
Abstract
Myhre syndrome (MS) is a rare genetic condition that presents with multiple genetic anomalies including cleft lip and palate and Eustachian tube dysfunction. These patients are at a high risk for airway scarring from intubation and mucosal inflammation. Hearing loss (conductive or mixed, of varying severity) is a common comorbidity in these patients, the exact etiology of which is still unclear. We present the cases of 2 unrelated children with MS who suffered progressive mixed hearing loss from fibrosis and obliteration of the middle ear spaces. Both patients had multiple sets of ear tubes that demonstrated early extrusion. The older patient underwent bone conduction implantation at age 11 which resulted in dramatic improvement of speech recognition and interactive skills. The other younger patient demonstrates a similar trajectory but has not yet undergone implantation. Otolaryngologists should take a cautious approach to surgery of the eardrum and middle ear to avoid unnecessary induction of fibrosis in this susceptible patient population. These cases highlight a newly described etiology for hearing loss and suggest a benefit to bone conduction implantation.
Introduction
Myhre syndrome is a rare genetic condition with multiple features including cleft palate and Eustachian tube dysfunction. Hearing loss is a common comorbidity in patients with Myhre syndrome, the etiology of which is still unclear. It presents as conductive or mixed, bilateral, and of any degree of severity. Phenotypically, it has not yet widely been classified, but the presence of thickened tympanic membranes, mastoid sclerosis, and bilateral enlarged vestibular aqueducts have been individually reported.
We report the case of 2 unrelated children with Myhre syndrome and variable histories of congenital heart, craniofacial, and musculoskeletal anomalies, who developed severe middle ear scarring after ear tubes. To our knowledge, the present cases are the first known reports of severe middle ear fibrosis as an etiology for hearing loss in patients with Myhre syndrome.
Case Report
Case 1
A 12-month-old girl affected by Myhre syndrome was referred to our pediatric otolaryngology clinic for evaluation of chronic otitis media with effusion. The patient presented with history of cleft lip and palate, scoliosis, and coarctation of the aorta requiring open repair and was found to have presumed de novo deleterious variant in SMAD4 c.1499T>C (p.I500T). At 12 months of age, she was originally fit with hearing aids for a bilateral mixed hearing loss. She then underwent bilateral ear tube placement at 15 months of age during the second stage of her cleft palate repair.
Auditory brainstem response (ABR) was significant for moderate-severe mixed hearing loss on the right and moderate mixed hearing loss on the left. She later underwent several subsequent myringotomy and T-tube placements due to plugged or early extruding tubes. At 3 years of age, she underwent repeat sedated ABR which showed worsening in her hearing with the right ear progressing to moderate/moderately-severe mixed hearing loss and the left having moderately-severe/severe mixed hearing loss (Figure 1). In total, she had 7 sets of ear tubes before the age of 6.
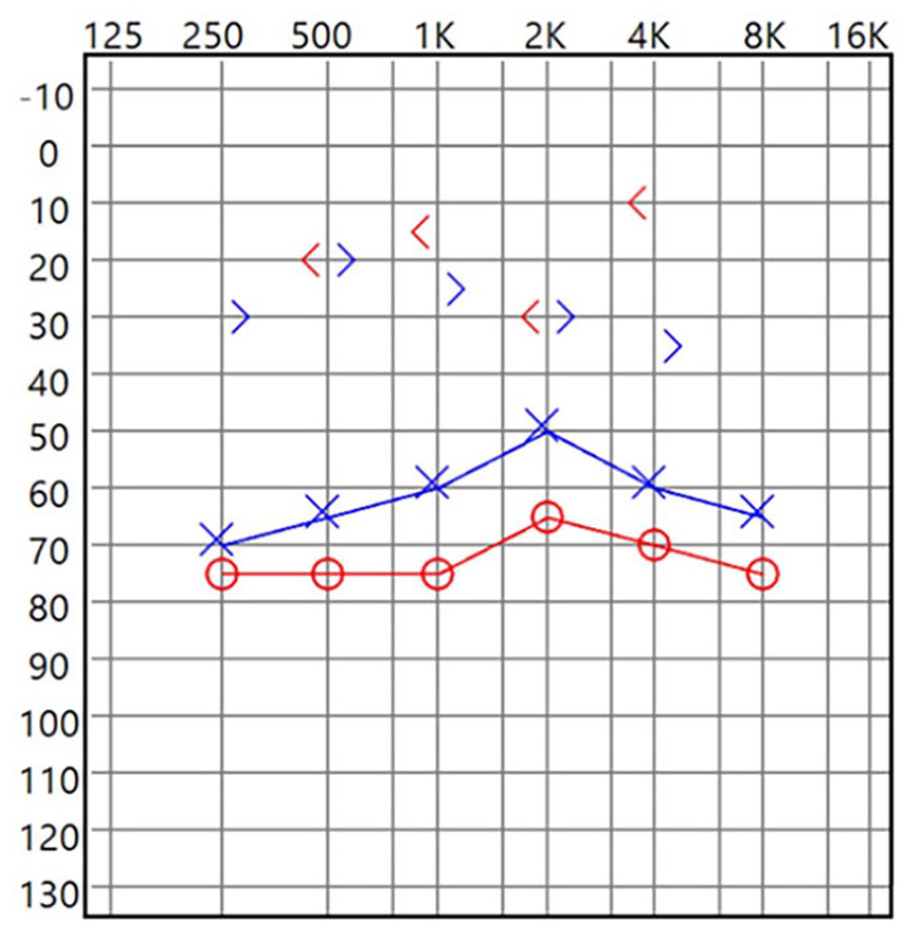
Preoperative audiogram.
She was then lost to follow-up due to family and health issues and re-presented at the age of 9 when she was noted to have a right-sided anterior perforation and posterior collapse with concern for cholesteatoma for which she underwent a tympanoplasty with cartilage graft. At this time, it was noted that the incus and malleus were rigid and malformed while the stapes superstructure was normal. The incus and head of malleus were removed to allow for improved visualization and t-tubes were placed at the conclusion of the case for improved ventilation. Postoperatively, she had early extrusion of her right tube. At age 11, she was taken back to the operating room for another attempted placement of ear tubes, at which time both middle ear spaces were found to be obliterated and scarred, making tube insertion impossible.
We then recommended proceeding with placement of a right-sided bone conduction hearing implant due to severe scarring of bilateral middle ear spaces as well as a worsening right ear canal scarring and bony stenosis. Computed tomography scan of temporal bone was obtained, which showed complete opacification of both middle ear cavities with severe scarring and sclerosis of the tympanic membrane as well as small sclerotic mastoids. The right tympanic membrane was replaced by bone (Figure 2); inner ear structures and facial nerves were normal bilaterally. She subsequently underwent implantation of right bone conduction implant (Med-El BCI 602) at the age of 13. After activation, she was noted to have a speech reception threshold of 20 dB, which was greatly improved from preoperative testing. While expressive communication is still limited due to developmental delay, her family has noted dramatic improvement in speech recognition and interactive skills.
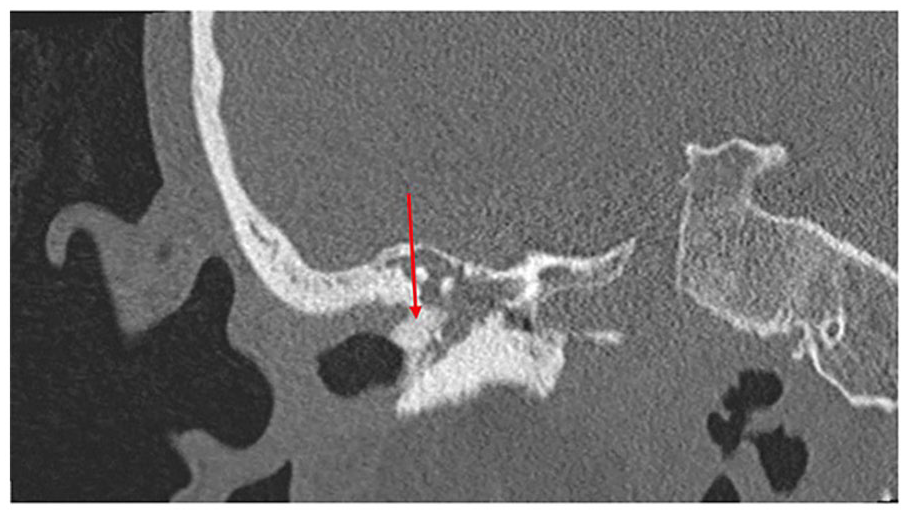
Coronal CT image demonstrating fibrosis and ossification of the right tympanic membrane (arrow). CT, computed tomography.
Case 2
A 20-month-old boy was referred to our clinic with multiple defining features of Myhre syndrome and a heterozygous pathogenic variant in SMAD4 c.1498A>G (p.Ile500Val). Notably, this is the same variant as case one expressed in modern nomenclature. He underwent ear tube placement and cleft lip repair at another facility, though bilateral ear tubes demonstrated occlusion on presentation. He would progress to have 4 sets of ear tubes in less than a 3 year period, all of which demonstrated early extrusion. While most recently his right TM appeared normal, his left TM appeared excessively scarred with possible effusion present. Follow-up audiograms supported the presence of worsening conductive hearing loss. After discussion with his mother, it was decided to avoid further ear tubes unless infection or retractions developed, as all tubes extruded early and the surgery may be inducing fibrosis. The family is tentatively planning for bone conduction implantation in the next few years.
Discussion
Myhre syndrome (MS, MIM 139210) is a rare autosomal dominant disorder characterized by a gain of function mutation in the SMAD4 gene. It was first described in 1981 by Myhre et al as a report of 2 unrelated males exhibiting growth and mental deficiencies, joint limitations, distinctive facial dysmorphias, muscular hypertrophy, and skeletal anomalies. 1 The clinical manifestations of this multisystem disorder are complicated by progressive, proliferative fibrosis affecting the heart (congenital heart defects, constrictive pericarditis, restrictive cardiomyopathy, long and short segment stenosis of the aorta and peripheral arteries, and arterial hypertension); lungs and airway (laryngotracheal narrowing, obstructive airway disease, restrictive pulmonary disease, and choanal stenosis); gastrointestinal tract (duodenal strictures, pyloric stenosis, severe constipation); and skin (sclerosis involving the hands and extensor surfaces).2-5 Facial dysmorphias include short palpebral fissures, a short philtrum, prognathism, or cleft lip and palate.
The SMAD4 gene encodes a protein that plays an important role in the signal transduction of transforming growth factor B (TGF-B) and bone morphogenetic proteins. The leading understanding of the pathogenesis underlying inheritance of this rare Mendelian gain of function trait was established via exome sequencing. Heterozygous de novo missense mutations in SMAD4 altering Ile500 in the MAD homology 2 domain are speculated to disrupt binding properties of the protein. 2 Only one case of Myhre syndrome with SMAD4 c.1486C > T (p.Arg496Cys) has been reported to date and was the only variant uniquely associated with familial recurrence. 6 The ability of the SMAD4 protein to integrate diverse signaling pathways underlies the expanded spectrum of phenotypes exhibited by patients with Myhre syndrome. 7 Early diagnosis of patients with Myhre syndrome is challenging and relies on observation of dysmorphic features in early childhood, but final diagnosis often occurs after clinical features become discernable in late childhood.
A less commonly studied manifestation of Myhre syndrome is hearing loss, the etiopathogenesis of which has not yet been deduced. Newborns with Myhre syndrome typically pass universal newborn hearing screening, with hearing loss becoming more apparent in early childhood through adulthood. 8 Detection of hearing loss in this population may be complicated by lack of established genetic diagnoses prior to presentation likely due to limited access to genetic testing and variability in phenotypic presentations. Lack of prompt hearing loss detection can contribute to further learning and behavioral difficulties, which may compound existing mild to moderate intellectual delay. It has been suggested that this hearing loss is a result of abnormalities in the cartilaginous and bony structures of the ossicular chain and bony labyrthinth. 9
Literature regarding specific hearing loss phenotypes in Myhre syndrome is minimal. Fifty-six known Myhre syndrome cases with reported hearing loss were evaluated by Yang et al and findings supported age of hearing loss onset in SMAD4 variant positive patients with Myhre syndrome to range from 6 months to 58 years old. Of these patients, conductive hearing loss accounted for 26.8% of cases (15 of 16), mixed hearing loss 17.9% of cases, and sensorineural hearing loss represented 10.8% of cases. Of the 56 cases, 30.4% suffered bilateral hearing loss, 1.8% reported unilateral hearing loss, and the remaining 67.8% of cases lacked this level of phenotypic information. 10
A case of a 58-year-old patient diagnosed with Myhre syndrome presenting with thickened tympanic membranes, mastoid sclerosis, and otospongiosis who underwent right-sided cochlear implantation (CI) is, to our knowledge, one of the first reports of hearing loss intervention in this subset of patients. Although the implant failed to evoke postoperative electrical potentials via medium-frequency electrodes, the speech perception in noise increased from 0% preimplantation with hearing aids to 50% postimplantation, suggesting a positive impact on quality of life and opportunity for intervention in these patients. 9
To our knowledge, the present cases are the first known reports of severe middle ear fibrosis as a cause for hearing loss in patients with Myhre syndrome. These cases demonstrate clinical progression of the disease with progressive ear tube placement and subsequent early extrusion. Operative treatment of patients with Myhre syndrome is to be approached carefully to prevent unnecessary induction of fibrosis of the airway, or in this case, the middle ears. The female patient’s success following bone conduction argues for alternative management of mixed hearing loss for this subset of patients with middle ear fibrosis, which will eventually be performed in the male patient as well. Early intervention and hearing enhancement for these patients should enhance learning ability and allow an opportunity for prevention of developmental delay.
Footnotes
Acknowledgements
The authors have no acknowledgments.
Author Contributions
Alia Tayara: Investigation, writing—original draft, writing—review; Laura G. Hendon: Conceptualization, investigation, writing—review and editing; Shelby C. Barrera: Conceptualization, investigation, writing—original draft, writing—review and editing, visualization, supervision; and Jeffrey D. Carron: Conceptualization, validation, writing—review and editing, supervision.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Statement
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent/Patient Consent
N/A.
Trial Registration Number/Date
N/A.
Data Availability Statement
All data underlying the results are available as part of the article and no additional source data are required.