
Abstract
Choristomas are rare benign developmental lesions composed of histologically normal tissue located in an abnormal (ectopic) anatomical site. Their occurrence in the middle ear is uncommon and may mimic more common pathologies such as chronic otitis media or cholesteatoma, often leading to diagnostic challenges. We report a 16-year-old female who presented with chronic left ear discharge and hearing loss for 8 years. Examination revealed bilateral grade I microtia with a narrow external auditory canal. Pure tone audiometry showed bilateral conductive hearing loss, and high-resolution computed tomography demonstrated a soft-tissue mass in the left middle ear and mastoid with ossicular erosion. A transmeatal exploratory tympanotomy was performed, revealing an encapsulated small brown soft-tissue mass limited to the tympanic cavity complete excision done. Histopathological examination confirmed a mixed heterotopic tissue choristoma composed of skin, adnexal structures, fibroadipose tissue, muscle, cartilage, and salivary gland elements. The postoperative course was uneventful, with hearing improvement and no recurrence at 6-month follow-up. This case highlights the importance of considering middle-ear choristoma in the differential diagnosis of chronic otorrhea and conductive hearing loss to avoid misdiagnosis and unnecessary aggressive treatment.
Introduction
Choristoma and hamartoma are both benign developmental lesions composed of histologically normal tissue. They differ fundamentally in their site of origin as hamartomas are disorganized overgrowths of native tissues, while choristomas are normal tissues in abnormal (ectopic) locations.1,2 Thus, although both entities are nonneoplastic developmental malformations, their classification depends on whether the constituent tissues are normally present at the affected site.
Middle-ear choristoma are very rare lesions and most commonly consist of ectopic salivary gland tissue, representing the majority of reported cases.3 -5 Other forms of choristomas, which are less frequently, are composed of single heterotopic tissues such as glial or cartilaginous elements.6 -8 The rarest middle-ear choristoma type is the mixed heterotopic tissue form, characterized by the presence of multiple ectopic tissue types within the same lesion. 9 These lesions are consistently characterized as developmental heterotopias arising from embryologic misplacement rather than neoplastic processes.5,10
Middle-ear choristomas often present with long-standing otorrhea and conductive hearing loss, mimicking chronic otitis media or cholesteatoma. 11 Because of this overlap, a preoperative diagnosis is rarely established, and radiologic findings are often nonspecific. The final diagnosis relies on histopathological confirmation following surgical excision.12,13
Due to their rarity and overlapping features with other middle-ear pathologies may represent a diagnostic challenge for otologists. Therefore, documenting new cases and raising awareness of this entity are valuable for expanding understanding of its clinicopathological spectrum and assisting clinicians in differentiating it from more common middle-ear lesions.
Here we report a rare case of a mixed heterotopic tissue choristoma of the middle ear in a 16-year-old female, initially suspected to have cholesteatoma, and highlight the clinical, radiologic, intraoperative, and histopathological features that guided the final diagnosis.
Case Presentation
A 16-year-old female patient, known to have a psychiatric illness, presented to the otology clinic with a long-standing history of left-sided ear pus and progressive decreased hearing for 8 years. There was no tinnitus, vertigo, otalgia, or any prior history of ear surgery, trauma, or family otologic disease.
On physical examination, the patient had bilateral microtia grade I according to Marx classification. 14 Otoscopic examination revealed narrow external auditory canals on both sides. The left ear showed purulent discharge covering the tympanic membrane, while the right ear demonstrated an intact tympanic membrane with a similarly narrow canal. There were no facial nerve deficits, mastoid tenderness, or cervical lymphadenopathy.
Pure-tone audiometry revealed bilateral conductive hearing loss with an average air–bone gap of 40 dB (Figure 1). High-resolution computed tomography (CT) of the temporal bones demonstrated a soft-tissue density lesion occupying the tympanic cavity and mastoid air cells on the left side. There was bony erosion of the attic and ossicular chain (Figure 2). The right ear appeared radiologically unremarkable.
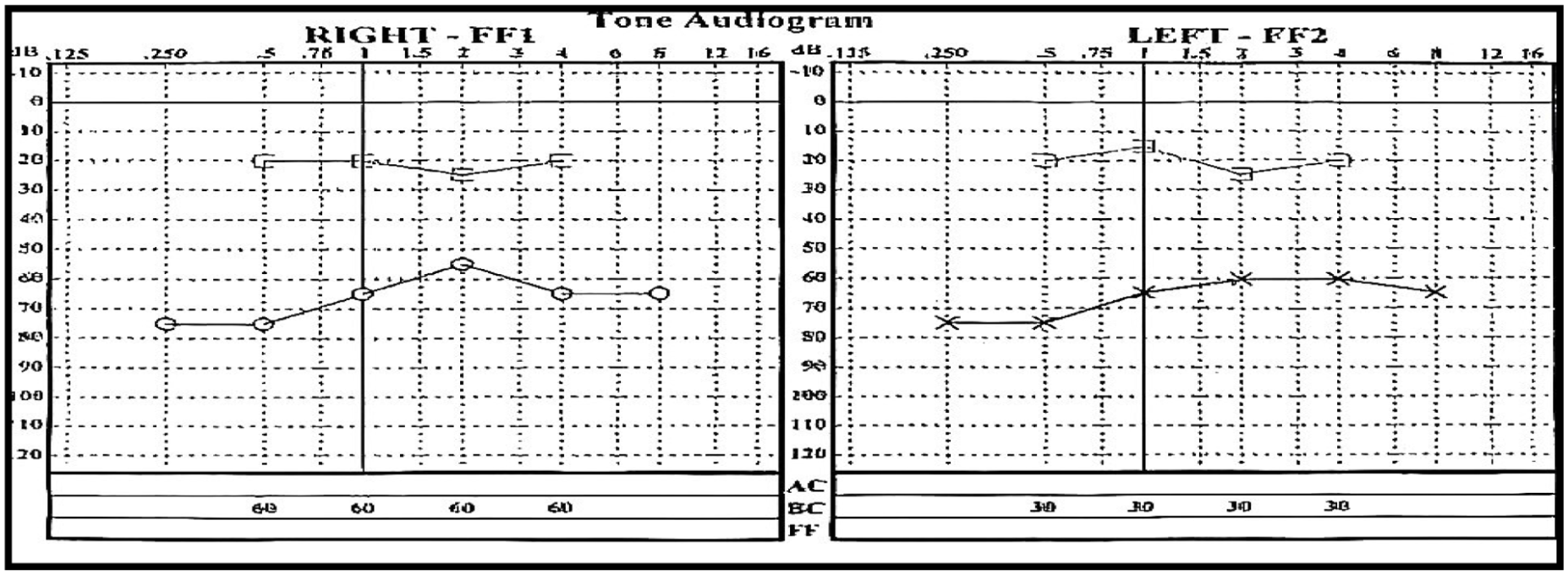
Preoperative pure tone audiogram demonstrating bilateral conductive hearing loss, with an average ABG of 40 dB on the left side. ABG, air–bone gap.
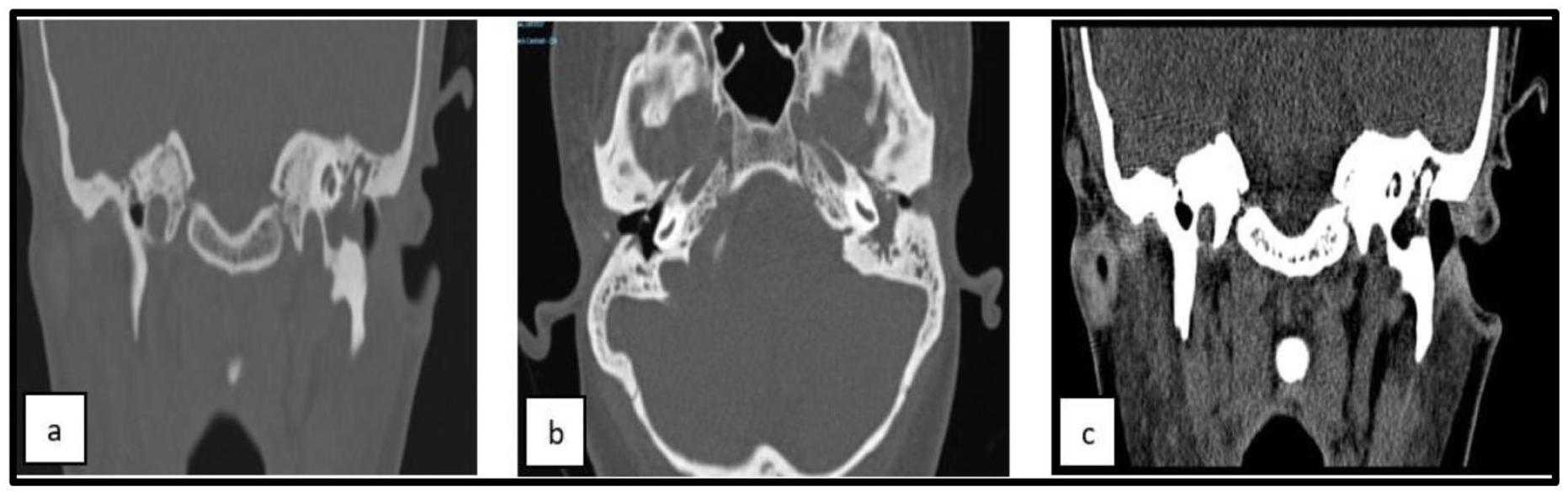
HRCT of the temporal bone. (a, b) Coronal sections and (c) axial section demonstrating middle-ear structural details. HRCT, high-resolution computed tomography.
Based on these findings, cholesteatoma was suspected, and a left middle-ear exploration was performed with the aim of complete lesion excision and ossicular chain reconstruction. The procedure was performed via a postauricle approach. Intraoperatively, the tympanic cavity was filled with a brownish soft-tissue mass limited to the middle ear. The long process of the incus was eroded, whereas the malleus and stapes were intact. The mass appeared atypical for cholesteatoma and was therefore completely excised and sent for histopathological analysis. A tragal cartilage graft was harvested for tympanic membrane reconstruction. The operation and postoperative course were uneventful.
Histopathological examination revealed a disorganized admixture of skin, skin appendages, fibroadipose tissue, salivary gland elements, skeletal muscle, and cartilage, findings consistent with a mixed heterotopic tissue choristoma (Figure 3).
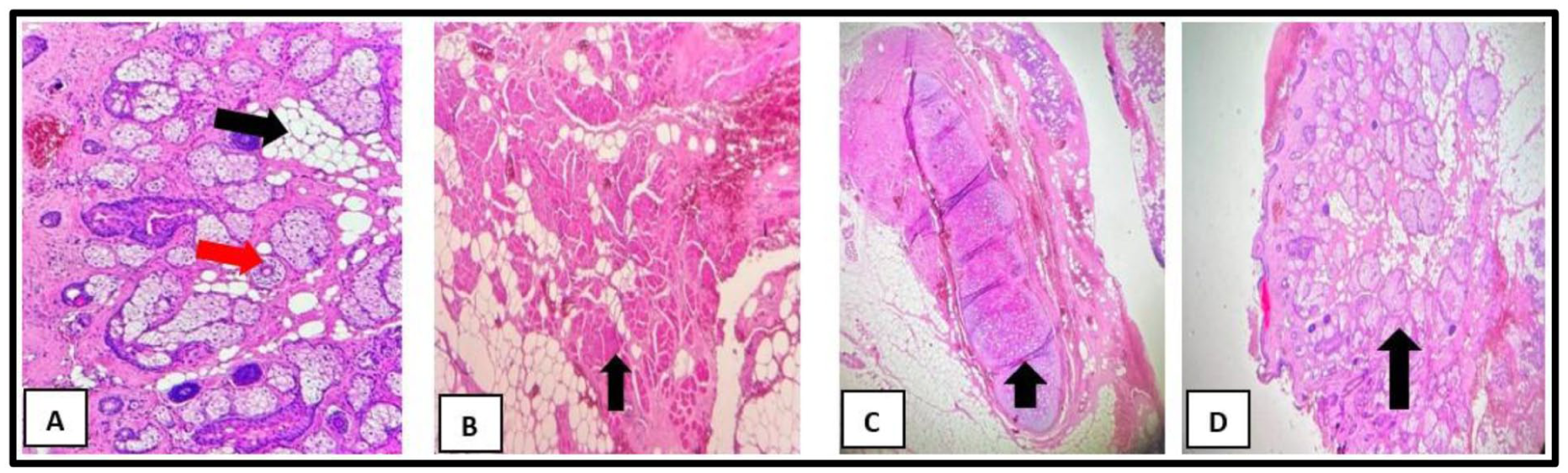
Photomicrographs of the excised lesion with hematoxylin and eosin (H&E). (a) Transverse section showing hair follicles (red arrow) and mature adipocytes (black arrow). (b) Well-differentiated skeletal muscle fibers (black arrow). (c) Lobules of mature cartilage (black arrow). (d) Developing salivary gland tissue (black arrow).
The patient was followed for 6 months with no evidence of recurrent ear discharge. A follow-up magnetic resonance imaging (MRI) study is scheduled at 1 year postoperatively to confirm the absence of recurrence, after which a second-stage ossiculoplasty is planned.
Written informed consent for the publication of this case report and accompanying images was obtained from the patient’s legally authorized representative, as the patient was a minor at the time of treatment.
Discussion
This case report describes a rare middle-ear mixed heterotopic tissue choristoma in a 16-year-old female with bilateral microtia who experienced 8 years of left-sided ear discharge, progressive conductive hearing loss, and bilateral microtia. Initially misdiagnosed as cholesteatoma, the exact diagnosis was established postsurgery, revealing a diverse tissue collection. Successful complete excision and ossicular reconstruction improved her hearing and resolved symptoms.
Despite the rare association between microtia and middle ear, heterotopic lesions are rare, Adam et al similarly hypothesized that bilateral microtia with aural atresia and rhabdomyomatous lesions containing salivary and muscle tissue might represent a novel clinical association with possible teratogenic or genetic origins. 15 Furthermore, other developmental heterotopic lesions, such as hypothalamic hamartomas, have been linked to multiple congenital anomalies, including poly- and syndactyly, cleft or high-arched palate, and nasal abnormalities. 16 These observations support the concept that aberrant embryologic development and tissue migration may underlie both congenital auricular malformations and heterotopic lesions of the middle ear. Accordingly, a high index of suspicion for middle-ear choristoma should be maintained in patients with congenital ear anomalies or persistent unexplained otorrhea.
Baget et al discussed a rare case of middle-ear lesion in a 5-month-old infant, which was similar to cholesteatoma and associated with ear discharge and conductive hearing loss, with the diagnosis confirmed histopathologically. 13 While CT imaging was performed in both cases, our patient demonstrated more extensive bony erosion of the attic and ossicular chain.
Surgically, we performed a transmeatal approach, followed by tympanic membrane reconstruction with a tragal cartilage graft, and partial ossicular reconstruction using cartilage fragments after removal of the incus. In contrast, Baget et al achieved tumor excision through a canal wall-up tympanoplasty, reflecting minimal ossicular involvement.
Postoperative outcomes were favorable in both cases, with no complications or evidence of recurrence. However, Baget et al noted widening of the mesotympanum and hypotympanum on follow-up CT imaging. 13
Similarly, canal wall up mastoidectomy with tumor excision was done in Baek et al which reported a comprehensive radiologic profile of a highly uncommon fibrous hamartoma of infancy (FHI) in a 26-month-old child, in which the diagnosis was significantly dependent on advanced imaging; preoperative temporal bone CT showed total opacification of the middle ear and mastoid air cells, while preserving the ossicles without erosion. 17 Furthermore, MRI elucidated the mass’s characteristics by displaying a unique heterogeneous architecture with signal intensities suggestive of intermingled fat and fibrous components, features that were essential in refining the differential diagnosis. The conclusive diagnosis was validated postoperatively through histological examination, which revealed the characteristic triphasic pattern of FHI, corroborated by immunohistochemistry (positive for CD34 and CD117).
Osipov et al reported 2 adult cases, aged 49 and 88 years, exhibiting cutaneous choristomatous lesions of the ear presenting as pedunculated nodules or papules. 18 Osipov’s diagnosis was based primarily on excisional biopsy and histopathological analysis, revealing distinctive cystic formations lined by stratified squamous epithelium, containing sebaceous glands, and surrounded by mesenchymal stroma. Unlike our study, Osipov’s cases lacked imaging specifics due to the superficial nature of skin tumors. Osipov’s cases were successfully treated with smooth recovery and no reported lesion recurrence during follow-up, with straightforward excisional biopsy of cutaneous nodules, eliminating the need for reconstructive surgery, as in our study.
Sanei et al reported a 49-year-old female exhibiting pain and mixed hearing loss, with high-resolution CT imaging indicating a highly aggressive temporal bone mass resulting in substantial bone destruction extending from the dura to the temporomandibular joint. 19 The diagnostic process was difficult, as an initial frozen section analysis by error indicated squamous cell carcinoma, leading to a radical resection; the conclusive diagnosis of ectopic meningothelial hamartoma (type II meningioma) was ultimately verified postoperatively through detailed histological examination, revealing meningothelial whorls and definitive immunohistochemistry demonstrating epithelial membrane antigen positivity and negativity for other markers such as cytokeratin and S-100. It’s important to note that the hamartoma age patterns vary dramatically by anatomical location, ranging from predominantly infant-onset to adult-onset conditions. 20
Overall, preoperative misdiagnosis is common, including conditions such as carcinoma or cholesteatoma, underscoring the need for histopathological examination, which varies significantly, as simple mixed tissue choristomas may be identified through histology alone, whereas fibrous, 17 or meningothelial 19 subtypes necessitate immunohistochemistry for validation. The preferred treatment is total surgical excision, generally employing conservative, anatomy-preserving techniques (e.g., transmeatal, canal-wall-up mastoidectomy). The extent of ossicular involvement directly influences the need for staged ossicular reconstruction, as demonstrated in the present case.
The key strengths of this case report are the comprehensive reporting of a rare middle-ear mixed heterotopic tissue choristoma from presentation through diagnosis, management, and follow-up, with audiometric outcomes. What distinguishes the present case is not only the rarity of middle-ear choristomas but the complex mixed heterotopic composition of the lesion. Unlike the majority of reported middle-ear choristomas, which typically consist of a single ectopic tissue type, most commonly salivary gland tissue, this lesion demonstrated a composite admixture of multiple heterotopic tissues, including salivary gland elements, skin and adnexal structures, skeletal muscle, fibroadipose tissue, and cartilage.
The main limitations of this case report are that the final long-term outcome is pending, as the patient had only a 6-month follow-up, and a crucial follow-up MRI is scheduled 1 year postoperatively to confirm the absence of recurrence before the second-stage ossiculoplasty can be performed.
Conclusion
Middle-ear choristomas constitute a range of benign lesions whose clinical manifestations frequently mimic more prevalent conditions, resulting in diagnostic challenges. Successful outcomes are achieved through total surgical excision, with the surgical approach and degree of reconstruction tailored to the individual’s intraoperative observations and the lesion’s specific characteristics. Collaboration between otologists and pathologists is essential for accurate diagnosis, effective management, and optimal long-term outcomes for these patients.
Footnotes
Ethical Considerations
The study was approved by the Institutional Review Board of Aseer Health Cluster Hospitals, Branch Aseer Health of Ministry of Health, Aseer region, Saudi Arabia (IRB: IRB-E31-2025). All procedures were conducted in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments.
Consent for Publication
Written informed consent for publication of this case report and accompanying images was obtained from the patient’s legally authorized representative, as the patient was a minor at the time of treatment. All reasonable efforts were made to protect patient confidentiality, and no identifying information is included in this publication.
Author Contributions
All authors read and approved the final manuscript and agree to be accountable for all aspects of the work.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article. Additional data are available from the corresponding author upon reasonable request, subject to appropriate ethical and legal considerations.