
Abstract
Crystal structures are naturally represented as graphs, making Graph Neural Networks (GNNs) a powerful tool for capturing complex atomic interactions and geometric relationships. This review summarizes recent advances in GNNs-based representation learning for crystal materials, with a specific focus on addressing the critical challenge of passive symmetry. We critically analyze existing frameworks by classifying them into asymmetric and symmetric paradigms, evaluating how they address periodic invariance and geometric completeness through graph construction and architectural design. We also compile key datasets and benchmarks to provide a systematic performance comparison of representative models. Finally, we discuss unresolved challenges, including the trade-off between architectural rigor and computational efficiency, modeling complex non-ideal systems, and predicting high-order tensorial properties, highlighting promising directions for future research.
Keywords
1. Introduction
Accurate property prediction of crystalline materials is critical for industries such as semiconductors and renewable energy.1–7 Since properties are determined by crystal structures, characterizing these structures effectively is a central research focus.8–10 Conventional trial-and-error methods are often slow, costly, and labor-intensive, 11 limiting the efficient discovery of new materials. To overcome these issues, computational simulations have become essential. 12 Methods such as quantum mechanics-based density functional theory (DFT),13,14 molecular dynamics simulations,15,16 and finite element analysis17,18 have demonstrated effectiveness in predicting the physical and chemical properties of materials and guiding experimental design. However, these simulation methods often struggle to model complex and dynamic material environments, and their high computational complexity reaching up to O(n3) or even O(n7), 19 limits their practicality in new materials development.
Recently, machine learning (ML) has advanced rapidly in materials science,20–28 giving rise to material representation learning techniques. 29 Unlike traditional simulation-based approaches, ML-based methods can rapidly predict crystal properties by automatically learning latent features.4,30–32 This reduces the reliance on domain experts while improving computational efficiency and flexibility. 5 Moreover, these methods can capture complex nonlinear relationships in the high-dimensional materials data that are often beyond the reach of conventional techniques. 33 However, standard ML methods often fail to fully capture material microstructures, particularly regarding the passive symmetry of crystals.
To address this, GNNs have recently been introduced into materials science.34–40 GNNs are well-suited for processing graph-structured data, 41 and since chemical structures such as molecules and crystals can be naturally represented as graphs, they offer a natural framework for modeling atomic interactions. By learning representations of atoms (nodes) and bonds (edges), GNNs capture material patterns more effectively than traditional ML schemes.3,42,43 Consequently, GNNs have become a powerful tool for property prediction. Yet, designing GNNs that strictly respect crystal passive symmetry remains a major challenge. 44 Capturing passive symmetry is fundamentally important because it ensures that the predicted material properties remain invariant under coordinate transformations (such as rotation and translation), thereby adhering to the intrinsic physical laws of the crystalline state. Without explicit symmetry constraints, models are forced to relearn these invariances from vast amounts of data, often resulting in data inefficiency and poor generalization to unseen structures. While recent solutions have made progress, balancing computational efficiency with rigorous symmetry handling remains an active area of research.
Alongside the advances in macroscopic property prediction, the domain of Machine Learning Interatomic Potentials (MLIPs) for molecular dynamics has been revolutionized by a distinct class of strictly equivariant architectures. Foundational frameworks, such as e3nn,45–48 have catalyzed the emergence of state-of-the-art models, notably NequIP,49,50 MACE,51,52 and Allegro.50,53,54 Unlike descriptor-based models that rely solely on scalar distances or angles, these architectures utilize irreducible representations (irreps) of the O (3) group and spherical harmonics to perform message passing directly on geometric tensors. While this tensor-based approach elegantly preserves directional information and achieves unprecedented local accuracy for microscopic forces and energies, it introduces substantial computational overhead due to expensive tensor products. Although some pioneering macroscopic frameworks, have recently begun integrating similar tensor product operations to rigorously capture directional information and achieve geometric completeness, scaling these operations across infinite periodic graphs remains highly computationally demanding.
Therefore, rather than focusing on microscopic MLIPs, this review precisely scopes its discussion on computationally scalable GNNs frameworks designed for macroscopic properties. This review analyzes the issue of passive symmetry in crystal materials and surveys various GNNs-based frameworks within this scope. We also compile key datasets and benchmarks to provide a systematic performance comparison of representative models. Finally, we discuss unresolved challenges and outline future directions to further enhance the symmetry-awareness and utility of GNNs in materials science.
2. Passive symmetry challenge
Accurately capturing passive symmetries is a central challenge in predicting crystalline material properties. 29 These symmetries fundamentally encompass SE(3) invariance, SO(3) equivariance, and periodic invariance. This review focuses on GNNs-based methods that learn from geometric inputs, such as atomic coordinates and interatomic angles. In this section, we introduce the three principal passive symmetries of crystals and further discuss the critical concept of geometric completeness, which addresses the distinction between rigid-body symmetry and reflection invariance (chirality).
2.1. SO(3) equivariance of crystal materials
The SO(3) group comprises all rotations in three-dimensional space. A unit cell is the smallest periodically repeating unit of a crystal. For a crystalline material, SO(3) equivariance implies a specific consistency constraint: if the crystal’s unit cell is rotated, any vector or tensor properties derived from it (such as interatomic forces or dielectric tensors) must rotate accordingly. 55
In materials representation learning, this property ensures that predictions depend only on the material’s structure, not on its orientation in the coordinate system. This is vital for modeling directional properties, such as band structures or mechanical deformations. By enforcing SO(3) equivariance, models avoid errors caused by arbitrary coordinate choices, thereby improving predictive accuracy. Figure 1 illustrates this concept, showing an original unit cell (a) and its SO(3) rotated counterpart (b), which corresponds to a 270° rotation around the yellow axis as indicated by the pink arrow. SE(3) and SO(3) operations of crystal materials.
The structure of crystal materials will be described using the lattice basis vectors and the unit cells they form. Following the definition by Yan et al.,19,56 the following relationship can be obtained. For a unit cell with n atoms, the crystalline material
For a crystal material The SO(3) equivariance requirement states that after any rotation transformation, the representation of the crystal material and its corresponding physical and chemical properties should change correspondingly. That is, for any rotation matrix


2.2. SE(3) invariance of crystal materials
The Special Euclidean group, SE(3), consists of all translation and rotation operations in three-dimensional space. It is a subgroup of the Euclidean group, E(3), which includes translations, rotations, and reflections. The key distinction is that SE(3) excludes reflections. Furthermore, the SO(3) group (discussed in Section 2.1) is a subgroup of SE(3) containing only rotations. Due to its coverage of all translation and rotation transformations, SE(3) is crucial in domains like computer graphics,57–59 materials science,19,60–62 and robot kinematics. 63
SE(3) invariance dictates that rigid transformations (rotations and translations) of the unit cell must not alter the crystal’s intrinsic scalar properties. Quantities such as formation energy, band gap, and total energy must remain constant regardless of the crystal’s position or orientation in space.19,61 Critically, strictly distinguishing SE(3) from E(3) is essential for geometric completeness. E(3) invariance inherently enforces reflection invariance. Consequently, models built on E(3) invariance cannot distinguish chiral structures from their mirror images. To address this, an ideal framework should strictly satisfy SE(3) invariance, handling rigid transformations, while maintaining sensitivity to chirality. In summary, SE(3) invariance ensures that scalar physical properties remain unaffected by rigid motions, whereas SO(3) equivariance ensures that vector or tensor properties transform consistently with the structure. Figure 1(b) and (c) illustrate the SE(3) operations of rotating the unit cell (specifically, a 270° rotation around the yellow axis indicated by the pink arrow) and translating it (along the direction indicated by the purple arrow), respectively.
The following will provide the mathematical definition of the SE(3) invariance of crystal materials.
For a crystal material Then the mathematical definition of SE(3) invariance is:

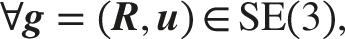


2.3. Periodic invariance of crystal materials
Periodic invariance implies that a crystal’s physical properties are independent of how its unit cell is defined. Since a crystal is an infinite periodic structure, there are infinite ways to select a repeating unit cell to represent it. While these selections change the numerical coordinates and lattice vectors, they describe the exact same physical material.56,64 Therefore, a robust model must produce identical predictions regardless of this mathematical choice.
Formally, we define this principle as follows:
The periodic invariance requires that for any possible representation ( Ensuring this invariance is vital for practical applications. Without it, a model might predict contradictory properties for the same material simply because the input structure was defined differently. This consistency is essential not only for static property prediction but also for tasks involving dynamic transformations and stress analysis. Figure 2 illustrates it using a two-dimensional example. The colored boxes represent different valid methods for selecting a unit cell from the same atomic arrangement (colored circles). It is evident that while the unit cell selection alters the mathematical description, the underlying crystal structure and its properties remain unchanged.


Periodic transformation of crystal materials.
2.4. Geometric completeness and chirality
While SE(3) invariance, SO(3) equivariance, and periodic invariance form the fundamental passive symmetries of crystals, a critical issue arises when considering the completeness of the representation, particularly regarding chirality. This challenge stems from the distinction between the SE(3) group and the E(3) group.
Most conventional crystal descriptors, such as bond lengths and angles, are scalars. Since scalars do not change under reflection, models relying on them are inherently E(3)-invariant. While this ensures stability, it creates a flaw: the model cannot distinguish a crystal from its mirror image (enantiomer), as both generate identical graphs. This is problematic for chiral crystals, where left-handed and right-handed structures are distinct physical entities with potentially different properties.
Therefore, a rigorous representation learning framework must satisfy geometric completeness. This concept requires the model to be strictly SE(3)-invariant while remaining sensitive to reflection. In simple terms, geometric completeness ensures the mapping from structure to representation is unique: distinct structures must map to distinct graphs. This property allows the model to avoid the “false symmetry” of reflection invariance, enabling the accurate distinction of chiral materials.
3. Framework for GNNs-based crystal material representation learning
Following the symmetry principles defined in Section 2, a robust representation learning framework must enforce SE(3) invariance, SO(3) equivariance, and periodic invariance. These constraints guide the design of the two core components of the framework: crystal graph construction and GNNs architecture design. 65
The crystal graph construction phase is the foundational step where molecules, crystal unit cells, and even complex polycrystalline systems are mapped into graph structures consisting of “atoms (nodes)” and “chemical bonds or spatial interactions (edges).” Crucially, this phase determines whether the passive symmetry constraints, especially periodic invariance, are rigorously preserved or compromised by implementation choices (e.g., neighbor selection strategies). Subsequently, the GNNs architecture must effectively encode these features while maintaining symmetry consistency between local and global representations.
Section 3 first presents the general prediction pipeline. It then critically examines graph construction strategies, focusing on the integration of passive symmetries. Finally, we classify and analyze representative Crystal GNNs architectures, comparing their design features and compliance with symmetry constraints.
3.1. A pipeline for GNNs-based crystal property prediction
Figure 3 illustrates the standard pipeline for GNNs-based crystal property prediction, comprising three key stages: (i) (ii) (iii) A pipeline for GNNs-based crystal property prediction.
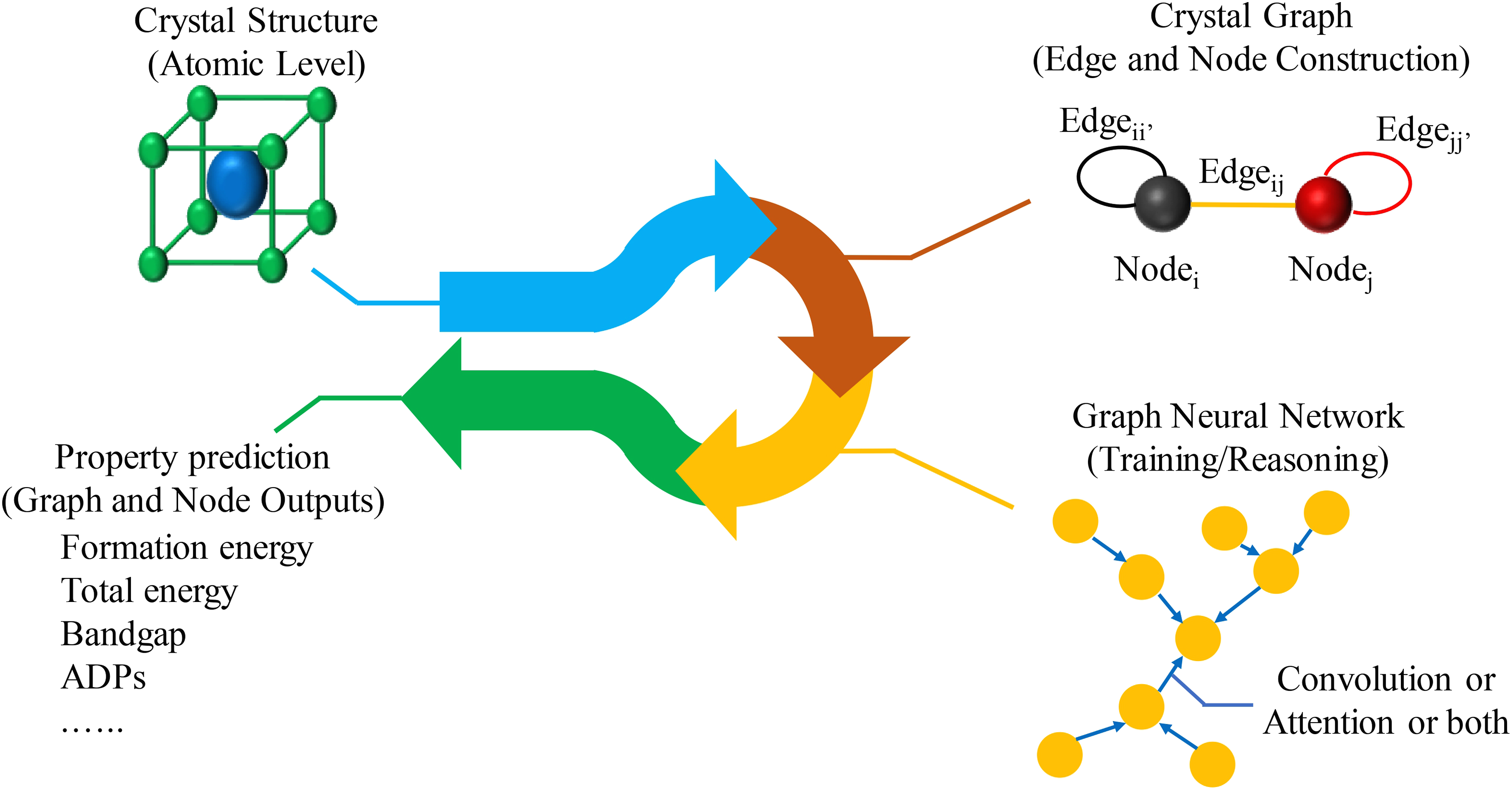
3.2. Evolution of crystal graph construction
Section 3.2 details the construction of crystal graphs in the pipeline. Crystal graph construction is a crucial step in translating physical entities into mathematical representations, and the quality of this construction significantly influences the performance of the crystal material representation learning framework. The fundamental elements of graph construction involve defining the nodes, edges, and the final mathematical representation of the graph. In molecular graph construction, the process is relatively straightforward: atoms serve as nodes, with their chemical properties as node features, and chemical bonds or spatial distances between atoms are represented as edge features.75–79 In crystal graph construction, however, the unique periodicity of crystal materials, along with their invariance to SE(3) transformations and equivariance to SO(3) transformations, presents a core challenge: integrating these constraints and features into the graph construction process.
3.2.1. Basic connectivity: nearest-neighbor multi-edge graphs
Prior work shows an evolution in crystal-graph construction from implicit representations to explicit modeling of SE(3)/SO(3) symmetries and periodicity. A seminal example, CGCNN, 34 introduced a multi-edge graph scheme coupled with nearest-neighbor search to encode the infinite periodicity of crystalline materials. Atoms in a unit cell are treated as graph nodes, with each node implicitly representing the atom and its infinite periodic copies along all lattice directions. The interaction between atom pairs needs to be defined by recording the Euclidean distance after considering the nearest neighbor condition or truncation condition, which will result in multiple edges representing the interaction between different periodic copies for the same pair of atoms.
Owing to its simplicity and efficiency, this multi-edge paradigm has been widely adopted in subsequent models, including MEGNET, 80 GATGNN, 81 iCGCNN, 82 and DeeperGATGNN. 83 However, it is critical to distinguish between the two primary neighbor selection strategies employed within this paradigm: radius-based cutoffs and fixed k-nearest neighbor (k-NN) search. Models like MEGNET typically employ a strict cutoff radius, where all atoms within a distance R c are connected. This strategy is deterministic and physically consistent. In contrast, standard implementations of CGCNN and GATGNN often rely on a fixed k-NN strategy for computational efficiency.
It is explicitly worth noting that this specific implementation choice, basing neighbor selection on hard cutoffs (fixed k), can violate periodic invariance in certain cases.
56
As shown in the left of Figure 4(a), the multi-edge construction with nearest neighbor search selects neighbors using a Voronoi face–sharing criterion. Each node connects to its neighbors via multiple edges, reflecting interactions with periodic copies of the same atomic species. Node features embed atomic properties (e.g., atomic number), while edge features encode geometric attributes (e.g., bond length). On the right of Figure 4(a), a counterexample is shown: nodes of the same color denote the same atomic species, and a central green atom is equidistant from four neighbors. Under a standard k-NN rule with a fixed cutoff k, the set of selected neighbors becomes ambiguous due to numerical tie-breaking dependence on atomic indexing. Consequently, different unit cell choices yield different graph topologies. Such local non-uniqueness disrupts global periodic consistency, alters the mapping to physical properties, and violates Equation (5) thereby failing to satisfy periodic invariance. Diagram of different crystal graph construction methods. (a) Basic connectivity: nearest-neighbor multi-edge graphs; (b) beyond connectivity: encoding higher-order geometry; (c) incorporating physical priors: Symmetry-aware graph construction; (d) beyond the unit cell: Graphs for long-range periodicity.
Crucially, this limitation is an artifact of the specific implementation adopted by these models (i.e., the use of hard cutoffs for computational efficiency) rather than a fundamental flaw in the graph representation itself. On the contrary, schemes based on deterministic physical criteria, such as a strict cutoff radius without neighbor count truncation (as seen in MEGNET) or an ideal nearest-neighbor graph including all equidistant neighbors, have been proven to strictly meet periodic invariance. 56 Therefore, the violations observed in the aforementioned works arise from the use of arbitrary neighbor truncation strategies in high-symmetry environments, rather than the multi-edge formalism itself. Nevertheless, since classical models like CGCNN and GATGNN default to these hard-cutoff implementations, they inherently fail to guarantee strict periodic invariance.
3.2.2. Beyond connectivity: Encoding higher-order geometry
Relying exclusively on pairwise distance information (i.e., two-body interactions) introduces limitations in capturing complex local geometric environments. To capture higher-order interactions, more geometric information has been incorporated into the graph construction process. For instance, ALIGNN 66 simultaneously considers distance and bond angle (three-body interaction), introducing the concept of a “line graph”. In a line graph, the edges of the original graph are converted into nodes, with edges in the line graph representing the relationships between corresponding edges in the original graph. This approach seeks to capture more intricate geometric relationships. Similarly, to encode three-body interactions, M3GNet 84 and CHGNet 85 employ advanced graph construction strategies based on cutoff radii. M3GNet incorporates bond angle information into its multibody computation module, while CHGNet introduces a novel dual-graph structure, using a secondary graph with “chemical bonds” as nodes and “bond angles” as edges to specifically model three-body interactions. As shown in Figure 4(b), these graph construction schemes that incorporate more high-order geometric information integrate bond angles as the main features into node or edge features. These graph construction strategies have made significant progress in enhancing the framework for representation learning of crystal materials to capture complex internal structural information of crystals.
Nevertheless, the effectiveness of these high-order representations in preserving global symmetry strictly depends on the underlying neighbor selection strategy. While geometric descriptors such as bond angles and bond lengths are inherently invariant to rigid-body rotation and translation, specific implementation choices can still compromise periodic invariance. This is particularly evident in models like ALIGNN, which typically define the graph topology using the k-NN algorithm (e.g., k=12) before computing bond angles. As detailed in Section 3.2.1, k-NN strategies introduce topological ambiguities in highly symmetric crystals where neighbor selection depends on arbitrary atomic indexing. Consequently, in the case of ALIGNN, different unit cell selections can lead to topologically different graphs, and thus different sets of computed bond angles, violating the strict Periodic Invariance (Equation (5)). In contrast, M3GNet and CHGNet utilize radius-based cutoffs, which theoretically preserve periodic invariance regarding connectivity, although they represent a different approach to balancing computational cost and symmetry constraints compared to the fully symmetric frameworks discussed in Section 3.3.2.
3.2.3. Incorporating physical priors: Symmetry-aware graph construction
Matformer, 56 a novel framework that sets six self-connection edges on a global node, with edge weights encoding the lengths of the three lattice basis vectors and the angles between them. This enables the model to directly capture macroscopic periodicity, or the periodic pattern, of the lattice. Additionally, this framework introduces two different graph construction strategies: one is a fully connected graph, and the other is an adaptive radius graph. In the latter strategy, the cutoff radius is dynamically determined by the distance of the k-th nearest neighbor (e.g., k=12). Crucially, Matformer includes all atoms within this adaptive radius, ensuring that equidistant neighbors are treated equally without arbitrary truncation. Both construction methods, combined with the lattice-encoding self-connections, have been designed to strictly satisfy Equation (5), ensuring the periodic invariance of crystal materials.
Building on this, ComFormer 19 further enhances the handling of crystal symmetries through a sophisticated “passive symmetry treatment”, ensuring the model’s geometric completeness. ComFormer offers two graph construction options: an SE(3)-invariant graph based on the two invariants of the bond-lattice vector angle and bond length, and an SO(3)-equivariant graph based on the invariant bond length and the equivariant bond vector. These options offer customized geometric representations for predicting and optimizing various material properties. Since the bond-lattice vector angle and bond length do not change with the overall rotation and translation of the crystal graph, nor do they vary with the selection of different unit cells, the SE(3)-invariant graph satisfies Equation (4) and Equation (5). Crucially, by explicitly encoding the angles relative to the lattice basis vectors, the graph construction in ComFormer ensures a bijective mapping between the crystal structure and its graph representation. This rigorously solves the “many-to-one” problem, thereby achieving geometric completeness for both SE(3)-invariant graph and SO(3)-equivariant graph. While iComFormer relies on complete invariant scalars, eComFormer utilizes equivariant bond vectors to explicitly capture directional information, fully realizing the three passive symmetries of crystal materials with higher expressivity, which will be discussed in Section 3.3.2.
Two recent works, PerCNet 86 and GCPNet, 87 have adopted similar strategies to address the issue of geometric completeness. PerCNet introduces more crucial dihedral angles to address the “many to one” problem (a lack of geometric completeness) existing in previous models (such as ALIGNN and GATGNN), where different crystal structures are mapped to the same crystal graph by the graph construction scheme. By resolving this problem, PerCNet achieves a geometrically complete representation. GCPNet, on the other hand, introduces the concept of “crystal pattern graphs”, which incorporate three elements: atomic types, bond types, and global descriptors, ensuring the passive symmetries of crystal materials from a global perspective. Figure 4(c) illustrates such symmetry-aware graph construction schemes that incorporating physical priors. These schemes utilize more geometric information of crystal materials, such as self-connection edges, the angles between bonds and lattice basis vectors, and dihedral angles. By combining this information with original node and edge features, these graph construction schemes can satisfy varying degrees of periodic invariance, SE(3) invariance, and SO(3) equivariance, while significantly improving geometric completeness.
3.2.4. Beyond the unit cell: Graphs for long-range periodicity
This section introduces graph construction strategies that focus on how to effectively handle and capture the long-range periodic interactions within crystals. These efforts leverage the “Ewald summation” concept from physics and computational materials science to address the long-range interactions arising from periodicity. For instance, PotNet 88 avoids the information loss caused by distance truncation by classifying the potential energy interactions between different atomic types in the lattice and introducing approximate functions to calculate the total sum of each type of interaction throughout the infinite lattice, thereby encoding the long-range periodic effects of the crystal into node representations. In contrast, Ewald-MP 89 more directly applies the decomposition idea of Ewald summation, decoupling the information transfer into two parts: short-range signals processed in real space and long-range signals efficiently modeled and filtered through Fourier transformation in reciprocal (frequency) space, achieving a clear separation and capture of periodic structural information. As shown in Figure 4(d), this scheme integrates potential energy terms into edge or node features, thereby enhancing the physical rationality of the graph construction strategy.
While the evolution of these schemes reflects ongoing efforts in constructing complete and physically rigorous crystal graph representations, it is crucial to distinguish between their theoretical validity and numerical implementation. Theoretically, methods based on Ewald summation are rigorously periodic and thus strictly satisfy Equation (5) (periodic invariance) by design, as they mathematically account for interactions across the infinite lattice. However, in practical implementations, the infinite summation series must inevitably be approximated via truncation or function fitting due to computational constraints. Consequently, the primary challenge for these methods lies not in a theoretical violation of symmetry principles, but rather in managing numerical stability and precision errors introduced by these necessary approximations.
Furthermore, graph construction strategies are often optimized for specific tasks. For instance, CartNet, 90 which is specifically designed for predicting Atomic Displacement Parameters (ADP) adopts a radius-based graph construction strategy. It relies solely on spatial distances when building graphs and disregards chemical bond information, focusing more on the packing effects of atoms within the lattice. This approach offers significant computational efficiency advantages. Crucially, unlike models relying on fixed k-NN logic, CartNet’s radius-based approach avoids neighbor ambiguity, thereby strictly satisfying Equation (5) and preserving the periodic invariance of crystal materials. Additionally, this method incorporates SO(3) data augmentation into the constructed graphs to align with the physical characteristics of ADP, satisfying Equation (2).
Regardless of the final graph construction strategy adopted, the crystal graph ultimately composed of nodes and edges needs to be encoded into a machine-readable mathematical form. The core of this is the adjacency matrix
3.3. GNNs architectures for learning on crystal graphs
The transformation of crystal graph representations into meaningful material properties relies on the design of Crystal GNNs. A key challenge in the architecture design of Crystal GNNs is how to accurately and completely capture the passive symmetry of crystal materials based on the input crystal graph. In this review, we classify Crystal GNNs into two categories: symmetric Crystal GNNs and asymmetric Crystal GNNs.
Crucially, this classification is not solely based on basic periodic invariance, but on a more rigorous criterion: whether the framework explicitly incorporates architectural designs to fully realize the passive symmetries. This entails strictly guaranteeing periodic invariance, while simultaneously achieving either geometric completeness (resolving the “many-to-one” ambiguity) or explicit global symmetry modeling (e.g., through lattice encoding or infinite summation), without relying on unstable local approximations.
3.3.1. Asymmetric crystal GNNs
Asymmetric Crystal GNNs primarily focus on characterizing local chemical environments but fail to strictly enforce the full spectrum of global symmetry constraints. These models generally fall into two sub-types based on their architectural choices.
The first sub-type includes foundational models focusing on local pairwise interactions. Early works such as CGCNN, 34 iCGCNN, 82 MEGNET, 80 GATGNN, 81 DeeperGATGNN, 83 and SchNet 91 laid the foundation. These models share similar input crystal graphs but demonstrate distinct architectural choices in message passing. SchNet pioneered the use of continuous-filter convolutions, where interatomic distances are expanded using radial basis functions (RBF) and processed by a filter-generating network to map continuous spatial positions to convolutional weights. In contrast, CGCNN and GATGNN introduce more refined control mechanisms. CGCNN specifically employs a gated convolution architecture involving a sigmoid-softplus unit, which dynamically regulates the information flow between atoms to mimic chemical bonding environments. GATGNN further advances this by utilizing attention scores to weight neighbors based on their impact. Critically, the symmetry compliance of this group varies by implementation. Models like CGCNN and GATGNN typically employ a fixed k-NN strategy for efficiency. As discussed in Section 3.2.1, this introduces topological ambiguities in high-symmetry crystals, directly violating periodic invariance (Equation (5)). Conversely, models like MEGNet typically use a radius-based cutoff, which avoids strict periodicity violations. However, they are still classified as asymmetric here because they conceptually reduce the infinite crystal to a localized chemical network, missing the explicit encoding of macroscopic lattice parameters required for global structural awareness.
The second sub-type includes models like ALIGNN, 66 M3GNet, 84 and CHGNet, 85 which integrate higher-order geometric information. These models employ advanced architectures to process edge features such as bond angles and bond lengths. ALIGNN integrates concepts from line graph GNNs, processing edge features with a line graph GNNs before handling node features with an ordinary GNNs to reason about crystal geometry from a third-order perspective. However, ALIGNN is classified as asymmetric because its graph construction relies on a fixed k-NN algorithm. This reliance causes the graph topology to change under periodic unit cell transformations, preventing it from achieving strict periodic invariance despite using invariant scalar features. In parallel, M3GNet and CHGNet utilize radius-based cutoffs and incorporate three-body interactions into their message passing (e.g., CHGNet’s dual-graph structure). While they technically satisfy periodic invariance regarding connectivity, these frameworks prioritize the capture of complex local atomic forces over geometric completeness. By not explicitly resolving the “many-to-one” mapping ambiguity, they remain susceptible to mapping distinct periodic structures to identical graph representations, placing them outside the strictly symmetric category.
3.3.2. Symmetric crystal GNNs
In contrast to conventional approaches, symmetric Crystal GNNs strictly guarantee periodic invariance and typically achieve either geometric completeness or explicit global symmetry modeling through rigorous architectural designs or physical formulations. These models can be broadly categorized into two streams: those based on geometric graph construction and those leveraging physics-informed global summation.
The first category focuses on geometric graph construction, unified by the explicit handling of crystal symmetries within their architectures. Matformer 56 exemplifies the integration of global periodicity into message passing. Unlike conventional models that rely solely on local neighbors, Matformer utilizes a specialized Transformer architecture with multi-head attention. Its key innovation lies in a multigraph construction with self-connecting edges that explicitly encode periodic lattice vectors (lattice lengths and angles). This design allows the network to intuitively capture macroscopic periodic patterns, ensuring strict periodic invariance and global structural awareness regardless of local connectivity details.
Taking geometric rigor further, ComFormer 19 employs a Transformer-based architecture that alternates between node-level and edge-level Transformers to maximize expressivity while ensuring stability. Crucially, ComFormer addresses the “many-to-one” ambiguity, where distinct structures map to identical graphs, thereby achieving geometric completeness. Unlike ALIGNN’s unstable k-NN topology, ComFormer utilizes a deterministic lattice-based graph construction. Notably, while sharing identical rotation-invariant inputs (bond lengths and angles) with ALIGNN, iComFormer distinguishes itself through topological rigor. As mathematically detailed in Supplemental Material A.2, ALIGNN’s k-NN construction is susceptible to topological instability, whereas iComFormer’s lattice-based strategy strictly guarantees the periodic invariance required for a symmetric classification (proven in Supplemental Material A.3). Within this framework, iComFormer, operates on SE (3)-invariant graphs using bond-lattice angles, while eComFormer advances this by utilizing SO(3)-equivariant vector representations. The use of bond vectors allows eComFormer to capture directional information, with node-level update layers designed to simultaneously satisfy SE(3) invariance and SO(3) equivariance. Following a similar pursuit of completeness, PerCNet 86 and GCPNet 87 utilize hybrid architectures combining convolutional and attention channels. PerCNet specifically introduces dihedral angles to resolve geometric ambiguities, while GCPNet employs “crystal pattern graphs” with global descriptors to enforce symmetry from a global perspective.
Beyond general property prediction, symmetry-aware designs are also tailored for specific physical quantities, as exemplified by CartNet. 90 Designed for predicting ADP, CartNet utilizes a radius-based graph construction to avoid neighbor ambiguity. However, because its architecture prioritizes computational efficiency by processing only pairwise spatial distances, it inherently lacks geometric completeness. Furthermore, while CartNet applies SO(3) data augmentation during training to help the model learn rotational equivariance, this augmentation does not retroactively grant the input distance graph the topological ability to resolve chirality. Nevertheless, CartNet is classified as a symmetric GNNs under our rigorous framework because it incorporates explicit symmetry modeling at the output level to align with the tensor properties of ADP: uniquely, its Cholesky Head enforces strict physical constraints to ensure the output matrix is always mathematically valid (symmetric and positive semidefinite).
The second category, represented by PotNet 88 and Ewald-MP, 89 achieves symmetry not through geometric descriptors alone, but by explicitly modeling the infinite periodic lattice through physics-based summation strategies. Unlike local graph methods that suffer from truncation boundaries, these models theoretically capture the complete periodic interaction field. PotNet incorporates infinite potential energy summation directly into the message passing and aggregation process. Ewald-MP adopts the Ewald summation architecture, decomposing interactions into real-space and reciprocal-space components. It employs Fourier transformation to efficiently model long-range signals in frequency space. Crucially, while practical implementations involve numerical approximations, the underlying theoretical framework of these models is based on rigorously periodic physics equations, classifying them as symmetric architectures.
Furthermore, Crystal GNNs can be classified into three types based on their message passing mechanisms: convolutional, attention-based, and hybrid. Crystal GNNs such as CGCNN, iCGCNN, MEGNET, and ALIGNN can be categorized as convolutional type, while GATGNN, Matformer, and ComFomer can be classified as attention type. Finally, PerCNet and GCPNet, which employ both convolution and attention mechanisms, can be classified as hybrid type.
Summary of methods employed in symmetric crystal GNNs.

3.4. A comparative analysis of framework paradigms
Comparative analysis of framework features of different material representation learning frameworks. 29

Table 2 highlights a fundamental divergence between asymmetric and symmetric paradigms regarding passive symmetry compliance. First, regarding periodic invariance: Conventional frameworks often prioritize efficiency or local expressivity over strict symmetry. For instance, CGCNN and ALIGNN rely on k-NN construction. As analyzed in Section 3.3, this introduces topological instability, leading to violations of periodic invariance. In contrast, symmetric frameworks (e.g., iComFormer, Matformer, PotNet) adopt deterministic strategies, such as lattice-based construction or global summations, to strictly guarantee the topological stability required by the crystalline state.
Second, regarding geometric completeness: While early frameworks satisfy basic SE(3) invariance, they often rely on scalar distances, implicitly imposing reflection invariance and failing to distinguish chiral structures. Advanced frameworks explicitly address this. Notably, eComFormer achieves geometric completeness alongside periodic invariance through vector-based message passing, while PerCNet incorporate chiral-sensitive descriptors (e.g., dihedral angles).
Essentially, the comparison of ALIGNN, iComFormer, and PerCNet, all utilizing third-order geometric information (bond angles), reveals a key design principle: High-order geometric descriptors alone are insufficient for symmetry compliance; they must be grounded on a topologically rigorous graph structure. ALIGNN leverages bond angles on an unstable k-NN topology (asymmetric), whereas iComFormer integrates them within a strictly invariant framework (symmetric).
Ultimately, designing a robust crystal learning framework requires satisfying two levels of symmetry: (1) Strict periodic invariance to ensure validity across infinite lattice transformations, and (2) either geometric completeness to uniquely distinguish chemically distinct structures (including chiral enantiomers), or explicit global symmetry modeling to capture macroscopic lattice behaviors and physical constraints. Furthermore, task-specific constraints often drive architectural specialization, as seen in CartNet, which enforces physical validity (positive semidefiniteness) for ADP prediction. This trend indicates a shift from generic graph models toward physics-informed architectures where architectural rigor aligns strictly with material laws.
4. Datasets and related metrics
This section surveys the core datasets, key physical quantities, and evaluation metrics used to benchmark Crystal GNNs. We also analyze the comparative performance of leading frameworks on these standard tasks.
4.1. Core dataset
Currently, the data utilized in the research of crystal material representation learning frameworks using GNNs primarily comes from high-throughput calculations based on DFT. The following introduces three key datasets.
4.1.1. Materials project dataset
Initially, a widely used dataset was the MP-2018.6.1 subset
80
derived from the Materials Project database.
92
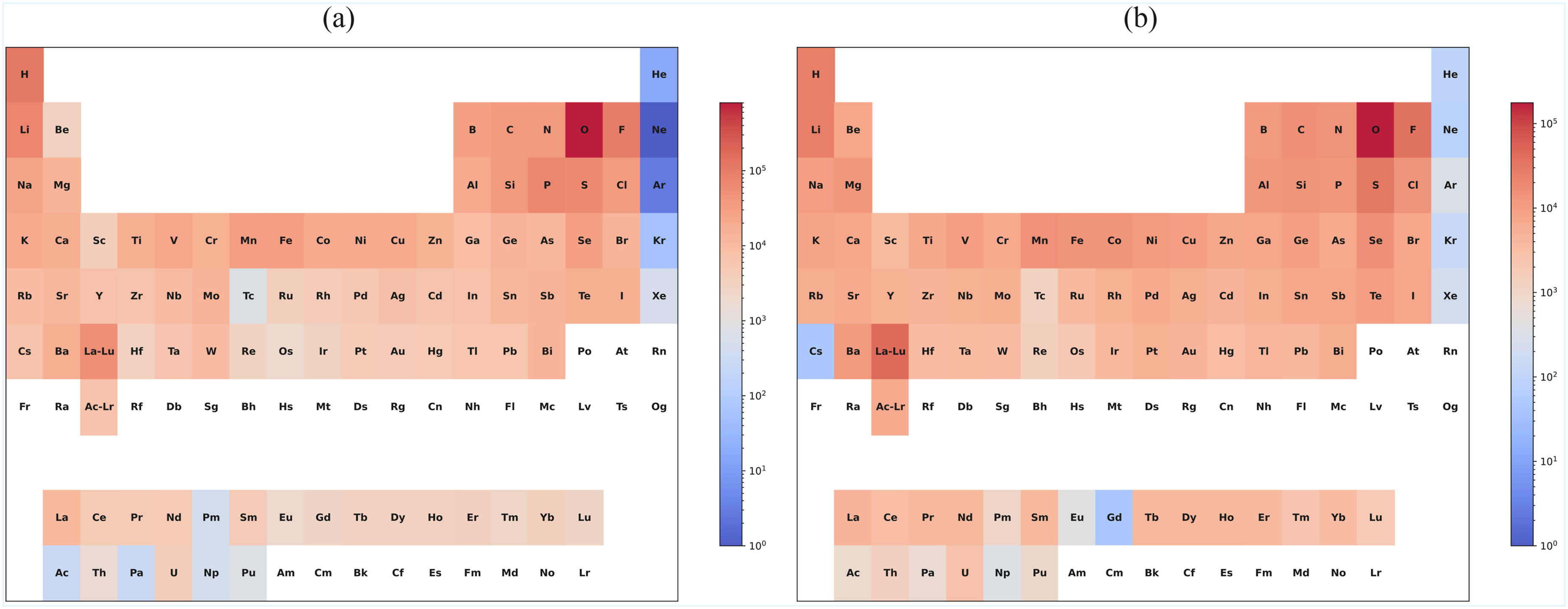
This dataset, covering four fundamental regression tasks (formation energy, band gap, bulk modulus, and shear modulus), provided a common platform for evaluating the performance of early models. (i) (ii) Heatmap of element distribution in the datasets. (a) Materials project dataset; (b) Jarvis dataset. Distribution map of data for different tasks in the materials project dataset.

4.1.2. JARVIS dataset
As research advances, the need for broader and deeper data has led to the development of more comprehensive data platforms, with JARVIS emerging as a prominent representative. JARVIS provides a vast testing ground, with its task set far surpassing early benchmarks, including up to 29 regression tasks and 10 classification tasks.66,93,94 This breadth allows researchers to more thoroughly evaluate the generalization ability of a model architecture. For example, in the JARVIS dataset, researchers can not only test the prediction of thermodynamic stability, such as formation energy, total energy, and distance to convex hull energy, but also evaluate the model’s ability to distinguish band gaps calculated by different DFT functionals (such as OPT and MBJ), reflecting the depth of its data and attention to computational details. (i) (ii) Distribution map of data for different tasks in the Jarvis dataset.

4.1.3. The Matbench dataset
To address the difficulty of comparing models across unstandardized tasks, Matbench 95 was introduced as a rigorous benchmarking suite. Unlike raw databases, Matbench is a curated collection of 13 tasks (10 regression, 3 classification) integrated from various sources. Its primary value lies in its standardized evaluation protocol. Instead of allowing random data splitting, Matbench provides fixed, nested cross-validation splits for every task. This design strictly minimizes “information leakage” and ensures that all models are evaluated under identical conditions. Consequently, despite containing fewer tasks than JARVIS, Matbench serves as a critical standard for ensuring fairness and reproducibility in comparative studies.
The data volume and common tasks of different datasets.

4.2. Key physical quantities and evaluation metrics
Model evaluation focuses on specific physical properties that determine a material’s practical value. Table 4 presents these quantities alongside their calculation formulas. (i) (ii) Key physical quantities for evaluating model performance. Key metrics to evaluate model performance.


For classification tasks (predicting material categories), Accuracy measures the overall proportion of correct predictions. However, given the class imbalance common in materials science, strictly relying on accuracy can be misleading. Therefore, Precision (the proportion of true positives among predicted positives), Recall (the proportion of true positives detected), and the F1 Score (the harmonic mean of Precision and Recall) are essential for a balanced evaluation. By systematically comparing models using these unified metrics, researchers can objectively quantify architectural advancements.
4.3. Performance comparison of mainstream frameworks
The test MAE results of different frameworks on the Jarvis dataset.

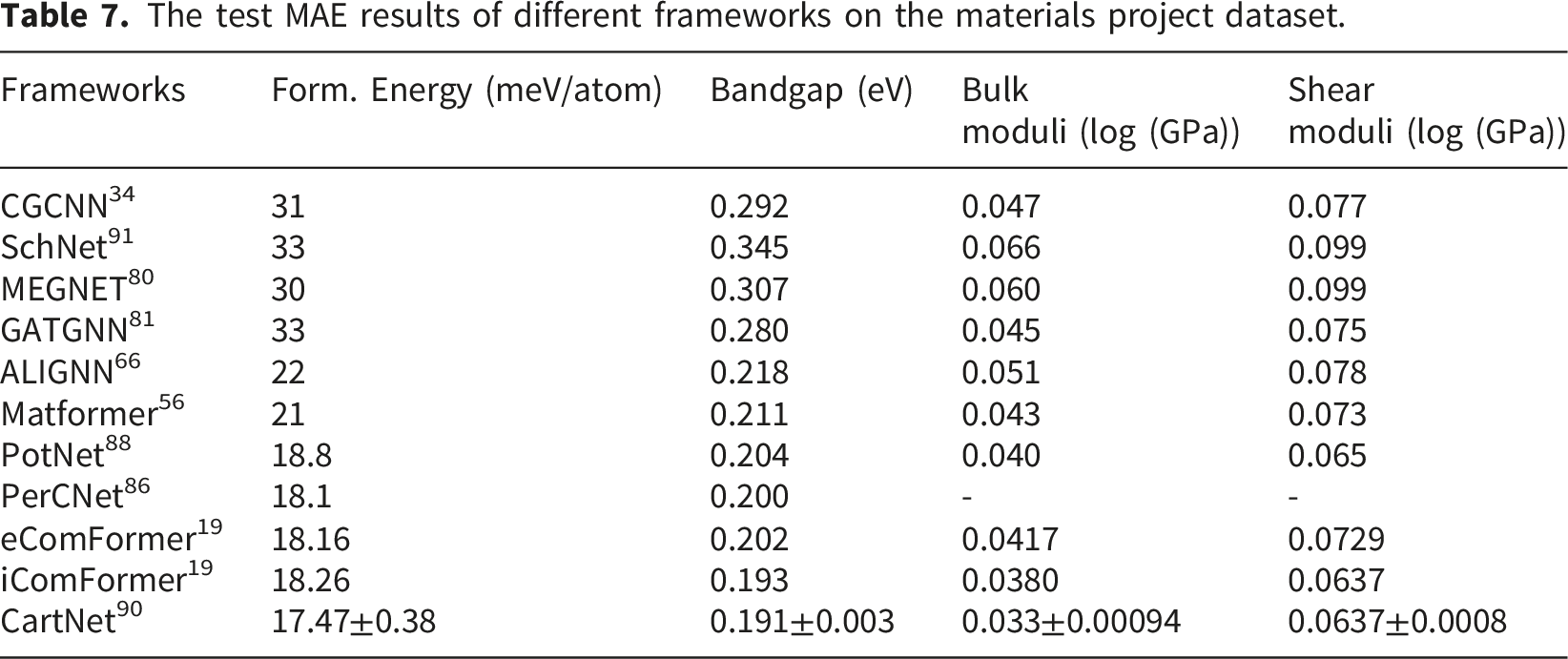
The test MAE results of different frameworks on the materials project dataset.
The data in Table 6 reveals a trajectory of performance improvement driven by architectural evolution and the refinement of graph construction strategies. Early models like CGCNN and SchNet exhibited relatively high MAEs, guiding subsequent optimizations. With the introduction of attention mechanisms and higher-order geometric information, frameworks such as GATGNN and ALIGNN achieved significant improvements. Recently, frameworks like CartNet have elevated performance to new heights. CartNet demonstrates superior performance across all five material property prediction tasks, with significantly lower prediction errors for formation energy, bandgap (OPT), and total energy compared to other models. This strongly demonstrates its exceptional generalization ability in capturing the “structure-property” relationship.
A noteworthy observation is that, although Transformer-based models such as Matformer and iComFormer perform slightly worse than CartNet, they still outperform earlier models with simpler crystal graph constructions that do not incorporate periodic patterns or equivariant information. This reveals an important insight: in crystal material property prediction, a framework tailored to the periodicity and passive symmetry of crystal structures possesses inherent advantages over general frameworks. This emphasizes that framework design should carefully consider inherent physical constraints rather than blindly pursuing complexity.
Table 7 displays the independent evaluation results on the Materials Project dataset. CartNet continues to deliver the best performance across most tasks, while other models exhibit similar trends compared to their previous versions. Although absolute error values differ across the two datasets due to varying material types and structural complexities, the relative performance ranking remains largely consistent. This suggests that effective frameworks possess strong generalization capabilities across diverse data environments.
Training time per epoch, total training time, and total testing time for some different frameworks on JARVIS formation energy prediction.

4.4. Discussion: The trade-off between learning and enforcing symmetry
The performance analysis in Section 4.3 highlights a critical divergence in how modern frameworks address crystal symmetries. Although symmetric architectures such as ComFormer and augmentation-based approaches like CartNet both achieve high accuracy, they represent fundamentally different paradigms with distinct trade-offs regarding training efficiency, data efficiency, and architectural rigidity.
Architecturally invariant models, including ComFormer and Matformer, incorporate symmetry as a hard inductive bias directly into the neural network architecture. This approach yields high data efficiency because the model is mathematically constrained to be invariant, meaning it does not require rotated versions of a crystal to learn that they are identical. However, this rigorous enforcement incurs a specific computational cost. The complexity per forward pass is often higher due to the overhead of specialized geometric operations, such as SO(3) equivariant tensor products or complex edge updates. These models are therefore ideal when training data is scarce or when strict physical guarantees of exact invariance are required.
In contrast, the paradigm of learning invariance via data augmentation, exemplified by CartNet, achieves best performance through a different mechanism. This strategy typically employs a computationally efficient backbone, such as a radius-based graph, trained on a dataset expanded by random SO(3) rotations. While the backbone itself might be lighter per step than an equivariant transformer, the model must learn approximate invariance from the data itself. Theoretically, achieving the same level of symmetry coverage as an invariant architecture requires a significantly larger effective dataset and potentially more training iterations. Therefore, the computational burden shifts from complex operators within the architecture to increased data volume and total training duration. A primary advantage of this paradigm is its flexibility, as it allows researchers to use simpler and more scalable backbone networks without designing complex equivariant layers. This flexibility offers a potential advantage for large-scale pre-training scenarios where data throughput is the primary concern.
Ultimately, the choice between these paradigms depends on the specific constraints of the application. For scenarios characterized by data scarcity or a requirement for theoretical rigor, architecturally invariant symmetric GNNs offer a robust solution. Conversely, for benchmarks where maximizing prediction accuracy is the priority and computational resources permit extensive training on augmented data, the augmentation-based approach currently yields superior empirical results.
5. Conclusion and outlook
This review has systematically investigated a range of GNNs-based representation learning frameworks for crystalline materials, analyzing their architectural distinctions and comparative performance. By leveraging graph-structured modeling and innovative network designs, these frameworks have demonstrated a remarkable ability to overcome the limitations of traditional methods particularly in capturing passive symmetry and higher-order geometric information within complex material systems. As a result, GNNs-based representation learning methods for crystal materials have emerged as highly promising tools in materials science, achieving remarkable success in predicting macroscopic properties of materials (such as band gap, formation energy, total energy, etc.) and accelerating the discovery of novel materials.
However, despite these advances, several critical challenges remain that hinder the broader adoption and performance of GNNs-based crystal models. First, the trade-off between symmetry enforcement and computational efficiency remains unresolved. As analyzed in Section 4.4, strictly symmetric models (e.g., ComFormer) rely on computationally expensive operations like tensor products or spherical harmonics to guarantee invariance, limiting their scalability to large-scale screening. Conversely, augmentation-based models (e.g., CartNet) sacrifice theoretical rigor for speed but suffer from low data efficiency. Second, current frameworks predominantly focus on perfect bulk crystals, neglecting the local symmetry breaking inherent in real-world materials (e.g., defects, grain boundaries). Third, most existing models are designed to output rotation-invariant scalars (e.g., energy), lacking the capability to predict high-order tensorial properties which are crucial for functional materials.
To address these issues, future research should prioritize the following technical directions: (i) (ii) (iii) (iv)
In summary, the field is transitioning from the initial phase of demonstrating feasibility to a new phase of rigorous physical alignment. Achieving fully efficient, interpretable, and generalizable material modeling will require continued interdisciplinary effort. With growing computational resources and richer material datasets, these methods are poised to significantly accelerate the intelligent design and discovery of new materials, opening up new frontiers in computational materials science.
Supplemental material
Supplemental material - Representation learning of crystal materials using Graph Neural Networks: Passive symmetry challenges and advances
Supplement material for Representation learning of crystal materials using Graph Neural Networks: Passive symmetry challenges and advances by Jie Cui, Chenglei Han, Jiyao Liang, Lin Li and Fei Wang in Science Progress.
Footnotes
Acknowledgements
Shaorong Fang and Tianfu Wu from Information and Network Center of Xiamen University are acknowledged for the help with the GPU computing.
Author contributions
Jie Cui: Conceptualization, Data curation, Investigation, Visualization, Writing – original draft, Writing – review and editing. Chenglei Han: Writing – review and editing. Jiyao Liang: Writing – review and editing. Lin Li: Funding acquisition, Supervision, Writing – review and editing. Fei Wang: Funding acquisition, Supervision, Writing – review and editing.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partly supported by the National Key R&D Program of China (2019YFB2205001); the National Natural Science Foundation of China (62371407); the State Key Lab of Processors, Institute of Computing Technology, CAS - (CLQ202402); and the Shandong Province Natural Science Foundation, China (ZR2024MF073).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data will be made available on request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.