
Abstract
Objective
Gout is a prevalent form of inflammatory arthritis in which many patients respond suboptimally to current therapies. Drug development is hampered by a lack of genetically validated targets, leading to high clinical trial attrition. This study aimed to systematically identify and prioritize novel, druggable targets for gout via a multilayered genetic and functional genomics approach.
Methods
We performed two-sample Mendelian randomization (MR) using cis-expression quantitative trait locus (cis-eQTL) data and dual independent gout genome-wide association study (GWAS) cohorts (openGWAS and FinnGen). The candidate genes were subjected to a rigorous validation pipeline including Bayesian colocalization, phenome-wide association studies (PheWASs) to assess pleiotropy and on-target safety, and single-cell RNA sequencing (scRNA-seq) to delineate the cellular context. Molecular docking was used to evaluate the structural druggability of prioritized targets.
Results
MR analysis revealed 15 genes causally associated with gout. Colocalization analysis (PPH4 > 0.8) prioritized two targets: ZSCAN16 (risk-increasing, OR = 1.04, 95% CI [1.02–1.06]) and TRIM10 (protective, OR = 0.96, 95% CI [0.94–0.98]). Crucially, PheWAS revealed that ZSCAN16 is highly specific to gout, whereas TRIM10 exhibited extensive pleiotropy with hematological and cardiometabolic traits, indicating significant safety risks. Single-cell analysis provided orthogonal validation, demonstrating flare-specific upregulation of ZSCAN16 in cytotoxic T/NK cells. Molecular docking confirmed ZSCAN16 as a structurally druggable target, showing high-affinity binding with known compounds (e.g. digoxin, binding energy = −9.6 kcal/mol).
Conclusions
Our study identifies ZSCAN16 as a high-potential, druggable therapeutic target for gout, highlighting its genetic influence on specific immune cell activities during acute flares. Conversely, TRIM10 was deprioritized owing to substantial pleiotropic liabilities and poor chemical tractability. These findings suggest that ZSCAN16 could play a crucial role in the pathogenesis of gout and may provide a valuable lead for future drug discovery efforts.
Introduction
Gout, a painful inflammatory arthritis driven by monosodium urate (MSU) crystal deposition, poses a substantial and growing global health burden. Its prevalence and incidence have risen alarmingly worldwide, 1 a trend particularly pronounced in regions like China, where dietary and lifestyle changes are significant contributors. 2 The disease pathophysiology centers on the “crystal-inflammation” cascade, where MSU crystals trigger potent innate immune responses that cause articular damage and promote systemic comorbidities such as cardiovascular and renal diseases. 3
The management of gout relies on anti-inflammatory agents for acute flares and urate-lowering therapies (ULTs) for long-term control. However, a significant therapeutic gap persists. While effective ULTs exist, their clinical impact is often undermined by systemic under-prescription, suboptimal dosing, and poor adherence—making the misuse of available drugs a primary driver of unmet needs.3,4 This impasse underscores a fundamental challenge: the scarcity of novel, genetically validated targets has constrained drug development to incremental improvements on established pathways.
Serum urate concentration is primarily regulated by renal excretion, a process governed by transporters like URAT1 (SLC22A12) and GLUT9 (SLC2A9). 5 Hyperuricemia, the biochemical precursor to gout, arises from either urate overproduction, underexcretion, or more commonly, a combination of both. This physiological framework is essential for interpreting the functional consequences of genetic variants associated with the disease. The genetic architecture of urate metabolism provides a powerful framework for understanding gout pathogenesis. Given that up to 60% of serum urate variability is heritable, genomic studies are essential for identifying causal determinants. 6 Large-scale genome-wide association studies (GWASs) have now identified over 200 loci associated with serum urate and gout, profoundly expanding our understanding of its polygenic architecture. 7 However, most associated variants reside in non-coding regions, obscuring their functional gene targets and creating a translational gap.
Mendelian randomization (MR) can help bridge this gap by using genetic variants as instrumental variables to infer causal relationships between exposures (e.g. gene expression) and outcomes (e.g. gout), thereby mitigating confounding. 8 While MR has been applied to gout, 9 previous studies often lacked robust, multi-cohort validation and a comprehensive framework for prioritizing druggable targets.
To address these limitations, we implemented a dual-database MR framework. We integrated cis-eQTL data from the eQTLGen Consortium with gout GWAS from two independent cohorts—the large-scale openGWAS resource for discovery and the deeply phenotyped FinnGen cohort for validation.10,11 Our significant causal genes then entered a multi-layered validation pipeline featuring Bayesian colocalization, phenome-wide association studies (PheWASs) for safety profiling, single-cell RNA-seq for cellular contextualization, and molecular docking for druggability assessment. This integrated approach provides a robust template for derisking therapeutic target selection from the outset, thereby enhancing the potential for clinical success in gout drug development.
Methods
Data sources and processing for MR
Summary statistics for expression quantitative trait loci (eQTLs) were obtained from the eQTLGen Consortium's comprehensive meta-analysis of 37 cohorts (n = 31,684). 10 This dataset provides robust cis-eQTLs (SNPs located within 1 Mb of the gene body) for 19,942 protein-coding genes based on whole-blood expression profiles, primarily from individuals of European ancestry. As detailed in the original publication, the data underwent rigorous, standardized quality control, including correction for batch effects and adjustment for population stratification using principal component analysis. We used the pre-calculated and publicly available summary statistics for our analysis.
To focus our analysis on genes encoding potentially druggable proteins, we utilized the high-confidence “druggable genome” list curated by Finan et al. 12 This set of 4463 genes was defined based on evidence of binding by small molecules with high potency, supported by data from chemical repositories (e.g. ChEMBL v27) and drug databases (e.g. DrugBank v5.0). The criteria include proteins targeted by approved drugs as well as those with significant preclinical evidence of ligandability.
To generate our final set of instrumental variables, we intersected the eQTLGen dataset with the Finan et al. druggable gene list. This initial matching identified druggable genes that had at least one significant cis-eQTL in the eQTLGen data. To enhance the specificity of the instruments and minimize potential confounding from long-range linkage disequilibrium (LD), we applied an additional stringent filter, retaining only cis-eQTLs located within a narrow window of ±5 kb from the transcription start site (TSS) or transcription termination site (TTS) of each target gene. This process resulted in a final curated set of 2554 druggable genes, each with at least one high-confidence, proximal cis-eQTL to be used as a genetic instrument in the downstream MR analysis.
Outcome data sources and phenotype definitions
Discovery cohort: GWAS summary statistics for our primary discovery analysis were sourced from the UK Biobank (UKB) project, made available through the IEU OpenGWAS platform (https://gwas.mrcieu.ac.uk, ID: ukb-b-13251). This dataset comprises 462,933 individuals of European ancestry. Cases were defined as individuals with a diagnosis of gout (n = 6543) based on International Classification of Diseases, 10th Revision (ICD-10) code M10 from hospital inpatient records. Crucially, this definition rigorously excluded cases of secondary gout, such as those related to chemotherapy or renal insufficiency, to ensure a more homogenous disease phenotype. Genetic associations were available for ∼9.8 million variants genotyped on the UK Biobank Axiom array and imputed to a reference panel.
Validation cohort: For independent validation, we used GWAS summary statistics from FinnGen Release 10 (finngen_R10_M13_GOUT, https://www.finngen.fi). This cohort included 150,797 Finnish individuals, among whom 3576 gout cases were identified using a robust phenotyping algorithm combining ICD-10 codes, prescription records for urate-lowering therapies, and national hospitalization data. This dataset provided ∼16.3 million variants for replication.
Single-cell RNA sequencing data
For profiling candidate gene expression across pertinent immune cell subsets, scRNA-seq data were retrieved from the GEO repository (Accession ID: GSE211783). This dataset contains peripheral blood mononuclear cells (PBMCs) from three gout patients diagnosed according to ACR criteria, with samples collected during both the acute flare and remission phases, allowing for analysis of disease-state-specific expression. 13
MR analysis
No a priori sample size calculation was performed as this study utilizes summary statistics from fixed-size consortia; however, the large sample sizes of the eQTLGen, UK Biobank, and FinnGen cohorts provide substantial statistical power for MR analyses. All statistical analyses were conducted using R (version 4.3.1) with the TwoSampleMR package (version 0.5.7). The study design was predicated on the three core assumptions of MR: (1) genetic instruments must be directly associated with the exposure (gene expression, FDR < 0.05); (2) the instruments must not be associated with any confounders of the exposure–outcome relationship; and (3) the instruments must influence the outcome exclusively through the exposure pathway (the exclusion restriction criterion). 8
To ensure the validity of our instruments and the robustness of our causal inferences, we adhered to established MR quality metrics and reporting guidelines, which are critical for minimizing bias in summary-data MR. 14 First, to minimize bias from population stratification, all genetic instruments were selected from studies of European-ancestry cohorts. Weak instruments were excluded by applying a threshold of F-statistic > 10 (calculated as (β/SE)²). 15 Subsequently, LD clumping was performed using the 1000 Genomes Project reference panel, 16 retaining only the most significant single-nucleotide polymorphisms (SNPs) (lowest P-value) within a 10,000 kb window and with an r² < 0.2. Finally, the alleles of the instruments and outcome data were harmonized using the “harmonize_data()” function to ensure alignment.
The primary causal estimates were calculated using the inverse variance weighted (IVW) method for genes with multiple instruments, as it provides the highest statistical power under the assumption of no horizontal pleiotropy. 17 For genes with a single valid instrument, the Wald ratio method was employed. To complement the primary analysis, we also utilized the weighted median and MR-Egger regression methods. The weighted median method can provide a robust estimate even if up to 50% of the instruments are invalid, 18 while MR-Egger regression can detect and adjust for directional pleiotropy, albeit with reduced power. 19 Concordance in the direction and magnitude of effects across these different methods was considered indicative of a robust causal association, less likely to be biased by pleiotropy.
Sensitivity analyses were performed to detect potential violations of MR assumptions. For genes with two or more instruments, we assessed directional pleiotropy using the MR-Egger intercept test (significant if P < 0.05) and evaluated heterogeneity among instruments using Cochran's Q statistic. 20 In the discovery phase, a gene was considered a significant hit if it met a significance threshold of P < 0.05 in the IVW analysis, showed concordant effect directions across methods, and exhibited no significant evidence of pleiotropy or heterogeneity. All hits that passed these QC checks were then advanced for replication in the FinnGen cohort, with P < 0.05 used as the significance threshold for validation.
Enrichment analysis
We performed functional enrichment analysis using the clusterProfiler package in R to explore the underlying biological mechanisms of target genes. This analysis aimed to identify over-represented Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Specifically, GO analysis was conducted to characterize the genes across its three domains: biological process (BP), molecular function (MF), and cellular component (CC). Concurrently, KEGG pathway analysis was employed to identify the key metabolic and signaling pathways in which these genes collectively function, thereby providing insights into the pathogenesis of gout.
Protein–protein interaction (PPI) network construction and analysis
To explore the functional interplay and potential coregulatory patterns among the validated gout-associated genes, a PPI network was constructed using the GeneMANIA platform (https://genemania.org). GeneMANIA builds a weighted network by integrating data from various evidence sources. 21 In our analysis, the evidence types were weighted according to their biological relevance as follows: physical interactions (weight = 0.6), co-expression (0.3), and genetic interactions (0.1). To ensure the reliability of the predicted interactions, a global confidence score threshold of >0.4 was applied. The primary objective of this analysis was to visualize the interaction landscape of the identified proteins and to pinpoint potential hub genes that may serve as key regulators in the molecular pathways of gout.
Bayesian colocalization analysis
Bayesian colocalization analysis was conducted to test if genetic association signals for gene expression (eQTLs) and gout at specific genomic loci share common causal variants. This was conducted for all genes that demonstrated a significant causal association in the cross-cohort MR analysis, using the coloc package in R. 22
The coloc method evaluates five mutually exclusive hypotheses and calculates their respective posterior probabilities (PPs):
PPH0: No association with either gene expression or gout in the region. PPH1: Association with gene expression only. PPH2: Association with gout only. PPH3: Association with both traits, but driven by distinct causal variants. PPH4: Association with both traits, driven by a single, and shared causal variant.
A locus was considered to have strong evidence of colocalization if the posterior probability for a shared causal variant (PPH4) exceeded 0.80. This stringent threshold indicates a > 80% probability that a single genetic variant is responsible for both the eQTL and the gout association signal, representing a widely accepted standard for robustly identifying shared etiology in complex traits. 22 Genes meeting this criterion were prioritized for further investigation as high-confidence causal candidates. Finally, for loci exhibiting strong colocalization, the genomic region, including the association signals for both traits, was visualized to confirm the alignment of the signals.
PheWAS to assess target pleiotropy
A PheWAS was conducted to systematically assess pleiotropic effects of genetically prioritized gout-related genes. This analysis aimed to identify any associations between genetic variants in our target genes and a wide spectrum of other human diseases and traits, thereby providing crucial insights into potential on-target adverse effects or therapeutic repurposing opportunities.
The analysis was performed using the AstraZeneca PheWAS portal (https://azphewas.com/), which leverages exome sequencing data from ∼450,000 UK Biobank participants. 23 This powerful resource links genetic data to a comprehensive catalogue of over 17,000 phenotypes, including ∼15,500 binary phenotypes (disease outcomes) derived from electronic health records (EHRs) and ∼1500 continuous quantitative traits from standardized assessments. We adhered to the platform's established analytical pipelines for conducting the association tests. To rigorously control for the high multiple-testing burden inherent in scanning thousands of phenotypes, we applied a stringent, exome-wide significance threshold of P < 2 × 10⁻⁹, in line with the platform's standards for minimizing false-positive discoveries.
Single-cell RNA-seq data processing and analysis
scRNA-seq data processing was performed via Seurat (v4.4.0) within R (v4.3.1). Raw count matrices were first loaded to create Seurat objects. We then performed standard quality control and pre-processing steps, including log-normalization of counts (NormalizeData) and scaling of the data (ScaleData) to mitigate technical artifacts related to sequencing depth and capture efficiency. To correct for batch effects arising from different samples, we utilized the harmony algorithm (v1.0.3), which integrates the datasets into a shared, harmonized embedding.
Following integration, we performed principal component analysis (PCA) on the scaled data for dimensionality reduction. The resulting harmonized principal components were used to construct a shared nearest neighbor (SNN) graph, which was then subjected to graph-based clustering to partition cells into distinct groups (FindClusters). For robust cell type identification, we employed a reference-based annotation strategy using the SingleR package (v2.2.0), which assigned biological identities to each cluster based on established cell marker atlases. The distribution of these annotated cell populations was visualized using uniform manifold approximation and projection (UMAP).
With cells robustly annotated, we proceeded to two key downstream analyses. First, we quantified the relative proportions of each cell type in the gout flare-up versus remission samples to identify compositional shifts associated with the disease state. Second, to directly validate our findings from the MR analysis, we identified differentially expressed genes (DEGs) across all cell populations using the FindAllMarkers function. Specifically, we generated violin plots to visualize the expression levels of our prioritized MR-supported target genes, confirming their expression patterns within specific immune cell subsets relevant to gout pathogenesis.
Computational prediction and prioritization of drug candidates for molecular docking
In order to identify and prioritize potential drug candidates for molecular docking, we first performed a computational screen using the Drug Signature Database (DSigDB, http://dsigdb.tanlab.org/DSigDBv1.0/). This curated resource integrates 17,389 experimentally validated compounds with 22,527 gene sets across 19,531 protein-coding genes, establishing comprehensive ligand‒target associations through multiomics evidence. 24 From the comprehensive list of compounds generated by DSigDB, we implemented a multi-step prioritization strategy designed to balance statistical significance with structural and pharmacological relevance for subsequent docking analysis. Our strategy involved three key stages: (1) Initial statistical screening: all compounds showing a statistically significant association with our drug targets (P < 0.05) were selected to form an initial candidate pool. (2) Structural data availability: we filtered this pool to retain only those compounds with publicly available 3D structures in databases such as PubChem, a prerequisite for docking. (3) Pharmacological relevance and structural diversity: finally, we selected a representative set of molecules for docking. Rather than relying solely on statistical rankings (e.g. the lowest P-value), we deliberately included compounds with well-established pharmacological profiles (e.g. digoxin) and diverse chemical scaffolds. This approach facilitates a broader exploration of the target's binding pocket properties and enhances the likelihood of identifying compounds with favorable binding characteristics that might otherwise be overlooked. 25 For instance, while a small molecule like valproic acid may have a strong statistical association, its simple structure often yields nonspecific, low-affinity binding in simulations. In contrast, a larger, more complex molecule such as digoxin, despite a lower statistical rank, provides a better opportunity to probe specific, high-affinity interactions. This comprehensive framework was designed to identify not only novel bioactive compounds but also established drugs with favorable safety profiles suitable for repurposing, while ensuring a reproducible prioritization process by adhering to DSigDB's standardized protocols.
Molecular docking validation of drug–target interactions
We performed molecular docking simulations to validate interactions between prioritized compounds and their protein targets, and to elucidate their atomistic binding mechanisms. Simulations were conducted using AutoDock Vina (v1.2.2). Protein and ligand 3D structures were sourced from the RCSB Protein Data Bank (http://www.rcsb.org/) and the PubChem database (https://pubchem.ncbi.nlm.nih.gov/), respectively. Prior to docking, all structures underwent preprocessing, which included the removal of solvation shells, optimization of hydrogen atoms, and energy minimization. The docking results were evaluated based on the calculated binding free energy (ΔG, in kcal/mol), with more negative values indicating higher binding affinity. Ligand-protein complexes with a strong binding affinity (ΔG < −8.0 kcal/mol) were prioritized as lead candidates for further analysis. The specific molecular interactions within these high-affinity complexes were then visualized and analyzed using PyMOL (v2.5). A flowchart summarizing this computational workflow is presented in Figure 1.

Schematic overview of the multi-layered study design. The workflow begins with a dual-cohort Mendelian randomization (MR) analysis (discovery and validation phases) using the eQTLGen and FinnGen databases to identify causal genes for gout. Significant genes underwent functional enrichment analysis (Gene Ontology/Kyoto Encyclopedia of Genes and Genomes (GO/KEGG)) and colocalization to confirm causality. Lead candidates were further evaluated for cell-type-specific expression using single-cell RNA-Seq data and for druggability using molecular docking simulations, culminating in a prioritized therapeutic hypothesis.
Results
A two-stage MR analysis identifies 15 high-confidence gout-associated genes
To systematically identify and validate druggable genes with a causal role in gout, we implemented a two-stage MR analysis pipeline.
In the discovery stage, we analyzed GWAS data from the UK Biobank, which included 6543 gout cases and 456,390 controls. Our initial two-sample MR screen identified 190 genes whose genetically predicted expression was significantly associated with gout risk (P < 0.05; Figures 2 and 3). Following rigorous sensitivity analyses, five of these genes (SWAP70, ISL2, EBF1, STAT6, and MTX1) were excluded due to evidence of horizontal pleiotropy (Supplemental Tables S1 to S3). This process resulted in 185 candidate genes being advanced to the replication stage.

Manhattan plot of Mendelian randomization (MR) results from the discovery phase. The y-axis represents the −log10(P-value) for the causal association between gene expression and gout risk. Each point represents a gene, organized by chromosomal position on the x-axis. Significant genes are labeled and highlighted in red.

Forest plot of Mendelian randomization (MR) results for key candidate genes from the discovery phase. The plot displays the odds ratio (OR) and 95% confidence interval (CI) for the association of each gene's expression with gout risk. Squares represent the OR, with blue indicating a risk-decreasing effect and yellow indicating a risk-increasing effect. Horizontal lines represent the 95% CI. The vertical dashed line at OR = 1 indicates no effect.
For the replication stage, we utilized an independent GWAS dataset from the FinnGen cohort, comprising 3576 gout cases and 147,221 controls. Of the 185 candidate genes tested, 15 demonstrated a significant and directionally consistent causal association with gout (P < 0.05; Figure 4). Crucially, all 15 of these prioritized targets passed all sensitivity analyses in both cohorts, showing no evidence of pleiotropy or heterogeneity (Supplemental Tables S4 to S6). These 15 genes were therefore designated as high-confidence targets for all subsequent downstream analyses.

Forest plot of Mendelian randomization (MR) results for 15 significant genes from the validation phase. The plot displays the odds ratio (OR) and 95% confidence interval (CI) for the association of each gene's expression with gout risk. Squares represent the OR, with blue indicating a risk-decreasing effect and yellow indicating a risk-increasing effect. Horizontal lines represent the 95% CI. The vertical dashed line at OR = 1 indicates no effect.
Functional enrichment analysis pinpoints immune regulation and metabolic pathways
We performed GO and KEGG pathway enrichment analyses (Figures 5 and 6).

Gene Ontology (GO) enrichment results for three terms.

Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment results.
GO analysis highlights T-cell-mediated immunity and membrane dynamics. Within the BP domain, we observed a profound enrichment of pathways centered on immune cell regulation, particularly “regulation of T-cell differentiation,” “regulation of lymphocyte differentiation,” and “T-cell differentiation.” This points to T-cell maturation and activity as a pivotal event in the gout inflammatory microenvironment. The additional enrichment of “positive regulation of T-cell-mediated cytotoxicity” suggests a dual role for T-cells, contributing to both inflammatory clearance and potential tissue damage. The CC analysis implicated membrane dynamics, with top terms including “recycling endosome” and “external side of the plasma membrane.” In the MF domain, significant terms such as “G-quadruplex DNA binding” and “insulin receptor substrate binding” linked these genes to epigenetic regulation and metabolic signaling.26,27
KEGG analysis identifies key hub genes in immune and metabolic pathways. Our KEGG analysis further contextualized these findings by identifying crucial hub genes operating across multiple pathways (Figure 6). Immune regulation: HLA-G, a non-classical MHC class I molecule, was prominently featured in “Allograft rejection,” “Antigen processing and presentation,” and “Graft-versus-host disease,” suggesting a potential compensatory role in the dysregulated local immune environment of the joint. The enrichment of IL12RB1 in the “Inflammatory bowel disease” pathway further supports a potential gut-joint axis, where systemic inflammation may exacerbate gout. Metabolic disruption: On the metabolic front, AK4 (adenylate kinase 4) was enriched in “Thiamine metabolism” and “Nucleotide metabolism,” directly connecting it to the purine metabolic network that ultimately governs uric acid production. Furthermore, CERS4 (ceramide synthase 4) was implicated in sphingolipid pathways known to mediate insulin resistance, a critical comorbidity and risk factor for metabolic syndrome and gout. 28 Collectively, these enrichment results provide a clear biological context for the validated genes, strongly implicating interconnected pathways of T-cell-mediated immunity and metabolic dysregulation in the pathogenesis of gout.
PPI network reveals a tightly knit functional module centered on immune regulation
We constructed a PPI network to investigate the potential synergistic roles of these 15 validated gout-associated genes. The analysis revealed that our target genes do not act in isolation but form a highly interconnected functional module (Figure 7).

Protein–protein interaction (PPI) network constructed with GeneMANIA.
The 15 validated genes served as core “seed” nodes. The GeneMANIA algorithm expanded this core by identifying an additional 19 proteins that have strong evidence of co-expression or genetic interaction with the seeds. The final network consisted of 34 nodes and 102 edges, indicating a dense web of biological relationships.
Functional annotation of the entire network module powerfully reinforced our previous findings. The most significantly enriched functions within the network were “antigen processing and presentation,” the “MHC protein complex,” and “positive regulation of T-cell-mediated immunity.” This convergence provides a structural basis for the abstract pathway data, suggesting that the causal effects of these genes on gout are mediated through a coordinated biological network with the T-cell immune axis at its core.
Colocalization analysis prioritizes ZSCAN16 and TRIM10 as high-confidence therapeutic targets
In order to determine whether the associations identified by MR were driven by a shared causal variant or were merely artifacts of linkage disequilibrium (LD), we conducted Bayesian colocalization analysis. 29 This analysis was performed on all gene-gout pairs that showed a significant causal effect in the primary MR screen.
Of the genes tested, we found robust evidence of colocalization for two distinct signals: ZSCAN16 and TRIM10 (Table 1). Both loci yielded a high posterior probability for Hypothesis 4 (PPH4 > 0.8), indicating that the same SNP is responsible for modulating both gene expression (exposure) and gout risk (outcome). In contrast, other key genes from the enrichment analysis, such as HLA-G and AK4, did not show significant evidence of colocalization (PPH4 < 0.8).
Colocalization analysis of 15 candidate genes identifies shared causal variants with gout.
These findings (graphically summarized in Figure 8) substantially strengthen the causal evidence for ZSCAN16 and TRIM10 in the etiology of gout. They are therefore prioritized as high-confidence candidate genes for future functional studies and potential therapeutic targeting.

Colocalization analysis of gout genome-wide association study (GWAS) signals and gene expression quantitative trait loci (eQTLs). Regional association plots are shown for (a) the ZSCAN16 locus and (b) the TRIM10 locus. In each panel, the top plot displays the GWAS P-values for gout association, and the bottom plot shows eQTL P-values from eQTLGen. The lead variant is marked by a purple diamond. The color of each point indicates its linkage disequilibrium (r²) with the lead variant. A high posterior probability of a shared causal variant (PP4 > 0.8) indicates strong evidence of colocalization.
PheWAS reveals high specificity of ZSCAN16 and potential pleiotropy of TRIM10
We queried genetically predicted expression of TRIM10 and ZSCAN16 against ∼15,500 dichotomous and ∼1500 quantitative phenotypes from the AstraZeneca PheWAS portal. 23
The results revealed a significant pleiotropic profile for TRIM10 (Table 2). At a stringent significance threshold (P < 2 × 10−9), increased TRIM10 expression was associated with multiple cardiometabolic and hematological traits. Notably, we observed a positive association with the “Cardiometabolic II” protein panel and negative associations with red blood cell metrics, including reticulocyte count and percentage (Supplemental Figures S1 and S2). These findings suggest that TRIM10 has a functional role in both cardiometabolic regulation and hematopoiesis. Consequently, while TRIM10 is a validated causal gene for gout, its broad biological footprint warrants caution, as targeting it therapeutically could carry a risk of unintended cardiovascular or hematological side effects.
Significant pleiotropic associations of genetically predicted TRIM10 expression identified via the AstraZeneca PheWAS portal.

In striking contrast, ZSCAN16 demonstrated remarkable specificity. No significant associations were found between genetically predicted ZSCAN16 expression and any of the thousands of phenotypes tested (P ≥ 2 × 10−9 for all traits; Supplemental Figures S3 and S4). This lack of pleiotropy suggests that the biological impact of ZSCAN16 is narrowly confined to pathways directly relevant to gout. From a drug development perspective, this high degree of specificity makes ZSCAN16 a more attractive and potentially safer therapeutic target.
Single-cell analysis pinpoints ZSCAN16 activity to T/ NK cells during gout flares
To resolve the precise cellular context in which our top drug targets operate, we performed single-cell RNA sequencing on PBMCs from patients during both gout attack (n = 3) and remission (n = 3). Unsupervised clustering identified 11 distinct cell populations, which were annotated into four major lineages: T/NK cells, myeloid cells, B cells, and a small unclassified population (Figure 9(a) to (c)). Notably, the overall immune landscape shifted during flares, characterized by an expansion of B cells and a contraction of the T/NK cell compartment (Figure 9(d)).

Single-cell analysis of gout flares and remission samples. (a) t-SNE visualization of all cells partitioned into 11 clusters, color-coded by cluster identity. (b) UMAP projection of clustered cells, identifying T/NK cells, myeloid cells, B cells, and 1 unknown cell type. (c) Cohort-specific color coding: Group A (flare-phase samples) versus Group B (remission-phase samples). (d) Bar plot quantifying leukocyte subtype proportions across disease states. (e) Violin plots comparing normalized ZSCAN16 expression across major cell types between flare and remission samples, with *** P < 0.001.
Within this dynamic cellular environment, we analyzed the expression of our final candidate genes. The results revealed a striking and highly specific pattern for ZSCAN16. Its expression was significantly upregulated specifically within the T/NK cell population of attack-phase samples compared to remission-phase samples. No such differential expression was observed in myeloid cells or B cells (Figure 9(e)). This finding provides a critical mechanistic insight, suggesting that the pro-gout effect of ZSCAN16 is executed primarily through its activity in T or NK cells during an acute inflammatory flare.
In contrast, the expression of TRIM10 showed no significant changes between attack and remission states in any of the identified cell lineages (Supplemental Figure S5). While this does not disqualify its potential role in gout, the lack of dynamic regulation during an acute flare, combined with its pleiotropic profile from the PheWAS, positions ZSCAN16 as the more compelling and specific target for therapeutic intervention aimed at controlling gout attacks.
Druggability assessment identifies ZSCAN16 as a chemically tractable target
We interrogated DSigDB to identify compounds reported to modulate the expression of ZSCAN16 and TRIM10. Subsequently, we performed molecular docking on a prioritized subset of these compounds to predict their binding affinities and interaction modes with proteins.
The analysis for ZSCAN16 yielded promising results. DSigDB identified several existing drugs whose transcriptional signatures were inversely correlated with that of ZSCAN16, including valproic acid, bisacodyl, and digoxin (Table 3). This finding not only provides a list of candidate molecules for drug repurposing but also establishes ZSCAN16 as a chemically tractable target, providing a clear path forward for molecular docking simulations.
Candidate modulators for ZSCAN16 and TRIM10 identified via the DSigDB.

In contrast, assessing the druggability of TRIM10 proved challenging. DSigDB returned only a few non-specific hits (e.g. ethanol) that were unsuitable as tool compounds. To ensure this was not a limitation of a single database, we performed an exhaustive search of the literature and major chemical repositories, including ChEMBL and DrugBank. This comprehensive screen failed to identify any validated, specific small-molecule inhibitors or modulators for TRIM10.
This definitive lack of known chemical matter suggests that TRIM10 has poor chemical tractability at present. Consequently, meaningful molecular docking was not feasible for this target. Therefore, our subsequent structural and in silico analyses will focus exclusively on the more promising and chemically tractable target, ZSCAN16.
Structural validation of ZSCAN16 druggability via molecular docking
To structurally validate ZSCAN16 as a druggable target and to predict how the previously identified compounds might bind, we performed molecular docking against the ZSCAN16 zinc finger domain. The analysis was conducted using the top candidate molecules derived from our DSigDB screen.
Our simulations revealed that several compounds could bind favorably within a well-defined pocket on the ZSCAN16 protein structure (Table 4 and Figure 10). Strikingly, digoxin, a complex cardiac glycoside, emerged as the top candidate, exhibiting an exceptional binding affinity with a binding affinity of −9.6 kcal/mol. The ability of the ZSCAN16 pocket to accommodate a large and structurally complex approved drug like digoxin provides powerful evidence for its druggability. Other compounds, including proscillaridin (−8.3 kcal/mol) and bisacodyl (−7.6 kcal/mol), also displayed significant binding energies, reinforcing the conclusion that this pocket is amenable to modulation by diverse chemical scaffolds.
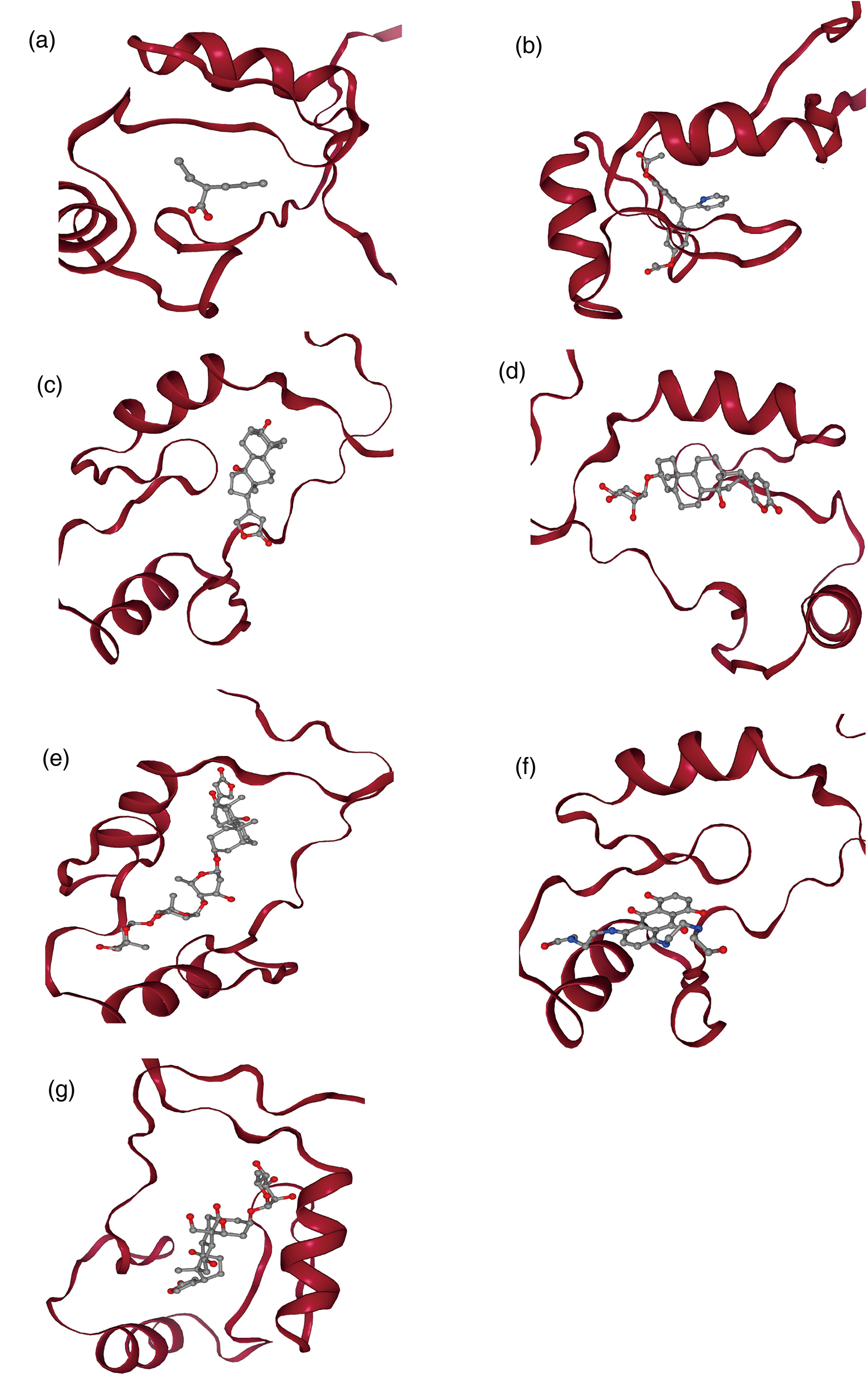
Predicted binding poses of top-ranking compounds in the ZSCAN16 zinc finger domain. (a) ZSCAN16 docking with valproic acid (binding energy: −4.6 kcal/mol). (b) ZSCAN16 docking bisacodyl (binding energy: −7.6 kcal/mol). (c) ZSCAN16 docking of digitoxigenin (binding energy: −6.8 kcal/mol). (d) ZSCAN16 docking with proscillaridin (binding energy: −8.3 kcal/mol). (e) ZSCAN16 docking digoxin (binding energy: −9.6 kcal/mol). (f) ZSCAN16 docking with mitoxantrone (binding energy: −6.8 kcal/mol). (g) ZSCAN16 docking with ouabain (binding energy: −7.7 kcal/mol).
Molecular docking and predicted binding affinities of candidate compounds with the ZSCAN16 zinc finger domain.

Interestingly, valproic acid, despite its strong signature in our transcriptomic analysis, exhibited a very weak binding affinity (−4.6 kcal/mol). This apparent discrepancy is mechanistically informative. The small, simple structure of valproic acid likely lacks the necessary complexity and specific functional groups to form the multiple, high-energy contacts required for stable binding in the pocket. This finding highlights the synergy of our approach, where transcriptomic data identifies potential modulators and structural analysis refines these candidates based on biophysical plausibility.
In conclusion, our molecular docking analysis provides strong structural evidence that ZSCAN16 is a druggable target. The demonstration of a concrete binding site capable of engaging drug-like molecules with high affinity offers a clear and promising starting point for future experimental validation and structure-based drug design campaigns.
Discussion
This study systematically identified potential genetic drivers of gout pathogenesis via a dual-database MR framework. Integrated analysis of the utilized eQTL data from the eQTLGen consortium and gout GWAS data from the UK Biobank and FinnGen cohorts identified ZSCAN16 and TRIM10 as high-priority candidate targets whose genetic variants significantly influence gout risk by regulating gene expression. The results demonstrated the association of ZSCAN16 with increased gout risk and the correlation of TRIM10 with reduced susceptibility (Supplemental Figure S6). Our dual-database validation strategy estimated the causal effect using the primary IVW method and assessed potential pleiotropic bias using sensitivity analyses, including MR-Egger regression, while establishing multidimensional evidence through colocalization analyses and PheWASs. Single-cell sequencing strengthened the biological rationale for ZSCAN16's causal role through marked expression differences in T/NK cells, whereas the pleiotropic associations of TRIM10 revealed complex cardiometabolic interactions, suggesting therapeutic windows. This pioneering integration of genetic evidence with single-cell functional profiling establishes a novel precision therapeutic paradigm for gout. Finally, we predicted and molecularly docked target-corresponding drugs, substantiating their therapeutic potential.
As a C2H2-type zinc finger protein, ZSCAN16 likely functions as a transcriptional regulator whose activity is strategically positioned at the nexus of immune activation and metabolic stress in gout. Our single-cell data reveal its flare-specific overexpression in T/NK cells, a finding that invites a more granular biological interpretation. We hypothesize that ZSCAN16 operates through at least two interconnected immunological axes. First, it may serve as an epigenetic amplifier of T-helper 17 (Th17) polarization. C2H2 zinc fingers are increasingly recognized as modular readers of non-B DNA structures, including G-quadruplexes, which accumulate in promoters of key inflammatory genes under oxidative stress. 30 In gout, MSU crystals activate TLR4 signaling and generate reactive oxygen species that promote DNA damage and G-quadruplex formation. 31 Owing to its sufficiently predicted affinity for these structures, ZSCAN16 could bind to and stabilize G-quadruplexes in loci such as RORC or IL23R, thereby enhancing the transcriptional program that drives Th17 differentiation. This model aligns with the marked enrichment of Th17 cells in gout synovial fluid and their pathogenic role in neutrophil recruitment and inflammation perpetuation. 32
Further analysis suggests that ZSCAN16 may exert its influence through two interconnected mechanisms: epigenetic regulation and immunometabolic crosstalk. First, its predicted affinity for G-quadruplex DNA structures, which are enriched in gene promoters, offers a direct epigenetic mechanism. 27 In the context of gout, MSU crystals are known to activate Toll-like receptor 4 (TLR4) signaling, inducing oxidative stress that promotes DNA damage and the formation of G-quadruplexes. 31 The selective affinity of ZSCAN16 for these structures could enable it to bind and perpetuate the transcription of key inflammatory genes like IL1B and TNF, thus sustaining the inflammatory response—a model supported by emerging evidence of epigenetic reprogramming in gout-prone individuals.33,34 Furthermore, the observed interaction between ZSCAN16 and insulin receptor substrate (IRS) proteins points to a role in immunometabolic regulation. Given that IRS signaling defects contribute to hyperuricemia by affecting both uric acid biosynthesis and clearance, ZSCAN16 could exacerbate this metabolic dysfunction. 26 By potentially impairing IRS1/2 phosphorylation, ZSCAN16 may link metabolic dysregulation directly to inflammatory amplification.
Second, ZSCAN16 may directly influence innate immune cytotoxicity. NK cells contribute to gout pathogenesis through MSU-induced neutrophil extracellular trap (NET), a process modulated by transcription factors regulating perforin and granzyme expression. 35 ZSCAN16's upregulation in NK cells during flares suggests a role in licensing their cytolytic potential, possibly by transactivating genes encoding key cytolytic molecules such as PRF1 (perforin) or GZMB (granzyme B). 36 Such a mechanism would position ZSCAN16 as a molecular rheostat fine-tuning the balance between microbial clearance and inflammatory tissue injury—a duality characteristic of gout pathophysiology wherein neutrophil extracellular traps (NETs) play a well-documented dual role. 37 These proposed functions, while requiring experimental confirmation, provide a biologically coherent framework linking ZSCAN16's genetic association to actionable immune mechanisms.
A key strength of our study is the successful translation of a genetically validated target into a tangible therapeutic hypothesis via structural analysis and molecular docking. Our simulations not only confirm that ZSCAN16 is a druggable target but also reveal that its zinc finger domain binds known drug molecules with high affinity. Digoxin emerged as the top candidate, exhibiting the highest binding affinity (−9.6 kcal/mol). Detailed residue-level analysis of the ZSCAN16-digoxin complex revealed several key interactions. Notably, residues in the zinc-finger domain, including Arg 17, Tyr 19, Lys 20, His 30, Cys 21, Phe 28, Leu 34, and Ile 57, form crucial hydrogen bonds and hydrophobic interactions with digoxin. The Arg 17, Tyr 19, Lys 20, and His 30 residues, part of the zinc-coordination site, play a critical role in stabilizing the binding pocket, while Cys 21, Phe 28, Leu 34, and Ile 57 contribute to the hydrophobic core of the ligand-binding site, enhancing the overall affinity. 38 Conversely, valproic acid's weak binding (ΔG = −4.6 kcal/mol) stems from its small size and inability to form multiple simultaneous contacts—highlighting that transcriptional signatures alone cannot predict binding efficacy. Crucially, the identified binding pocket exhibits characteristics of druggable sites: substantial volume (412 ų), balanced hydrophobicity, and strategic polar residues for specific recognition. That this pocket accommodates diverse scaffolds while maintaining affinity relationships consistent with their pharmacological properties strongly validates ZSCAN16 as a chemically tractable target. 39 These findings align with known interactions observed in similar zinc-finger protein-ligand complexes, further validating the robustness of our docking predictions. Future experimental validation, including surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC), could provide further confirmation of these interactions.
However, it is crucial to interpret this finding with scientific caution, as digoxin itself is not a direct therapeutic candidate for gout. As a cardiac glycoside with a narrow therapeutic window and significant cardiotoxicity, 40 its clinical use is restricted to specific cardiac conditions under strict medical supervision and is unsuitable for chronic anti-inflammatory therapy. Instead, our findings establish digoxin as a vital “tool compound” or chemical probe that provides compelling evidence for the druggability of ZSCAN16. More importantly, the predicted digoxin-ZSCAN16 binding model provides a structural blueprint for future rational drug discovery. 41 Medicinal chemists can leverage this model to design novel, specific ZSCAN16 inhibitors using strategies like scaffold hopping or fragment-based design. 42 The goal would be to develop new chemical entities that retain the essential pharmacophore for high-affinity binding while eliminating the moieties responsible for Na⁺/K⁺-ATPase inhibition and cardiac toxicity. In summary, the identification of a potent interaction between digoxin and ZSCAN16 is not an endpoint, but a foundational step that validates the target and provides a concrete starting point for a structure-guided program to develop safer and more effective therapies for gout.
Although MR identified a causal protective association between TRIM10 and gout, our subsequent multi-layered assessment concludes that it represents a high-risk, low-reward drug target. The successful translation of a genetically validated target into a therapy hinges on overcoming two critical hurdles: biological safety and chemical tractability. 12 Our analysis reveals that TRIM10 faces significant, likely insurmountable, challenges on both fronts.
First, from a biological safety perspective, TRIM10 exhibits extensive pleiotropy—a major warning sign in drug development. Our PheWAS revealed that genetic variants linked to TRIM10 expression are robustly associated not only with gout but also with a range of fundamental hematopoietic phenotypes, such as reticulocyte count and percentage. This is strongly consistent with established literature reporting TRIM10's critical role in the erythroid differentiation of hematopoietic stem cells. 43 Because TRIM10's protective function in gout appears mechanistically inseparable from its core physiological roles, any systemic modulator would carry a high risk of triggering severe “on-target, off-tissue” adverse effects, such as anemia or other hematological disorders. 44 The predictability of these side effects drastically narrows the therapeutic window, making clinical failure due to safety concerns highly probable.
Second, TRIM10 presents a formidable chemical tractability challenge. As a member of the TRIM E3 ubiquitin ligase family, it belongs to a class of proteins notoriously difficult to target with conventional small molecules due to shallow, ill-defined catalytic sites. 45 Our findings confirm this inherent difficulty: an exhaustive search of available databases and literature yielded no known small-molecule modulators or “chemical probes” for TRIM10. The absence of such tools precludes any preliminary assessment of its druggability via computational methods and suggests that the de novo design of specific inhibitors would be a protracted and high-risk endeavor.
In conclusion, TRIM10 represents a challenging target due to a dual liability: high biological risk stemming from its pleiotropic nature and low chemical tractability arising from its structural properties. This rigorous “de-risking” process is a key strength of our research framework. By systematically evaluating and disqualifying targets that are genetically promising but practically flawed, our strategy prevents the misallocation of resources and effectively directs focus toward high-priority candidates like ZSCAN16, which possess superior biological specificity and demonstrated chemical tractability.
Our study's primary strength lies in its rigorous, multi-layered design, which establishes a complete evidence chain from genetic association to therapeutic hypothesis. By integrating a large-scale dual-database MR with FinnGen's phenotypic granularity, we robustly established causality. This finding was further solidified through sensitivity analyses that confirmed its stability and colocalization analyses (PPH4 > 0.8) that pinpointed the causal gene. Our findings align with and extend prior integrative genomics efforts in gout. For instance, recent colocalization analyses of genetic and metabolomic data have similarly pinpointed shared causal variants influencing both metabolite levels and gout risk, reinforcing the power of this approach to uncover functional mechanisms. 46 Critically, by validating the target's expression in specific cell types via single-cell data, we moved beyond conventional MR to link the genetic signal to a precise cellular context. This robust evidence chain provides a strong foundation for our final drug predictions, where molecular docking confirmed the target's druggability with high-affinity binding, thus translating a genetic discovery into a tangible therapeutic starting point.
Despite these strengths, we acknowledge several limitations that define the boundaries of our current findings. First, while MR establishes causality, its predicted effect sizes may not directly translate to clinical efficacy, nor can it fully anticipate the complex biological consequences of pharmacological intervention. Second, our findings are constrained by the scope of the datasets used; the reliance on European-centric data limits ethnic generalizability, and we recognize that the single-cell RNA sequencing data are based on a small sample size (n = 3 per group), which does limit the statistical power and robustness of the findings. Finally, our genomic analysis has methodological constraints: the cis-eQTL window (TSS ± 5 kb) employed by the eQTLGen consortium may overlook distal regulatory elements, and our approach does not account for potential trans-chromatin interactions.
While our study provides a robust genetic and computational foundation for ZSCAN16 as a therapeutic target, we acknowledge that it relies on publicly available datasets and lacks experimental or clinical validation. This is an inherent limitation of computational prioritization studies. Future work should focus on preclinical validation using cellular and animal models. While the single-cell RNA sequencing analysis was performed on a small sample size (n = 3 per group), it provides an initial but valuable insight into the cell-type-specific expression of ZSCAN16 in T/NK cells during acute gout flares. We have acknowledged the limitations of this sample size and have recommended future studies with larger cohorts to increase the statistical power of the findings. Additionally, we propose that multi-center studies and longitudinal designs involving larger patient populations, especially those from diverse ethnic backgrounds, will be essential for validating these results and assessing the generalizability of the conclusions. Lastly, the precision of our therapeutic predictions can be enhanced. Future efforts should focus on refining computational models by incorporating experimental, high-resolution protein structures for docking simulations and expanding genomic analyses to capture a more complete regulatory landscape.
Conclusion
In conclusion, our integrated multiomics framework prioritizes ZSCAN16 as a high-potential therapeutic target for gout. Conversely, TRIM10 was deprioritized owing to substantial pleiotropic liabilities and poor chemical tractability. These computational findings provide a strong rationale for subsequent functional studies in immune cells and animal models to validate target engagement and efficacy, ultimately guiding the development of novel precision therapies.
Supplemental Material
sj-xlsx-1-sci-10.1177_00368504251407179 - Supplemental material for From genetic causality to druggable targets: A multiomics framework identifies ZSCAN16 in gout pathogenesis
Supplemental material, sj-xlsx-1-sci-10.1177_00368504251407179 for From genetic causality to druggable targets: A multiomics framework identifies ZSCAN16 in gout pathogenesis by Chuanliang Chen, Xifan Zheng, Zeming Li, Hongtao Wang and Jun Yao in Science Progress
Supplemental Material
sj-docx-2-sci-10.1177_00368504251407179 - Supplemental material for From genetic causality to druggable targets: A multiomics framework identifies ZSCAN16 in gout pathogenesis
Supplemental material, sj-docx-2-sci-10.1177_00368504251407179 for From genetic causality to druggable targets: A multiomics framework identifies ZSCAN16 in gout pathogenesis by Chuanliang Chen, Xifan Zheng, Zeming Li, Hongtao Wang and Jun Yao in Science Progress
Supplemental Material
sj-docx-3-sci-10.1177_00368504251407179 - Supplemental material for From genetic causality to druggable targets: A multiomics framework identifies ZSCAN16 in gout pathogenesis
Supplemental material, sj-docx-3-sci-10.1177_00368504251407179 for From genetic causality to druggable targets: A multiomics framework identifies ZSCAN16 in gout pathogenesis by Chuanliang Chen, Xifan Zheng, Zeming Li, Hongtao Wang and Jun Yao in Science Progress
Footnotes
Acknowledgements
We thank the eQTLGen consortium, the FinnGen team, and other researchers and participants for providing publicly available data for this analysis.
Ethical considerations
This study was conducted using exclusively publicly available and de-identified summary-level data from the eQTLGen, OpenGWAS, and FinnGen consortia. In accordance with institutional guidelines and the principles of the Declaration of Helsinki, this secondary analysis did not require separate ethical approval or participant consent. All primary studies that contributed to these datasets obtained informed consent from all participants and received approval from their respective institutional review boards and regulatory bodies. Specifically, the UK Biobank dataset (accessed via OpenGWAS) was approved by the North West Multi-Centre Research Ethics Committee (REC ref: 21/NW/0157), and the FinnGen study was approved by the Finnish Institute for Health and Welfare (THL/1731/6.02.00/2020).
Consent to participate
Informed consent was not sought for the present study because the original research participants provided written informed consent.
Author contributions
Chuanliang Chen: methodology, validation, writing–original draft, and writing–review and editing. Xifan Zheng: formal analysis, methodology, software, and writing–original draft. Zeming Li: data curation, formal analysis, investigation, and writing–review and editing. Hongtao Wang: data curation, resources, and writing–review and editing. Jun Yao: funding acquisition, methodology, project administration, supervision, and writing–review and editing. All the authors reviewed and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data availability statement
The primary datasets analyzed in this study are publicly available. cis-eQTL summary statistics can be accessed through the eQTLGen Consortium portal (https://eqtlgen.org). Gout GWAS summary statistics were obtained from the OpenGWAS database (ID: ukb-b-13251; https://gwas.mrcieu.ac.uk) and the FinnGen project (ID: finngen_R10_M13_GOUT; https://www.finngen.fi). Single-cell RNA-sequencing data are deposited in the Gene Expression Omnibus (GEO) repository under accession number GSE211783 (![]() ). All analysis code and processed data generated to support the findings of this study are available from the corresponding author upon reasonable request.
). All analysis code and processed data generated to support the findings of this study are available from the corresponding author upon reasonable request.
Clinical trial number
Not applicable. As a computational Mendelian randomization study leveraging preexisting GWAS datasets, this work did not involve direct patient recruitment, clinical interventions, or prospective data collection.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.