean (sds) serum concentrations of alanine aminotransferase that were slightly but significantly higher than those of control animals (p < .05). Glucose and alanine aminotransferase concentrations in the two SHR and SD Imb treatment groups were similar.
Serum Cardiac Troponin
Cardiac troponin I (cTnI), measured by the Erenna ultrasensitive immunoassay, was detected in all evaluated 23 control and 44 treated animals (Table 3). Baseline levels of cTnI were higher in SHRs than in SD rats. In addition, mean concentrations of cTnI tended to be higher in SHRs treated with 50 or 100 mg/kg Imb than in the corresponding SD groups. An apparent dose-related increase in cTnI in each of the treatment groups paralleled the increase in lesion severity. However, none of the increases in mean cTnI concentrations differed significantly from control or from each other because of the large variability in individual cTnI values.
Functional Categorization of Statistically Significant Changes in Gene Expression in the Rat Heart after Imatinib Treatment
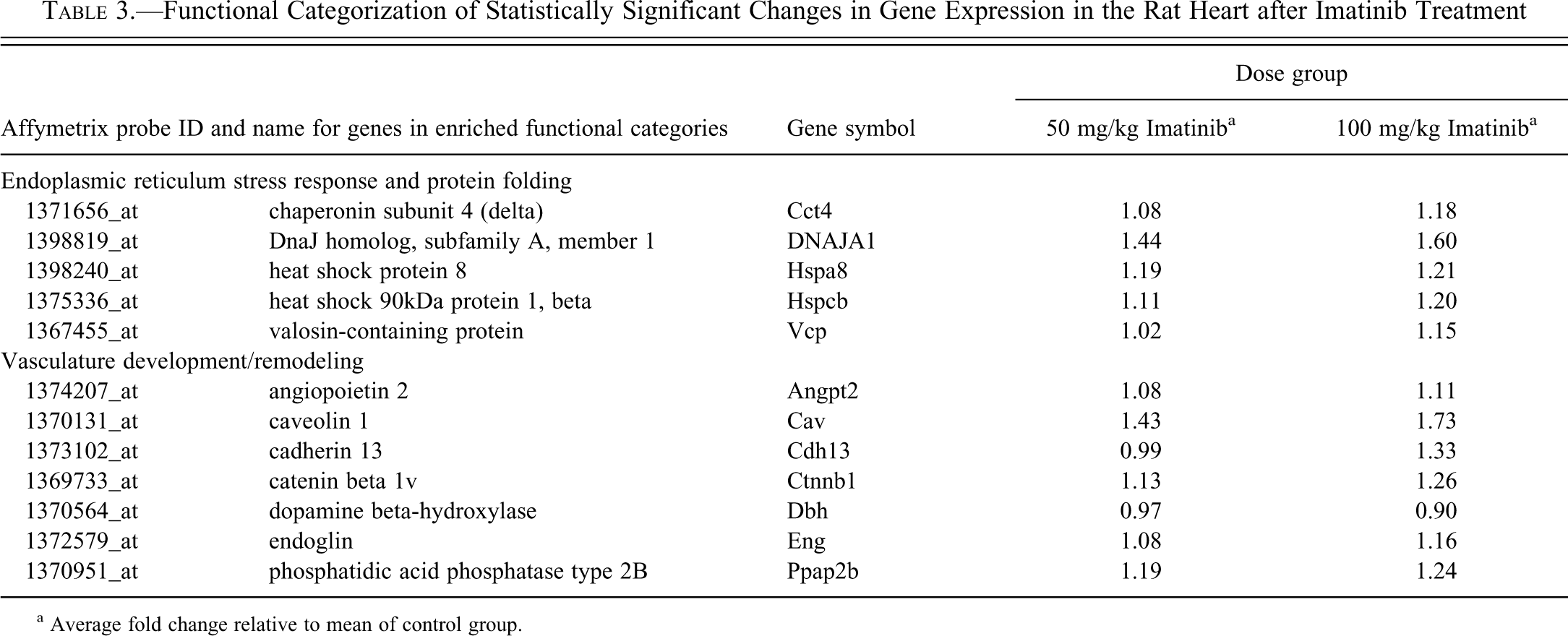
a Average fold change relative to mean of control group.
Microarray Analysis
Statistically significant changes in gene expression in the heart between Imb-treated and control groups of SHRs in experiment 2 were few and limited. The average change in expression in either the 50- or 100-mg/kg treatment groups compared with controls was less than twofold for all of the 92 differentially expressed genes identified at a .001 significance level. This expression profile was enriched in genes associated with the endoplasmic reticulum stress response and protein folding (Table 3). Dose-related increases in cardiac expression were also observed for several genes associated with vascular development and remodeling, a functional category overrepresented in the 92 genes affected by Imb (Table 3).
In Vitro Results
Effect of Imb Alone and in Combination with Dexrazoxane on LDH Release in Myocytes
Cytosolic LDH release is a widely used measure of drug-induced damage to myocytes (Adderley and Fitzgerald 1999; Hasinoff 2010; Hasinoff, Patel, and Wu 2007). Using LDH release as a biomarker of cell viability, we examined the ability of Imb to damage myocytes 5 to 7 d after isolation, when the myocytes would be essentially nonproliferating (Li et al. 1996). At 72 h, all concentrations of Imb between 5 and 40 µM significantly increased LDH release (p < .01 for 5 µM and <.001 for all other concentrations, respectively) up to 2.5-fold above that in untreated control myocytes (Figure 4A). Imb also significantly increased LDH release at 10, 30, and 40 µM at 48 h (Figure 4B). While the results shown were averages (±SEM) from a single experiment measured in four replicate wells, they were typical of three other separate myocyte isolations (data not shown).
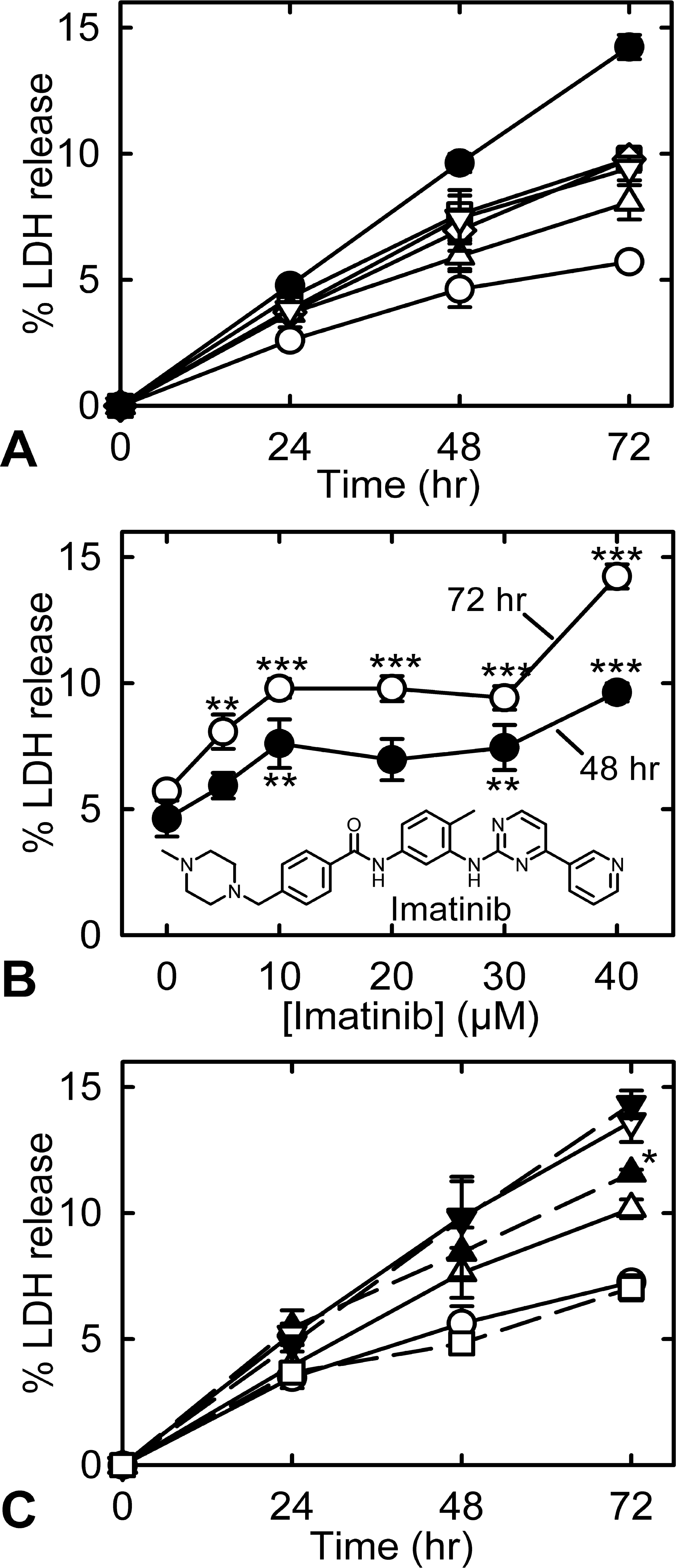
(A) Plot of cumulative percentage lactate dehydrogenase (LDH) release from myocytes that were untreated (^) or continuously treated with 5 µM (Δ), 10 µM (□), 20 µM (⋄), 30 µM (▽), or 40 µM (•) imatinib. (B) Plot of percentage LDH release at 48 h (•) or (^) 72 h after treatment with imatinib. Significance relative to the untreated controls: *p < .05, **p < .01, and ***p < .001. Where standard error bars are not seen, they are smaller than the size of the symbol. The structure of imatinib is shown in the inset. (C) Plot of cumulative percentage LDH release from cardiac myocytes that were untreated (^) or treated with either 30 µM imatinib (△) or 40 µM imatinib (▽) alone or pretreated with 100 µM dexrazoxane and 30 µM imatinib (m), or 100 µM dexrazoxane and 40 µM imatinib (.), or with 100 µM dexrazoxane (□) alone. The data were from an experiment in which the percentage LDH release was measured in four separate wells. At 72 h, the dexrazoxane pretreatment slightly, but significantly (*p < .05), increased LDH release compared with 30-µM imatinib-treated myocytes. Where error bars (±SEM) are not seen, they are smaller than the size of the symbol.
Pretreatment with 100 µM dexrazoxane was unable to reduce Imb-induced LDH release caused by exposure to either 30 or 40 µM Imb (Figure 4C). Dexrazoxane pretreatment slightly increased LDH release (p < .05) caused by 30 µM Imb treatment. While the data shown were from a single experiment, the results were typical of those obtained in experiments using two other separate myocyte isolations (data not shown).
Effect of Imb on Myofibrillar Damage and Induction of Apoptosis
Myocytes treated with Imb were scored for damaging effects on their morphology by a pathologist blinded to group assignment. Treatments between 5 and 20 µM Imb for 72 h produced no significant damage (Figure 5A). However, more and highly significant (p < .0001) myocyte damage was seen at 30 µM than in untreated control myocytes. The Imb-treated myocytes displayed myofibrillar loss with highly disrupted and disorganized sarcomeric α-actinin (Figure 6A, B). The morphology of the damage is similar to that we previously saw with doxorubicin (Hasinoff, Schroeder, and Patel 2003), a cardiotoxic anticancer drug highly damaging to myocyte morphology (Billingham and Bristow 1984; Gabizon et al. 2004; Lim et al. 2004). Myocyte scores for changes in nuclear morphology (characteristic of apoptosis) showed that treatment at all concentrations of Imb between 5 and 40 µM produced higher percentages of apoptotic nuclei (p < .001) than those of untreated control myocytes (Fig. 5B). Thus, Imb had strong apoptosis-inducing effects on myocytes under the conditions of this study.
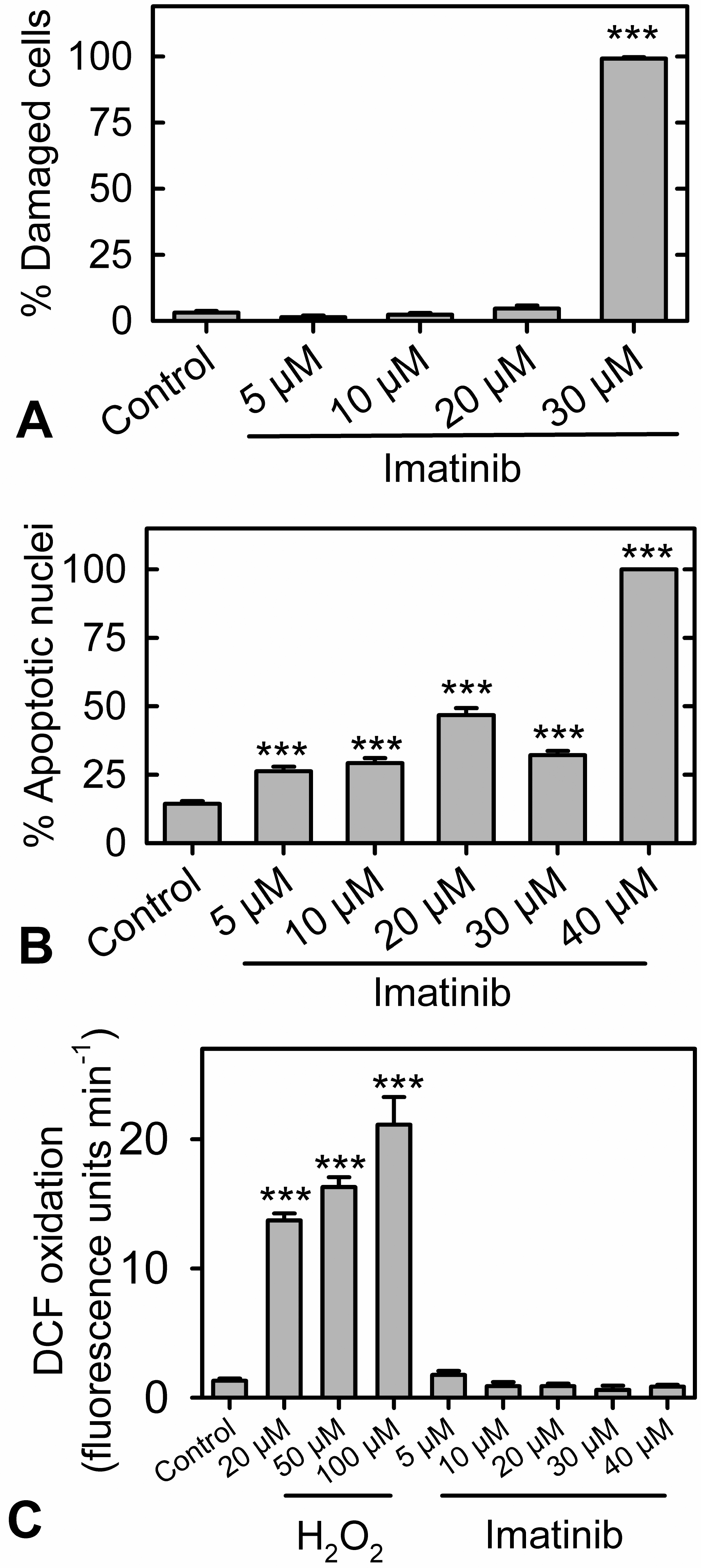
(A) As measured by anti-α-actinin antibody staining, imatinib damaged cardiac myocytes. Myocytes were continuously treated with the concentrations of imatinib indicated for 72 h and were scored blind for myofibrillar disorganization. (B) Scoring of nuclear-stained myocytes for changes in nuclear morphology characteristic of apoptosis showed that a 72-h imatinib treatment at all concentrations of between 5 and 40 μM significantly increased the percentage of apoptotic nuclei (p < .001) compared with untreated control myocytes. (C) In these experiments, 2′,7′-dichlorofluorescin-loaded attached
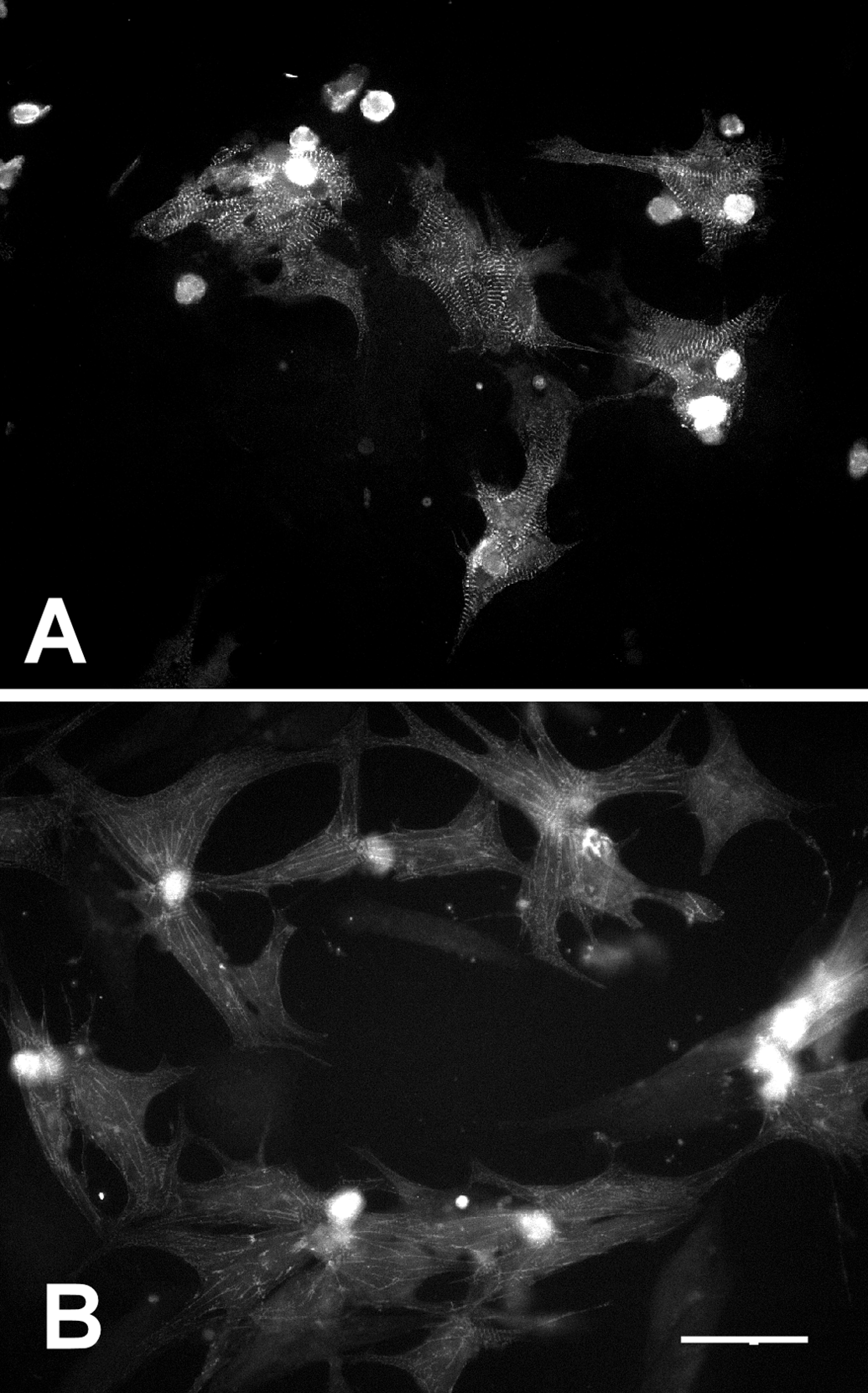
Epifluorescence photomicrographs of fixed attached α-actinin antibody-stained rat neonatal cardiac myocytes showing damage due to imatinib treatment. (A) Control untreated myocytes. (B) Myocytes treated with 30 μM imatinib for 72 h. While the untreated myocytes show well-defined sarcomeres, the imatinib-treated myocytes show highly disrupted sarcomeres. The bar in the lower right corner is 50 μm for both images.
Effect of Imb on Oxidation of Intracellular 2′,7′-dichlorofluorescin in Myocytes
To investigate whether Imb directly or indirectly produced oxidative stress in myocytes, the oxidation of 2′,7′-dichlorofluorescin loaded into myocytes was followed in a fluorescence plate reader. Although 2′,7′-dichlorofluorfluorescin is often used as a measure of reactive oxygen and nitrogen species production, some drugs can directly oxidize 2′,7′-dichlorofluorfluorescin (O’Malley, Reska, and Britigan 2004). Whereas exposure to H2O2 caused significant increases in the rate of 2′,7′-dichlorofluorescin oxidation (p < .0001), exposure to Imb at concentrations up to 40 µM did not (Figure 5C). Thus, at least over the short times of these experiments, the toxic effects of Imb on myocytes were not caused by oxidative stress.
Discussion
Experimental studies have reproduced certain features of the clinical Imb-induced cardiac toxicity. Kerkela et al. (2006) treated mice with Imb 200 mg/kg/d for 3 to 4 wk and reported alterations in left ventricular function (decreased left ventricular ejection fraction and dilatation). Also, transmission electron micrographs of cardiac tissue from mice treated with Imb 50 to 200 mg/kg daily for 3 to 6 wk showed morphologic changes in the sarcoplasmic reticulum (dilatation and membrane whorls) and mitochondrial abnormalities similar to those observed in patients (Kerkela et al. 2006). Impaired cardiac function in mice treated with 50 mg/kg Imb daily for 6 wk was also reported by Fernandez et al. (2007), who detected evidence of cardiac hypertrophy (increases in brain natriuretic peptide mRNA levels in the ventricles from mice treated with Imb).
Imb also elicits adverse cardiac effects in rats. Saad, Alkharty, and Arafah (2005) found that Imb induced mild, myocardial lesions in the hearts of Wistar rats treated with 30 mg/kg Imb for 10 d. Our study protocol included some similarities (in a 10-d experiment that included the 30-mg/kg-Imb dose) and differences (in a 14-d experiment that included higher Imb doses administered to both hypertensive and normotensive strains of rats) to that of Saad et al. (2006). In SHRs, we found that cardiac toxicity was the most consistent adverse effect of Imb treatment at 10, 30, and 50 mg/kg over 10 d. A unique observation was that myocardial lesions were present in animals from all treatment levels. All 10 animals from groups treated with the 30- or 50-mg/kg doses had similar and mildly severe myocardial alterations (mean lesion scores of 1.5). Our detection of cardiac alterations in animals dosed with 30 mg/kg Imb corroborates the findings of Saad, Alkharty, and Arafah (2005) and confirms the suitability of the rat model for investigating Imb-induced cardiotoxicity. Importantly, we also detected minimal-to-mild alterations in the hearts from three of five SHRs receiving 10 mg/kg of Imb. This dose appears to be a threshold because it is the lowest reported amount of Imb given to either mice or rats that induced changes myocyte morphology.
The characteristics of Imb-induced cardiac alterations observed under light microscopy and examination of ultra-thin, toluidine blue–stained slides was more comprehensive than reported previously and included necrosis, cytoplasmic vacuoles, and loss of myofibrils. These alterations were similar in both SHRs and SD rats. Wolf et al. (2010) have also reported myocyte necrosis in rats treated with higher doses of Imb for 28 d (120 or 200 mg/kg). In the present study, immunohistochemical analysis of hearts from SHRs treated with Imb revealed three types of alterations indicative of compromised myocyte viability: necrosis, autophagy, and apoptosis. Increased nitrotyrosine reactivity was observed in hearts from Imb-treated animals. The increased areas of reactivity were mainly in the cytoplasm of myocytes undergoing necrosis. The formation of powerful oxidants, such as peroxynitrite, could be a factor in the pathogenesis of Imb-induced myocyte necrosis (Mehlhorn et al. 2003). However, no evidence of reactive oxygen species generation was observed when isolated myocytes were exposed to Imb.
The in vivo and in vitro environments differ in that isolated myocytes were not exposed to certain potentially critical circulatory nitric oxide synthase–initiating factors (cytokines, such as tumor necrosis factor-α, interleukin-6, interleukin-2; Kooy et al. 1997). The two studies were similar regarding identification of a second type of myocyte alteration, apoptotic activity. The incidence of cardiac apoptosis, as determined by TUNEL staining, was greater in Imb-treated rats than control rats. Apoptotic activity was also greater in isolated rat neonatal myocytes exposed to a low (5 µM) concentration of Imb. These findings show for the first time that treatment with Imb can induce apoptotic myocyte death in vivo and also corroborate that of a report in which exposing isolated cardiac myocytes to 5 µM Imb induced several markers of apoptosis (Kerkela et al. 2006).
Positive staining with the LAMP-2 antibody indicated that Imb induced a third type of myocyte cell death. The presence of increased LAMP-2 is thought to be an indication of increased phagocytic activity (Saftig, Beertsen, and Eskelinen 2008). Formation of cytoplasmic vacuoles was a common myocyte alteration in the present study, and in many instances, the increased LAMP-2 staining appeared to be associated with these vacuoles. Autophagy has not been reported previously in Imb-based in vivo studies. However, TKI-induced autophagy has been identified as an important pathogenic mechanism causing cell death in H9c2 cardiac cells exposed to sunitinib (Zhao et al. 2010). Evidence of myocyte necrosis, apoptosis, and autophagy has been detected in the failing human heart (Kostin et al. 2003; Kostin 2005). Any or all of these three mechanisms of cell death could be involved in the development of heart failure seen in patients treated with either Imb or sunitinib (Kerkela et al. 2006; Chu et al. 2007; Tiribelli et al. 2008).
The cardiotoxic actions of Imb are thought to be mediated through suppression of fusion protein BCR-ABL activity, one of its tyrosine kinase targets (Force et al. 2007). The myocyte mitochondria have often been mentioned as a cellular site that is adversely affected by exposure to TKI. In mice, both Imb and sunitinib (a similar TKI) inhibit kinase pathways and induce changes in mitochondrial morphology sufficient to open the mitochondrial permeability transition pore. TKIs also initiate events upstream of the mitochondria, which result in activation of the cardiac endoplasmic reticulum stress response (Kerkela et al. 2006; Chu et al. 2007). Alterations in mitochondrial structure were found by transmission electron microscopy in human cardiac biopsy samples (Kerkela et al. 2006; Chu et al. 2007). Furthermore, exposure of rat myocytes to Imb was found to cause changes in the mitochondrial membrane potential and cell viability (Freebern et al. 2007). In the present studies, Imb-induced mitochondrial alterations were not detected by light microscopy. In contrast with our study with doxorubicin (Thompson et al. 2010), we observed no enrichment in the genes associated with mitochondrial function among the changes in gene expression associated with heart lesions in Imb-treated rats. However, the levels of several cardiac genes associated with the endoplasmic reticulum stress were higher in the hearts from SHRs treated with Imb.
One of the genes upregulated by Imb treatment in this study is DNAJA1A, an inhibitor of apoptosis induced by the C/EBP homologous protein (CHOP; Gotoh et al. 2004). CHOP is a proapoptotic transcription factor that regulates apoptosis initiated by endoplasmic reticulum stress (Minamino and Kitakaze 2010). Imb cardiotoxicity has been associated with activation of the endoplasmic reticulum stress response in vivo and in vitro, as measured by increased phosphorylation of eukaryotic translation initiation factor 2α (Kerkela et al. 2006). The endoplasmic reticulum stress response has been implicated in initiating myocyte death pathways, such as those detected in the present study (Minamino and Kitakaze 2010).
Expression of genes related to myocardial vascular development and remodeling was also elevated by exposure to Imb. These effects, which are similar to those reported by Wolf et al. (2010), may be the result of an interaction between Imb and one of its myocardial tyrosine kinase targets (the receptors for platelet-derived growth factor). In the cardiovascular system, platelet-derived growth factors are involved in the response of endothelial and smooth muscle cells to injury and other stressors (Raines 2004). These findings tend to indicate that Imb-induced cardiotoxicity involves multiple pathways, which may include alterations in endoplasmic reticulum stress or other downstream processes.
The Imb-induced changes in myocyte morphology detected in the present study appear to be analogous in certain respects to those observed in the hearts of animals treated long term with doxorubicin. In SHRs, doxorubicin induces a dose-related myocyte degeneration consisting primarily of intracellular vacuolization and loss of myofibrils (Herman et al. 1985). In the present study, somewhat similar changes in myocyte morphology were seen in rat hearts after Imb treatment. In vitro studies also suggest that the two agents can induce similar types of myocyte alterations. Isolated myocytes, when exposed to Imb, exhibited a loss of myofibrils and a disorganization of sarcomeric alpha-actinin. These changes are similar to those observed after exposing isolated myocytes to doxorubicin (Hasinoff, Patel, and Wu 2007).
Doxorubicin exerts several potentially cardiotoxic effects, of which the generation of reactive oxygen species is thought to be a major contributing factor (Sawyer et al. 2010). In experimental and clinical studies, dexrazoxane, an intracellular iron chelator, protects against doxorubicin cardiotoxicity (Herman, El-Hage, and Ferrans 1988; Lipshultz et al. 1997). This agent also attenuates isolated myocyte damage resulting from a 3-h exposure to doxorubicin (Bernabe et al. 2002; Hasinoff, Schroeder, and Patel 2003). As measured by oxidation of dichlorofluorescin, doxorubicin induces formation of reactive oxygen species in cardiac myocytes (Hasinoff, Schroeder, and Patel 2003; Sarvazyan 1996). Dexrazoxane pretreatment reduces both doxorubicin- and H2O2-induced dichlorofluorescin oxidation in cardiac myocytes (Hasinoff, Schroeder, and Patel 2003). In contrast, as we note in the present study, reactive oxygen species were not detected in isolated myocytes exposed to concentrations of Imb as high as 40 µM. We have previously shown that 100 µM dexrazoxane pretreatment reduced doxorubicin-induced LDH release in myocytes (Hasinoff, Schroeder, and Patel 2003). However, in the present study, exposing isolated myocytes to dexrazoxane did not protect against the toxic effects of Imb. The hearts of Imb-treated SHRs did show increased nitrotyrosine immunoreactivity in association with necrotic myocytes, indicating a potential role for the powerful oxidant peroxynitrite in the pathogenesis of this type of cell death. Formation of this oxidant could also stimulate the other pathways of cell death we detected here (apoptosis and autophagy; Nishida, Yamaguchi, and Otsu 2008; Nishida et al. 2009). Thus, the similarity of doxorubicin- and Imb-induced changes in myocyte morphology may be caused, in both instances, by reactive oxygen radicals that are generated by different pathways.
The possible role of hypertension in modifying the severity of Imb cardiotoxicity was explored in adult SHRs. Systemic arterial pressure is significantly elevated and reaches a peak by 12 to 14 wk of age in these rats (Herman et al. 1985). We compared the effects of a 14-d Imb treatment (at both 50 and 100 mg/kg) in SHRs and in normotensive SD rats. Both doses of Imb altered the myocardium in all treated animals. At both the 50- and 100-mg/kg doses, the effect of Imb on the heart was more profound in SHRs. This finding is not unique, in that SHRs are more sensitive than normotensive rats to the cardiotoxic effects of doxorubicin (Herman et al. 1985). Clinically, hypertension has been cited as a potential risk factor for doxorubicin-induced cardiotoxicity (Von Hoff et al. 1979). Our results suggest that hypertension may also be a risk factor for Imb cardiotoxicity.
An important issue in this and other experimental studies concerns the doses of Imb used and whether they are relevant to those used clinically. Peng et al. (2004) reported that the mean maximum concentration (sds) achieved in patients after a single oral dose of 400 mg Imb was 2.6 (0.79) µg/mL. The mean maximum concentration in rats treated orally with 120 mg/kg Imb was approximately 7.43 µg/mL (Wolf et al. 2010). We did not monitor Imb concentration after administering the various doses of Imb in the present studies. Because the cardiotoxic doses were between 58% and 88% less than the 120-mg/kg dose, we believe that the blood levels of Imb would be considerably lower and could be in the range of those found in clinical studies.
Concentration is also important in the degree to which Imb directly alters neonatal rat ventricular myocytes. Wolf et al. (2010) detected marked LDH release and increased myocyte apoptosis only after the Imb concentration reached 40 µM. Will et al. (2008) noted that Imb did not affect mitochondrial function at clinically relevant concentrations. In these studies, exposure was limited to 24 h. In our study, exposing myocytes to various concentrations of Imb progressively increased the release of LDH. Importantly, this release was statistically significant at a concentration as low as 5 µM after 72 h of exposure. These results are similar to those reported after exposure to 5-µM Imb for a 26 h, when release of cytosolic adenylate kinase was used as a marker of myocyte damage (Kerkela et al. 2006). In another study, exposing cardiac myocytes to a concentration of Imb below that achieved clinically (2 µM) for 72 h also caused significant release of LDH (Hasinoff 2010). In addition, the level of myocyte apoptosis was significantly higher than that observed in nontreated controls after 72 h of exposure to concentrations between 5 and 40 µM Imb. Thus, both in vivo and in vitro studies indicate that clinically relevant concentrations of Imb can alter the myocardium.
Despite the relatively consistent occurrence of myocyte alterations reported in this and other studies, the overall clinical relevance of these experimental findings remains an important question. The initial toxicity reported by Kerkela et al. (2006) included changes in both cardiac structure and function in patients treated with Imb. However, other studies found a low clinical incidence of cardiotoxic symptoms (Atallah et al. 2007; Verweij et al. 2007; Perik et al. 2008; Trent et al. 2010; Estabragh et al. 2011). Except for Estrabagh et al. (in press), these relatively negative studies were retrospective and did not consistently include comprehensive cardiovascular evaluations. As a result, part of the discrepancy in the occurrence of cardiotoxicity may be related to limitations of the diagnostic methods used to evaluate cardiovascular function in Imb-treated patients.
Even when a study specifically includes procedures for cardiac evaluation, diagnostic techniques, such as echocardiography or radionuclide ventriculography, may not always be sensitive enough to detect the early stages of myocyte injury or loss at a time when cardiac function has not yet deteriorated (Ganz et al. 1996). Newer, noninvasive technologies, such as monitoring serum concentrations of cardiac-specific biomarker proteins such as cTnI and cTnT do not rely solely on changes in function and are thought to be more sensitive for detecting early cardiac injury. In both clinical and experimental studies, cardiac biomarkers, such cTnI and cTnT, have provided evidence of early doxorubicin-induced myocardial injury (Cardinale et al. 2000; Herman et al. 1999).
Information is limited as to whether monitoring cardiac biomarkers might also be useful in detecting TKI-induced cardiotoxicity. Chu et al. (2007) found that levels of cTnI rose modestly in 12 of 68 (18%) patients treated with sunitinib. In the present study, we monitored cTnI levels with an ultrasensitive assay. With this assay, a baseline concentration of cTnI was detected in every rat. Control baseline cTnI levels were higher in SHRs than in SD rats. A dose-related increase in cTnI levels was found in both SHRs and SD Imb-treated rats. However, because of the variability in cTnI concentrations in the treatment groups, the differences in mean levels of cTnI were not statistically significant. Wolf et al. (2010) also reported that levels of cTnI increased in some Imb-treated rats. Serum concentrations of cTnI did increase after exposing SD rats to a single high dose of a different TKI (PF-04254644; Aguirre et al. 2010). In this instance, increases in heart rate and contractility preceded necrotic degeneration of the myocytes.
Other cardiac proteins, such as natriuretic peptides, have been suggested as potential biomarkers of TKI cardiotoxicity. In two clinical studies, BNP concentrations did not change markedly after Imb treatment (Perik et al. 2008; Tiribelli et al. 2008). Additional studies will be needed to determine whether cardiac biomarkers, such cTnI, cTnT, or BNP, can be as useful in diagnosing TKI-induced myocardial damage as they are in detecting numerous other cardiac conditions and thereby provide a more accurate assessment of the true incidence of Imb cardiotoxicity.
Limitations of the Study
The present study has several limitations that could be addressed in future experiments. The exact concentrations achieved at the various doses of Imb were not determined. Such an analysis would give a better indication of the correlation between Imb concentrations and incidence of cardiotoxicity. Cardiac alterations were detected at doses between 10 and 100 mg/kg in the present study. We did not determine a noncardiotoxic dose in either SHR or SD rats. We did not do ultrastructural analysis of the mitochondria in this study to verify the lack of mitochondrial gene expression changes after exposure to Imb. The number of animals used in certain groups was small. An increase in the number of rats evaluated in treatment and control groups would provide a more robust determination of whether cTnI or other substances would be useful biomarkers of Imb-induced cardiotoxicity.
Conclusions
In conclusion, Imb at low concentrations was found to exert cardiotoxic activity in both in vivo and in vitro experiments. The toxicity consisted of myocyte degeneration (cytoplasmic vacuolization and myofibrillar loss) and cell death (necrosis, apoptosis, and autophagy) after 10- to 14-d treatment. Myocyte lesions were more severe in SHRs than in SD rats. Dose-related increases in expression of several cardiac genes were observed. Isolated myocytes were adversely affected by exposure to low concentrations of Imb. The cardiotoxic actions of Imb appear to be complex, as indicated by the multiple types of alterations observed in the present study.
Footnotes
The authors declared no potential conflicts of interests with respect to the authorship and/or publication of this article. The authors declared the following source of financial support for the research and/or authorship of this article: Dr Hasinoff received a grant from the Canadian Institutes of Health Research, the Canada Research Chairs Program.
The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any agency determination or policy.