
Abstract
Objective
We applied Mendelian Randomization (MR) to investigate potential genetic targets involved in the pathogenesis of COVID-19, aiming to identify causal factors that may contribute to disease susceptibility and severity.
Methods
We aggregated multi-omics quantitative trait loci data and explored the pathogenic targets of COVID-19 using summary data-based Mendelian randomization, colocalization, and MR.
Results
We identified Latent transforming growth factor beta binding protein 2 (LTBP2) and α-1,3-N-acetylgalactosaminyltransferase (ABO) as primary targets for the risk of severe COVID-19. Elevated levels of LTBP2 (odds ratio (OR): 0.53, 95% CI: 0.39–0.70) were associated with reduced risk of severe COVID-19, while ABO (OR: 1.09, 95% CI: 1.06–1.11) were associated with increased risk of severe COVID-19. These effects were consistent with the expression of LTBP2 (OR: 0.58, 95% CI: 0.45–0.75) and the methylation of ABO (cg07241568: OR: 1.20, 95% CI: 1.14–1.26; cg24267699: OR: 1.11, 95% CI: 1.08–1.15) regarding the risk of severe COVID-19.
Conclusions
We have identified several potential genes related to COVID-19 risks, providing a basis for understanding the pathogenesis of COVID-19 and the development of candidate therapeutic drugs.
Introduction
The 2019 coronavirus disease (COVID-19) is an infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). 1 As of February 2024, there have been over 770 million confirmed cases of COVID-19 and over 7 million deaths (https://covid19.who.int). While government quarantine measures and vaccine development have reduced the incidence and mortality of COVID-19, 1 the widespread global variation of COVID-19 strains still results in significant heterogeneity among infected individuals in terms of symptoms, severity, and prognosis. Furthermore, the covert transmission by asymptomatic carriers continues to pose challenges for the prevention and treatment of COVID-19. 2
Host genetics determine the susceptibility, severity, and prognosis of COVID-19, and the COVID-19 Host Genetics Initiative has identified several risk loci associated with the disease. 3 Genome-wide association studies (GWASs) have provided abundant genetic information on COVID-19, 4 aiding in a deeper understanding of the genetic mechanisms underlying COVID-19 infection and progression. However, due to limitations in recruiting study participants and the lack of extensive clinical metadata, a comprehensive genotype-phenotype interpretation of the research findings is still not feasible. 5 Furthermore, GWAS cannot infer causal relationships, highlighting the urgent need to explore risk loci associated with COVID-19 infection and progression.
Mendelian randomization (MR) employs genetic variants as instrumental variables to infer causal relationships between exposure and outcome. MR is less susceptible to reverse causation and unknown confounding because genetic variants are randomly assigned at conception and are not influenced by the onset of disease. Therefore, MR has significant advantages in inferring causal effects, which may be difficult to achieve with traditional experimental techniques. 6 Studies using large-scale GWAS and molecular quantitative trait loci (QTL) data can explore the potential associations between gene expression and COVID-19 risk from the perspectives of expression. Previous studies have only explored the driving factors of COVID-19 through expression quantitative trait loci (eQTLs). However, in the context of rich genetics, the disease process of COVID-19 is heterogeneous and exhibits widespread environmental and temporal variability. Therefore, multi-omics analysis can help develop new and innovative effective drugs. This can provide appropriate targets for the prevention and treatment of COVID-19. Therefore, we employed MR to investigate the potential associations between gene expression, methylation, and protein abundance with the risk of COVID-19.
Method
Study design
Figure 1 illustrates the current research workflow. The current MR analysis utilized publicly available datasets including the COVID-19 Host Genetics Initiative (Release 7, https://www.covid19hg.org/), 3 FinnGen study, and other large-scale GWASs. The COVID-19 Host Genetics Initiative comprised GWAS data for very severe respiratory confirmed covid, hospitalized COVID-19, and population COVID-19. The GWAS data for COVID-19 confirmed population were obtained from the r10 release of FinnGen study. We employed the summary data-based Mendelian randomization (SMR) method to assess the relationship between gene expression, methylation, and protein abundance with the risk of COVID-19. The dataset from the COVID-19 Host Genetics Initiative served as the primary discovery dataset, with the FinnGen study dataset serving as a validation dataset to confirm our findings. Finally, the final candidate genes were determined by integrating the MR results from different omics. There was no overlap in samples between the exposure and outcome populations, and all were from European cohorts. The research process was based on the three major assumptions of MR: (1) a significant association between genetic variation and exposure (the correlation hypothesis); (2) genetic variation is unrelated to confounding factors (exclusivity hypothesis); and (3) genetic variation influences outcomes only through exposure (independence hypothesis). This study is reported according to the STROBE-MR statement (Supplementary STROBE-MR checklist table).

Flowchart of the study design. QTL: quantitative trait loci. eQTL: expression quantitative trait loci; mQTL: methylation quantitative trait loci; pQTL: protein quantitative trait loci; SMR: summary data-based Mendelian randomization; MR: Mendelian randomization; FDR: false discovery rate; PPH4: posterior probability of H4.
Sample and data
We used the integration of multi-omics evidence to elucidate the potential molecular networks underlying COVID-19 occurrence. QTLs can reveal associations between single-nucleotide polymorphisms (SNPs) and gene expression, methylation, and protein abundance. The eQTL dataset with blood gene expression within 1000 kb of the coding sequences was obtained from the eQTLGen Consortium, comprising 31,684 individuals. 7 The blood methylation quantitative trait loci (mQTL) dataset was extracted from the study by McRae et al., included 1980 individuals. 8 The dataset of protein quantitative trait loci (pQTL) was obtained from the UK Biobank - Pharma Proteomics Project (UKB-PPP), including plasma proteomic profiles of 54,219 UKB participants. 9 We employed the default genome-wide significance p-value threshold of 5 × 10−8 to select the most relevant eQTLs, mQTLs, and pQTLs as instrumental variables for the SMR analysis.
The summary GWAS data for COVID-19 outcomes were sourced from the COVID-19 Host Genetics Initiative and the FinnGen Study, with participants of European ancestry. The Release 7 of the COVID-19 Host Genetics Initiative includes very severe respiratory confirmed covid (severe COVID-19), hospitalized COVID-19, and population COVID-19. Severe COVID-19 included 13,769 cases and 1,072,442 controls; hospitalized COVID-19 included 32,519 cases and 2,062,805 controls; population COVID-19 included 122,616 cases and 2,475,240 controls. Since the FinnGen study was unable to access the summary data for very severe respiratory diseases, hospitalized COVID-19, and confirmed COVID-19, the comfirmed COVID-19 GWAS was used as the validation cohort, consisting of 2856 cases and 405,232 controls.
Measures of variables
COVID-19 severity was defined using three phenotypes provided by the COVID-19 Host Genetics Initiative: (1) very severe respiratory confirmed COVID-19 vs population, (2) hospitalized COVID-19 vs non-hospitalized, and (3) COVID-19 vs population (Table S1).
Data analysis procedure
SMR analysis (SMR v 1.3.1) was employed to assess the relationship between eQTLs, mQTLs, and pQTLs and the risk of COVID-19 (https://yanglab.westlake.edu.cn/software/smr/#Overview). 10 All analyses were conducted using the default settings of SMR. The odds ratio (OR) estimates for the association of QTLs with COVID-19 risk were calculated based on β coefficients. The effect size (β) of the variants reflects the direction of change in gene expression, methylation, and protein expression. To control for genome-wide type I errors, we adjusted p-values using the false discovery rate (FDR). The heterogeneity in dependent instruments (HEIDI) test was employed to distinguish pleiotropy from linkage disequilibrium (LD), where p-HEIDI < 0.01 was considered indicative of potential pleiotropy and thus excluded from the analysis.
For significant MR results, we investigated the shared causal variants between eQTLs, mQTLs, and pQTLs with COVID-19. Based on published studies, we selected a window of 100 kb for colocalization analysis. The posterior probability of hypothesis 4 (PPH4) was used as evidence of colocalization between QTLs and GWAS. For protein abundance, PPH4 ≥ 0.7 represents strong evidence of colocalization, while PP.H4 between 0.5 and 0.7 indicates moderate evidence of colocalization.11,12
Since proteins were the ultimate products of gene expression, further validation was needed to confirm the significant proteins identified in the SMR analysis. We used proteins from the UKB-PPP as exposures and employed traditional two-sample MR to validate the significance of these proteins. In the UKB-PPP, SNPs significantly associated with proteins (p < 5 × 10−8) were extracted, and LD r2 < 0.1 was used to determine the extent of LD between SNPs, with a clustering distance set at 100 kb. Additionally, SNPs and proteins within the major histocompatibility complex region (chr 6: 25.5–34.0 Mb) were excluded due to the complex LD structure. 13 Subsequently, we excluded weak instrumental variables based on the F-statistic (F < 10) and filtered out proteins exhibiting horizontal pleiotropy. The inverse variance weighted method was employed as the primary outcome of the MR analysis, followed by FDR correction. Therefore, proteins were included in subsequent multi-omics analyses only if they reached significance in both traditional two-sample MR and SMR. MR was performed using R software windows version 4.3.1
Omics evidence integration
Since significant proteins have already been identified based on SMR and two-sample MR, and the regulatory effect of proteins on phenotypes is significantly higher than that of gene expression or methylation, the integration of all multi-omics evidence needs to be based on these significant proteins. In other words, candidate genes need to have a causal association with COVID-19 at the protein level. Based on this principle, we categorized as follows: (1) primary targets were defined as proteins significantly associated with COVID-19 at the level of protein abundance (FDR-p value < 0.05), with a PPH4 > 0.7, and also significantly associated with COVID-19 at the level of methylation or gene expression (FDR-p value < 0.05); (2) secondary targets were defined as proteins significantly associated with COVID-19 (FDR-p value < 0.05) and with a PPH4 > 0.7, or proteins significantly associated with COVID-19 (FDR-p value < 0.05) with a PPH4 ranging from 0.5 to 0.7, and also significantly associated with COVID-19 at the level of methylation or gene expression (FDR-p value < 0.05). (3) Potential targets were defined as proteins significantly associated with COVID-19 at the level of protein abundance (FDR-p value < 0.05) with a PPH4 ranging from 0.5 to 0.7. The final candidate genes for COVID-19 were identified in the omics analysis.
Results
Gene expression and COVID-19
The results of the causal relationship between gene expression and COVID-19 outcomes can be found in Tables S2–S4. After FDR, HEIDI, and colocalization (PPH4 > 0.7), a total of 10, 15, and 10 genes were associated with severe COVID-19, hospitalized COVID-19, and population COVID-19 risk.
For severe COVID-19, ATP11A, latent transforming growth factor beta binding protein 2 (LTBP2), TNFAIP8L1, and NUCB1-AS1 were validated in the FinnGen study (Table S14). For hospitalized COVID-19, PPT2, STARD10, ZNF268, ATP11A, SLC22A31, TNFAIP8L1, and NUCB1-AS1 were validated in the FinnGen study (Table S15). For population COVID-19, NFKBIZ and NUCB1-AS1 were validated in the FinnGen study (Table S16). The specific gene effects are shown in Table 1.
The effect of gene expression on COVID-19 risk was validated by FinnGen.

OR: odds ratio; CI: confidence interval; PPH4: posterior probability of H4; FDR: false discovery rate; LTBP2: latent transforming growth factor beta binding protein 2.
Gene methylation and COVID-19
The results of gene methylation on the causal relationship with COVID-19 outcomes can be found in Table S5–S7. After FDR, HEIDI, and colocalization (PPH4 > 0.7), 21 genes and 30 CpG sites were identified for severe COVID-19, 33 genes and 67 CpG sites for hospitalized COVID-19, and 22 genes and 45 CpG sites for population COVID-19. In the FinnGen validation cohort, a total of 8 genes and 16 CpG sites were successfully validated for severe COVID-19, 7 genes and 15 CpG sites for hospitalized COVID-19, and 10 genes and 14 CpG sites for population COVID-19 (Table S17–19). The specific methylation site effect effects are shown in Table 2. For example, increased methylation at the cg02098565 site of the ACSF3 gene was associated with an increased risk of severe COVID-19 (OR: 1.5, 95% CI: 1.25–1.81), while increased methylation at the cg04308346 site was associated with a decreased risk of severe COVID-19 (OR: 0.68, 95% CI: 0.58–0.79) (Table 2). The same trend persisted in hospitalized COVID-19 and population COVID-19.
The association of gene methylation with the risk of COVID-19 was validated by FinnGen.
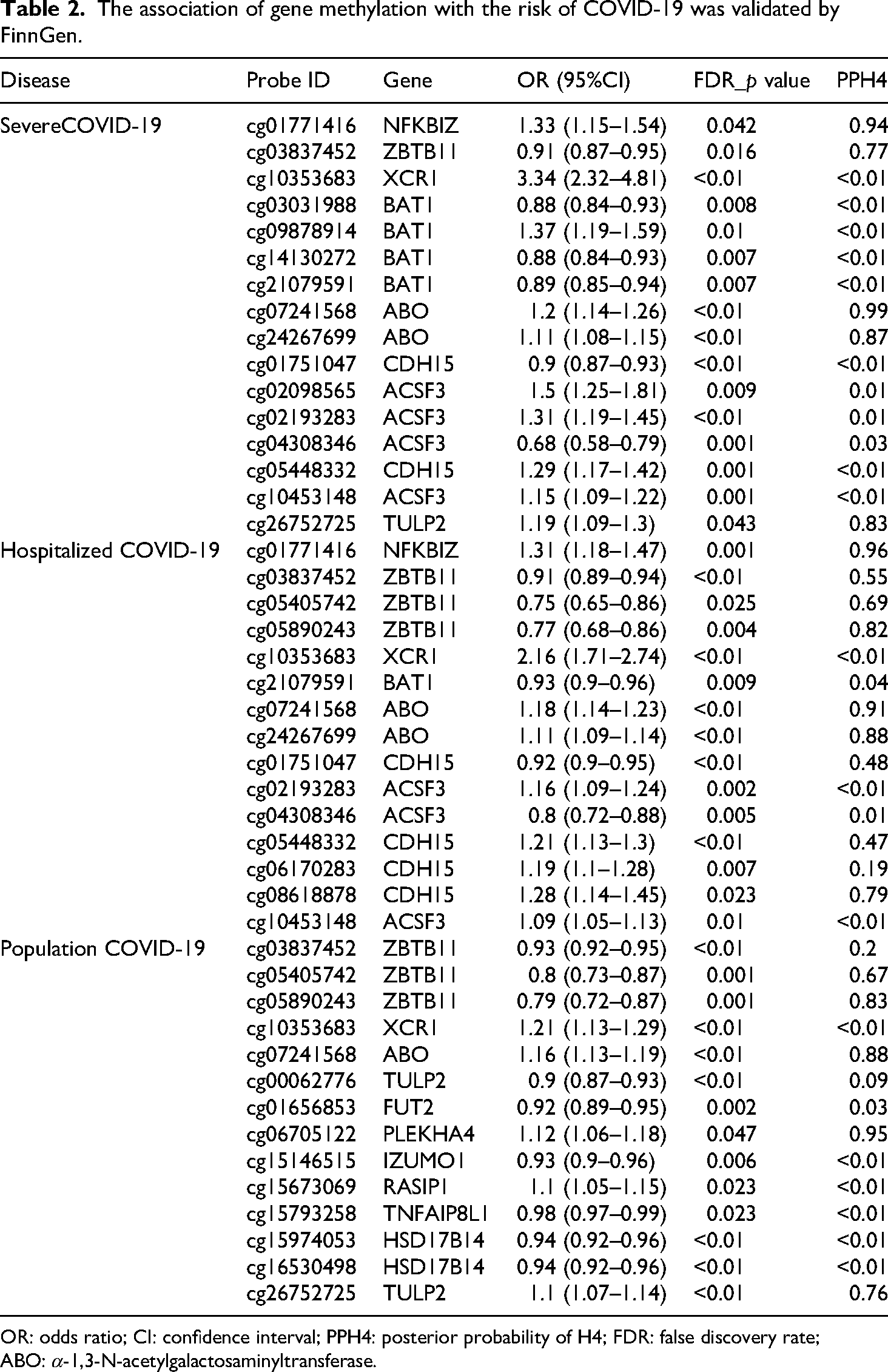
OR: odds ratio; CI: confidence interval; PPH4: posterior probability of H4; FDR: false discovery rate; ABO: α-1,3-N-acetylgalactosaminyltransferase.
Protein abundance and COVID-19
The protein abundance from the two-sample MR analysis is depicted in Figure S1 A-C. Following the identification of significant proteins through SMR, the results from SMR and the two-sample MR analysis were combined. While these proteins exhibited significance in both SMR and the two-sample MR analysis (Table S8–S13), there was a discrepancy in the effect direction of ACE2 between severe COVID-19 (OR: 1.29 vs. 0.82) and hospitalized COVID-19 (OR: 1.26 vs. 0.92) (Table 3); therefore, ACE2 was excluded from subsequent analyses. Eight, eight, and three proteins were associated with the risk of very severe respiratory disease, hospitalized COVID-19, and confirmed COVID-19, respectively (Tables S8–S10). We used colocalization to filter proteins. After colocalization filtering (PPH4 > 0.5), higher levels of SFTPD (OR: 0.91, 95% CI: 0.88–0.95) and LTBP2 (OR: 0.53, 95% CI: 0.39–0.70) were negatively associated with the risk of severe COVID-19, while elevated levels of GART (OR: 1.60, 95% CI: 1.23–2.07) and α-1,3-N-acetylgalactosaminyltransferase (ABO) (OR: 1.09, 95% CI: 1.06–1.15) were positively associated with the risk of severe COVID-19. Elevated levels of BTN1A1 (OR: 0.82, 95% CI: 0.73–0.91) and SFTPD (OR: 0.94, 95% CI: 0.91–0.96) were negatively associated with the risk of hospitalized COVID-19, while increased levels of RNF43 (OR: 1.38, 95% CI: 1.16–1.64) were positively associated with the risk of hospitalized COVID-19. Higher levels of leucine-rich repeats and calponin homology domain-containing 4 (LRCH4) (OR: 0.71, 95% CI: 0.60–0.83) were negatively associated with the risk of population COVID-19 (Table 4). Additionally, the direction of effect for these significant proteins remained consistent in FinnGen study (Table S20–S22).
Causal association of protein richness with COVID-19 risk.

MR: Mendelian randomization; SMR: summary data-based Mendelian randomization; OR: odds ratio; CI: confidence interval; FDR: false discovery rate; ABO: α-1,3-N-acetylgalactosaminyltransferase; LRCH4: leucine-rich repeats and calponin homology domain-containing 4.
Genetic prediction of association of genetically encoded proteins with the risk of COVID-19.

OR: odds ratio; CI: confidence interval; FDR: false discovery rate; PPH4: posterior probability of H4; LTBP2: latent transforming growth factor beta binding protein 2; ABO: α-1,3-N-acetylgalactosaminyltransferase; LRCH4: leucine-rich repeats and calponin homology domain-containing 4.
Integration of omics evidence
In all three COVID-19 outcomes, we identified 7 genes, 32 methylation sites, and 1 protein (Figure S1 D-F). These targets were significant across the three COVID-19 outcomes and exhibited consistent risk effects (Figure S1 G), indicating the stability of our findings and suggesting that common targets have similar effects on the risk of COVID-19. After integrating the multi-omics evidence and validating through FinnGen, we ultimately identified LTBP2 as a primary target for severe COVID-19, with overlapping effects observed in eQTLs and pQTLs. ABO was identified as a secondary target for severe COVID-19, with overlapping effects observed in mQTLs and pQTLs. For targets without overlapping effects, we identified SFTPD as a secondary target for severe COVID-19 and GART as a potential target for severe COVID-19. For hospitalized COVID-19, overlapping omics evidence was not found, but SFTPD was identified as a potential target for hospitalized COVID-19. For population COVID-19, overlapping omics evidence was not found, while LRCH4 was identified as a secondary target for population COVID-19. Additionally, for the omics evidence of severe COVID-19, we further examined the positive correlation between the expression of LTBP2 and ABO and their respective protein levels (Table S23), which is consistent with the risk effects of LTBP2 and ABO on severe COVID-19 (Table S7). Similarly, these associations were validated in FinnGen (Table S24), although not all associations reached statistical significance, the direction of all associations remained consistent, consistent with our observations across omics (Table S1, S4, S7).
Discussion
In this study, we used SMR combined with MR to explore the genetic drivers of COVID-19 for the first time. We found that LTBP2, ABO, SFTPD, and GART were associated with the risk of severe COVID-19. SFTPD was associated with the risk of hospitalized COVID-19, while LRCH4 was associated with the risk of population COVID-19. This provides evidence for potential mechanisms linking gene expression, methylation, protein abundance, and COVID-19. We propose suggestions to improve the problem-oriented drug discovery process, emphasizing best practices for the management of innovative drug development.
The spread and origin of COVID-19 are related to multiple factors, and traditional epidemiological models are inadequate in addressing viral mutations or changes in national containment policies. 14 Unlike previous studies, we incorporated pQTLs instead of solely eQTLs.15–18 Proteins are the molecules that directly perform biological functions, so pQTLs are closer to the ultimate biological effects of diseases. Moreover, our results differ from the targets obtained by Wu et al. using the cross-methylome omnibus test. 18 There is environmental variability in COVID-19. 19 In addition, we have analyzed the latest COVID-19 data. Compared to the previous version of the analysis, 18 our study has a larger sample size, enhancing our ability to identify reliable genetic associations and make causal inferences. Importantly, technological advancements in analysis provide new insights into drug development.5,20,21 We used SMR, which provided stronger statistical power and was validated through the MR method in the aforementioned study. Our full exploration of the genetic factors of COVID-19 will contribute to the development of new drugs and further reveal the pathogenic process of COVID-19. In addition, COVID-19 at different stages exhibits unique pathophysiology. 4 Therefore, we provide comprehensive multi-omics evidence based on different COVID-19 symptoms. The results also reveal distinct driving factors.
We identified an association between LTBP2 and the risk of severe COVID-19, with this relationship supported by combined eQTLs and pQTLs evidence. LTBP2 is critical for maintaining elastic fiber stability and has been implicated in both pulmonary and cardiac fibrosis. 22 In COVID-19, insufficient elastic components may exacerbate the excessive elastase-mediated degradation triggered by inflammatory bursts, which in turn reduces lung compliance and elevates the risk of severe disease. 23 This is consistent with our finding that reduced LTBP2 expression correlates with a lower risk of severe COVID-19. Notably, there is clear evidence that elastic tissue destruction elevates the risk of severe disease. 24 This is because the inability to maintain normal lung structure impairs lung function or causes respiratory distress, 25 progressing to persistent hypoxemia, multi-organ failure, and ultimately death. 26 Thus, the present study underscores the potential protective role of LTBP2 in preserving elastic fiber stability during severe COVID-19.
We found that ABO is involved in all three COVID-19 outcomes. ABO gene variations underpin the ABO blood group system and determine blood type. A COVID-19 GWAS showed that higher ABO expression correlates with increased risk of hospitalized COVID-19 (beta = 0.298). 27 Additionally, the ABO gene encodes the protein determining ABO blood type. In pQTLs analyses, we observed consistent risk effects of both gene expression and protein abundance on COVID-19, with this consistency maintained across multi-omics analyses. The precise mechanism by which the ABO protein influences COVID-19 risk remains unclear. However, carriers of the A and B alleles express glycosyltransferase activity, which converts the H antigen to A or B antigens, whereas individuals with blood type O lack this enzymatic activity due to a gene deletion (frameshift mutation). This underlies the differences in COVID-19 susceptibility across blood types. 5
Additionally, several observational insights may provide relevant links: for instance, naturally occurring anti-A antibodies could inhibit spike protein-mediated cell invasion via the ACE2 receptor—a hypothesized entry route for SARS-CoV-2. 28 Another potential explanation is that individuals with blood type A have a higher risk of cardiovascular disease—a known risk factor for COVID-19—whereas those with type O have a lower cardiovascular disease risk. 29 Additionally, we identified that gene-predicted ABO methylation sites (cg07241568, cg24267699) elevate the risk of severe COVID-19; these sites showed consistent risk effects at the protein abundance level, further reinforcing the robustness of our findings. Previous studies have shown that the gene-predicted ABO methylation site cg24267699 elevates the risk of coronary artery disease, 30 which may indirectly heighten the risk of severe COVID-19. However, clear evidence linking methylation of cg07241568 to an elevated risk of severe COVID-19 remains lacking. Nonetheless, our findings provide validation at the protein abundance level.
SFTPD protein abundance was negatively correlated with the risk of severe and hospitalized COVID-19, consistent with the findings of Palmos et al. 31 While there may be sample differences in blood protein exposure and COVID-19 outcomes, the consistent risk estimates from our current findings are noteworthy. Additionally, SFTPD downregulation impairs host immunity, potentially reducing lung gas exchange function. 32 Meanwhile, SFTPD colocalization was higher in severe COVID-19 than in hospitalized cases, indirectly indicating a more prominent role for SFTPD in severe disease. Thus, SFTPD holds potential for preventing severe COVID-19 and alleviating symptoms like respiratory distress.
We also identified that LRCH4 may exert a potential protective effect against COVID-19 in the population. Silencing LRCH4 can attenuate lipopolysaccharide-induced production of multiple cytokines and dampen innate immune responses in vivo. 33 Additionally, lipopolysaccharide contributes to the aggregation of the SARS-CoV-2 spike protein. 34 Immune dysregulation plays a critical role in the pathophysiology of SAR-CoV-2, leading to severe lung injury and respiratory failure. 35 This suggests that LRCH4 may alleviate the development of COVID-19 by regulating immune responses. However, the precise mechanism underlying LRCH4's role in COVID-19 pathogenesis remains unclear. While multi-omic evidence is lacking, given the regulatory effect of protein abundance on phenotypes, LRCH4 shows promise as a potential therapeutic target for COVID-19.
We identified GART as a potential target for COVID-19. Phosphoribosylglycinamide formyltransferase (GART) is critical to the purine biosynthesis pathway, which is essential for numerous cell functions, including signaling and proliferation. 36 Purines are involved in viral RNA synthesis, and reducing purine levels may decrease viral replication, thus inhibiting viral infection. 37 In COVID-19, GART is significantly upregulated compared to healthy controls, 38 consistent with our findings. Notably, GART's involvement in purine synthesis elevates the risk of COVID-19 onset. Moreover, dietary regulation of purines may aid in controlling COVID-19 infection. 37 These findings collectively suggest that GART could serve as a potential therapeutic target for COVID-19.
This study had several strengths. Firstly, our study only included samples of European ancestry, reducing bias caused by genetic backgrounds. Secondly, we utilized MR to provide genomic information on the susceptibility risk of COVID-19, 27 including genetic, methylation, and protein abundance data. Previous studies had only been able to provide summarized information on the transcriptome, without assessing the impact of protein abundance. Additionally, we employed the latest version (v7) of the COVID-19 Host Genetics Initiative, which includes a larger number of COVID-19 samples. This enabled us to attain sufficient statistical power to elucidate causal relationships. Thirdly, we utilized MR and SMR to assess the impact of protein abundance on the risk of COVID-19, ensuring the robustness of our findings. Finally, we integrated evidence from multiple omics levels, strengthening the causal association between these genes and the risk of COVID-19.
However, this study still has some limitations. Firstly, the susceptibility factors of these genetic loci for COVID-19 in other ethnicities remain unknown. Secondly, the GWAS did not control for all confounding factors, such as diabetes, obesity, cardiovascular diseases, among others, which could act as risk factors for COVID-19. Thirdly, although we conducted sensitivity analyses to reduce bias, it may not completely eliminate associations caused by horizontal pleiotropy. Moreover, further extensive research is needed to validate the impact of these genes on the risk of COVID-19.
Conclusion
In the context of continuous COVID-19 mutations, we have elucidated the heterogeneous molecular mechanisms of different symptomatic COVID-19 from a multi-omics perspective. And evidence for multi-level targets was proposed based on MR. This contributes to suggestions for improving the problem-oriented drug discovery process, emphasizing best practices for the innovative management of new drugs targeting COVID-19. The future goal is to develop new drugs or repurpose existing drugs targeting the pathogenic sites of COVID-19 to mitigate its pandemic.
Supplemental Material
sj-docx-1-sci-10.1177_00368504251385959 - Supplemental material for Identification of COVID-19 susceptibility genes by Mendelian randomization
Supplemental material, sj-docx-1-sci-10.1177_00368504251385959 for Identification of COVID-19 susceptibility genes by Mendelian randomization by Hao Wang, Keru Ma, Junpeng Wu, Ming Shan and Guoqiang Zhang in Science Progress
Supplemental Material
sj-docx-2-sci-10.1177_00368504251385959 - Supplemental material for Identification of COVID-19 susceptibility genes by Mendelian randomization
Supplemental material, sj-docx-2-sci-10.1177_00368504251385959 for Identification of COVID-19 susceptibility genes by Mendelian randomization by Hao Wang, Keru Ma, Junpeng Wu, Ming Shan and Guoqiang Zhang in Science Progress
Footnotes
Ethical considerations
This study did not require ethical approval as all analysis was based on publicly available aggregated statistics without access to individual data. The included GWAS studies obtained informed consent from the study participants and were approved by the relevant local ethics committees.
Consent to participate
This research has been conducted using published studies and consortia providing publicly available summary statistics. All original studies have been approved by the corresponding ethical review board, and the participants have provided informed consent. In addition, no individual-level data was used in this study. Therefore, no new ethical review board approval was required.
Authors’ contributions
HW and KM designed the study, analyzed and interpreted the data, and drafted the manuscript. HW, KM, JW, and MS analyzed and interpreted the data. QZ conceived and designed the study and revised the manuscript. The authors read and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Availability of data and materials
All data were from open access GWAS large data (Supplement table S1). The current MR analysis utilized publicly available datasets including the COVID-19 Host Genetics Initiative (Release 7, https://www.covid19hg.org/) and FinnGen study (![]() ).
).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.