
Abstract
Hepatocellular carcinoma (HCC) is one of the most common cancers in the world. The landscape of HCC’s molecular alteration signature has been explored over the last few decades. Even so, more comprehensive research is still needed to improve understanding of tumorigenesis and progression of HCC, as well as to identify potential biomarkers for the malignancy. In this research, a comprehensive bioinformatics analysis was conducted based on the publicly available databases from both the Cancer Genome Atlas (TCGA) program and the gene expression omnibus (GEO) database. R/Bioconductor was used to analyze differentially expressed genes (DEGs) between HCC tumor and normal control (NC) samples, and then a protein-protein interaction (PPI) network of DEGs was established through the STRING platform. Finally, the application of specific candidate genes as diagnostic or prognostic biomarkers of HCC was explored and evaluated by ROC and survival analysis. A total of 310 DEGs were detected in the HCC tumor samples. Thirty-six hub DEGs in the PPI network and 10 candidates of the 36 genes showed significant alterations in tumor expression, including CDKN3, TOP2A, UBE2C, CDC20, PBK, ASPM, KIF20A, NCAPG, CCNB2, CYP3A4. The 10-gene signature had relatively significant effects when distinguishing tumors from normal samples (sensitivity >70%, specificity >70%, AUC >0.8, p < 0.001). Eight candidate genes were negatively correlated with the overall survival rate of the patients (p < 0.05) and were all up-regulated in HCC tumor samples. The age and gender factors had no significant impact on the overall survival rate of HCC patients (p > 0.05), and the TNM stage status factor had a significant negative prognosis correlation (p < 0.05). This research provides evidence for a better understanding of tumorigenesis and progression of HCC and helps to explore candidate targets for disease diagnosis and treatment.
Keywords
Introduction
Hepatocellular carcinoma (HCC) is mainly caused by chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infection and the subsequent hepatic cirrhosis represents the second leading cause of cancer-related deaths and the fifth most common cancer in the world. 1 Overall, 44% of HCC cases were due to chronic hepatitis B infection, with the majority of cases occurring in Asia where the prevalence of viral hepatitis is higher. 2 What’s more, HCC is highly resistant to therapy and therefore difficult to treat. 3 Early diagnosis followed by surgery is a key factor for a favorable prognosis of HCC patients. However, most patients were at an advanced stage at the time of diagnosis and the 5-year survival rates still <12.5% in China. 4
It’s well accepted that the pathogenesis and progression of malignancies is a complex and multi-stage procedure.5,6 Over the last few decades, the proposal for precision medicine in the field of cancer diagnosis/therapy has advanced dramatically with the development of multi-Omics including genomics, transcriptomics, proteomics, or metabolomics, et al.7–9 For HCC, the next-generation sequencing studies have significantly contributed to the development of the landscape of molecular alteration signature. 3 Despite these studies, further studies are still needed to combine the current understanding of the biological characteristics of HCC with potential diagnostic and prognostic biomarkers to better assist clinicians in clinical decision-making and ultimately improve disease outcomes.
In this study, the global pattern of gene expression and potential diagnostic or prognostic biomarkers was analyzed and identified based on online public bioinformatics databases. Specifically, two datasets from the Cancer Genome Atlas (TCGA) program and the Gene Expression Omnibus (GEO) database of the National Cancer for Biotechnology Information (NCBI) were selected to obtain a more comprehensive gene expression signature and more convincing results. In addition, a protein-protein interaction (PPI) network was established and analyzed to better understand the underlying pathogenesis and progression of HCC.
Materials and methods
Sources of data
In this study, a comprehensive/global analysis of the HCC gene expression signature was conducted by combining two datasets, including dataset1 from TCGA program 10 and dataset2 from the GEO database. 11 Specifically, the dataset1 was downloaded through the UCSC Xena website (https://gdc.xenahubs.net/download/TCGA-LIHC.htseq_counts.tsv.gz, https://gdc.xenahubs.net/download/TCGA-LIHC.GDC_phenotype.tsv.gz, https://gdc.xenahubs.net/download/TCGA-LIHC.survival.tsv.gz), and the above includes data on RNAseq gene expression, phenotype, and clinical data from 374 HCC tumor samples and 50 normal control (NC) samples. The basic clinical characteristics of samples from TCGA was showed in Table 1. GSE76427 dataset2 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76427) from the GEO database contains information on gene expression of 115 HCC tumor samples and 52 NC samples.
Clinical characteristics of samples from the TCGA database.

SD: standard deviation; TCGA: the Cancer Genome Atlas.
Data processing and the differentially expressed genes analysis
The R/Bioconductor (R version 4.0.2; Bioconductor version 3.12) tool was used for data processing and analysis. Genes with differential fold change (FC) greater than 2 and p < 0.05 were identified as differentially expressed genes (DEGs) between tumor and control samples in both the dataset1 and dataset2, and the intersection of DEGs in both datasets was selected out as final DEGs in the HCC versus NC group for further study. The pheatmap toolkit on R platform was used to analyze the cluster heatmap of the DEGs.
Protein–protein interaction network construction and hub gene identification
The DEGs were uploaded to the STRING online analysis platform (https://string-db.org/, version: 11.0)12,13 for PPI network analysis, as the PPI network may help to better understand the underlying mechanisms of the disease. The criterion was as follows: organism: Homo sapiens, network type: full STRING network, meaning of network edges: evidence, active interaction sources: choose all, minimum required interaction score: highest confidence (0.900). The STRING online database also sets out the characteristics of the network including number of nodes, number of edges, average degree of nodes, et al. It is worth noting that nodes with higher degree values in the PPI network are often considered to be crucial hub genes in the disease. Thus, the degree values of all the nodes in the network were summarized and analyzed, to identify specific candidate nodes with higher degree values (degree greater than 20 in this study) for further analysis.
Receiver operating characteristics and survival analysis
Previously obtained DEGs with higher degree values in the PPI network were analyzed for receiver operating characteristics (ROC) and survival curves to evaluate the potential applications of the candidate DEGs as possible markers for HCC diagnosis and prognosis. The Medcalc statistical software (version 19.0.4) was used to analyze the candidate genes for the distinction between HCC tumor and NC samples, and the sensitivity, specificity and area under the curve (AUC) values were obtained. For survival analysis, the Kaplan-Meier survival curves were explored and visualized (using α = 0.05 as the test level) using the Survminer toolkits in R based on age, gender, TNM stage (clinical information was collected in the TCGA dataset) as well as the gene expression pattern of candidate genes in HCC subjects.
Results
Global analysis of DEGs in HCC tumor versus NC samples
As previously reported, genes with FC >2 and p < 0.05 were identified as DEGs between HCC tumors versus NC samples. As shown in Figure 1, there were 4249 DEGs (including 2940 up-regulated genes and 1309 down-regulated genes) and 440 DEGs (including 69 up-regulated genes and 371 down-regulated genes) in TCGA dataset and GEO dataset, respectively. Further, 310 overlapping DEGs (including 51 up-regulated genes and 259 down-regulated genes) were obtained from both datasets, and detailed information of the 310 final DEGs was provided in Supplemental Table 1. From the clustering pheatmap analysis and PCA (principal component analysis) (Figure 2) based on the expression of 310 DEGs, it was found that the HCC tumor and NC samples could be relatively differentiated.

Global analysis of DEGs in HCC tumor versus NC samples: (a–b) volcano plot of DEGs in TCGA dataset and GEO dataset, respectively, (c) the number of DEGs in both datasets, and (d) Venn diagram showing 310 overlapping DEGs combined from both datasets.

Bi-hierarchical cluster and PCA analysis of 310 DEGs in HCC tumor and NC samples based on the GEO dataset gene expression pattern: (a) the bi-hierarchical heatmap, the horizontal and vertical axis represents the samples and the DEGs, respectively and (b) the PCA score plots, each triangle or circle represents a sample.
PPI network reveals potential HCC candidate hub genes
The 310 DEG-based PPI network in HCC was constructed and analyzed using the online STRING platform as shown in Figure 3. After hiding the disconnected nodes, a total of 330 edges and an average node degree of 2.13 was obtained in the interaction network. It’s worth noting that the hub node genes in the PPI network may potentially associate or reflect the pathogenesis and progression of HCC. As a result, the hub genes with higher degree values were screened out. There were 36 hub DEGs (including 17 up-regulated DEGs and 19 down-regulated DEGs) with a degree greater than 20 in the network, as shown in Table 2.

The PPI network of DEGs in HCC based on the online platform of STRING.
The hub genes with degree value greater than 20 in the PPI network.
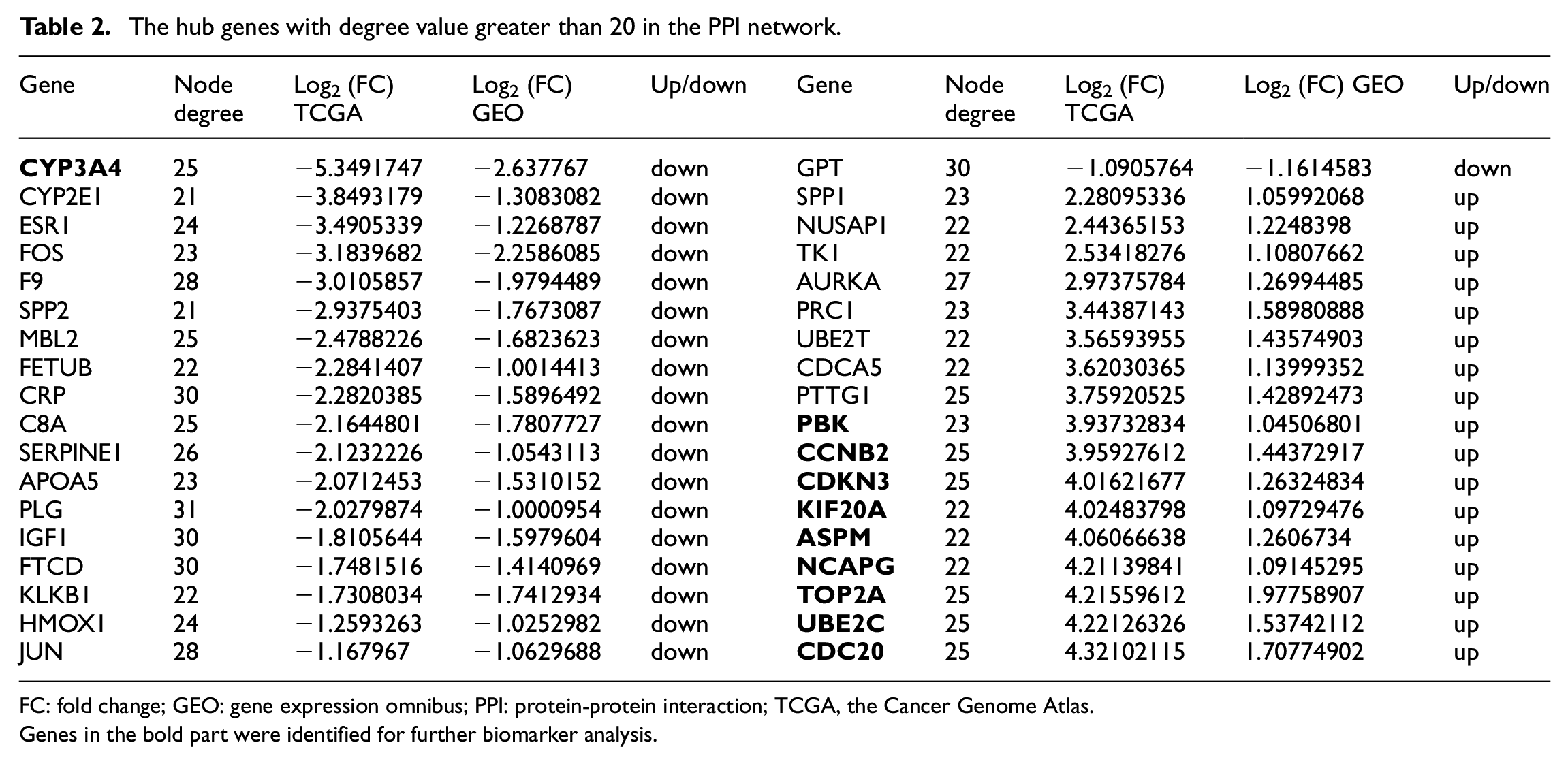
FC: fold change; GEO: gene expression omnibus; PPI: protein-protein interaction; TCGA, the Cancer Genome Atlas.
Genes in the bold part were identified for further biomarker analysis.
In addition, we supposed that if the hub genes in the PPI network show more significant expression alterations between HCC and NC samples, they may have potential applications as diagnostic or prognostic biomarkers for HCC. Based on this assumption, the candidate DEGs were identified as biomarkers, mainly considering the following two aspects: (1) the hub genes in the PPI network; (2) the most differentiated genes (DEGs with the top 50 fold changes). In the end, a gene signature of 10 DEGs were identified for further biomarker evaluation, including CDKN3, TOP2A, UBE2C, CDC20, PBK, ASPM, KIF20A, NCAPG, CCNB2, CYP3A4, and most of them were up-regulated in HCC (the bold part in Table 2).
Potential HCC diagnostic markers
On the basis of the previous results, the application of the above 10 candidate genes as novel biomarkers for HCC was evaluated. The results of the ROC analysis were presented in Figure 4 and Table 3. It was noted that all 10 candidate genes had relatively significant effects when distinguishing tumors from normal samples, particularly in the GEO dataset, as 9 of the 10 genes had an AUC value greater than 0.9 and a sensitivity and a specificity greater than 0.7, indicating that the 10-gene signature may act as potential diagnostic biomarkers for HCC.

ROC curves of the distinction between HCC tumor and NC samples based on 10 candidate genes.
Effects of the distinction between HCC tumor and NC samples based on 10 candidate genes.

AUC: area under curve; GEO: gene expression omnibus; HCC: hepatocellular carcinoma; NC: normal control; TCGA: the Cancer Genome Atlas.
Analysis of the prognostic factors in HCC patients
The TCGA program gathers clinicopathological annotation data and efforts to fully characterize molecular events in primary cancers, 14 making it possible to explore the prognostic factors for HCC and other malignant diseases. Table 1 summarized the basic clinical information for HCC patients in the TCGA dataset. Further, the survival curves based on factors such as gender, age, TNM stage status, and the 10 candidate gene signatures previously obtained were analyzed, to identify the potential biomarkers associated with HCC progression and prognosis. Figure 5 showed the Kaplan-Meier survival curves of the above factors, which demonstrates that age and gender factors had no significant impact on the overall survival rate of HCC patients (p > 0.05), and the TNM stage status factor had a significant negative prognosis correlation (p < 0.05). The eight candidate genes (CDKN3, TOP2A, UBE2C, CDC20, PBK, ASPM, KIF20A, NCAPG) were all negatively correlated with the overall survival rate of patients (p < 0.05) and were all up-regulated in HCC tumor samples (Table 2), while the remaining two candidate genes had no significant effects on the overall survival rate of HCC patients (p > 0.05).

Kaplan-Meier survival curves based on the candidate genes, age, gender and TNM stages of HCC patients: (a) Kaplan-Meier survival curves of 8 candidate genes that were negatively correlated with the HCC prognosis (p < 0.05), (b) Kaplan-Meier survival curves of 2 candidate genes which were not significantly correlated with the HCC prognosis (p > 0.05), and (c) Kaplan-Meier survival curves based on age, gender, and TNM stage status of patients. Among them, the values of age and gene expression were divided into high (high) and low (low) groups by median values. The horizontal axis represents the survival time (day), and the vertical axis represents the overall survival rate.
Discussion
As one of the most common malignant tumors in the world, it is critical for HCC clinical and scientific research to discover new molecular markers and potential diagnostic and therapeutic targets.15,16 To date unfortunately there are few prognostic indicators for patients with HCC represented mainly by liver function 17 or the development of drug-related adverse events during systemic therapy. 18 Therefore, the identification of genetic prognostic factors would be of great help for better management of patients. The aim of the TCGA program is to catalog and explore cancer-causing genomic alterations to establish a comprehensive “atlas” of cancer genomic signature. It is also worth noting that the TCGA platform has already integrated enormously standardized clinical information. The program can therefore provide publicly available datasets to help understand the underlying mechanisms of tumorigenesis and to improve approaches or standards for cancer diagnosis and therapy. Another well-known public database, the GEO datasets, is a gene expression compiling project initiated by NCBI, which aims to set up a gene expression database and online resources for the study of gene expression. This study analyzed the global gene expression characteristic of HCC by combing two datasets of TCGA and NCBI-GEO, respectively. Besides, the potential diagnostic or prognostic biomarkers for HCC were also identified by integrating gene expression signature and the clinical information about patients from TCGA.
To obtain more accurate results, the DEGs intersections shared between the two datasets were identified as final DEGs in HCC tumor versus NC samples. Ultimately, a total of 310 DEGs in HCC tumors were screened out (Figure 1 and Supplemental Table 1). Bi-hierarchical cluster and PCA analysis of DEGs showed relatively distinguishable effects between tumors and normal samples in general (Figure 2). Furthermore, the hub node genes, which may play an important role in the pathogenesis and progression of HCC, were characterized through the PPI network analysis based on DEGs (Figure 3 and Table 2). In addition, a 10-candidate gene signature was identified by integrating the hub node genes and the most significantly altered DEGs, since the candidate genes could be considered as potential disease biomarkers. Interestingly, the majority (9/10) of the candidate genes showed an up-regulation pattern in HCC tumor samples. The ROC analysis showed that 10 candidate genes ideally distinguished the tumor group from the NC group (Figure 4 and Table 3). Survival curves of the candidate genes indicated that eight candidate genes were significantly related to the overall survival rate of HCC patients, including CDKN3, TOP2A, UBE2C, CDC20, PBK, ASPM, KIF20A, NCAPG. All eight candidate genes were up-regulated in the tumor samples and their expression was negatively correlated with the HCC prognosis (p < 0.5, Figure 5(a)). The above results suggest that key candidate genes may function as cancer-prompting genes and have a potential application as diagnostic or prognostic biomarkers for HCC. In addition, the gender-based, age-based and TNM stage-based survival curves of patients in the TCGA dataset were analyzed to explore the prognosis-related factors of HCC. Results indicated that age and gender factors had little effect on the overall survival rate of patients (p > 0.5, Figure 5(c)), while advanced TNM stages reflected poor HCC prognosis (p < 0.5, Figure 5(c)) relatively.
In fact, biomarker detection is a relatively convenient and non-invasive approach to tumor diagnosis and prognostic estimation. More and more studies have shown promising results in HCC research,1,19,20 some of which were consistent with results in this study. Cyclin-dependent kinase inhibitor 3 (CDKN3) is a member of the protein kinase family. 21 Studies have shown that CDKN3 predicted poor prognosis in HCC and CDKN3 silencing reduced sensitivity to adriamycin but did not suppress the proliferation of HCC cells, indicating that CDKN3 may have a dual role in HCC development. 21 Topoisomerase II alpha (TOP2A) is considered to be an oncogene in various types of cancer and a target for cancer therapy, particularly in breast cancer.22–24 Overexpression of TOP2A was detected in most tumor tissues but not in matching non-tumor tissues in HCC. 22 In an early-stage HCC study, CDKN3 and TOP2A were highly expressed in very early HCC tissues compared to cirrhotic tissues and reflected poorer overall survival in patients. 25 Similarly, the ubiquitin-conjugating enzyme E2C (UBE2C) gene has been overexpressed in cancers and acts as a potential oncogene.26–29 In HCC, UBE2C could increase the proliferation, migration, invasion, and drug resistance of cancer cells. 30 Besides, recent studies have shown that several types of cancers exhibit high expression of non-SMC condensin I complex subunit G (NCAPG),31,32 and NCAPG induced HCC cell proliferation by regulating PI3K/AKT signaling. 33 In addition, overexpression of PDZ-binding kinase (PBK) promoted HCC cell metastasis by activating the ETV4-uPAR signal pathway. 34 Another study identified a 20 gene-based signature, the alteration of which could reflect pathological progression from liver cirrhosis to hepatocellular carcinoma, including TOP2A, CDC20 and KIF20A, et al. 35 CDC20,36,37 ASPM,38,39 and KIF20A 40 have also been reported to be over-expressed in HCC and may predict poor overall survival of patients. Collectively, these findings reveal that the above set of genes may act as potential oncogenes and therefore provide promising diagnostic and therapeutic biomarkers or targets for HCC.
One of the hub genes in the PPI network was cytochrome P450 3A4 (CYP3A4). Furthermore, results showed that CYP3A4 and CYP2E1 were down-regulated in HCC tumor samples, which was consistent with other related study. 41 Similarly, down-regulation of the CYP2C19 gene is linked to aggressive tumor potential and a lower recurrence-free survival rate in HCC. 42 As the major CYP isoform, CYP3A4 is primarily expressed in the liver and is an essential enzyme in the body. It is worth noting that CYPs are responsible for approximately 75% of drug metabolism as well as the metabolism of a wide range of dietary constituents and endogenous chemicals. 43 Capecitabine, for example, has recently been reported as a safe and effective systemic treatment for advanced HCC.44–46 Capecitabine can be metabolized by cytochromes P450 enzymes. In this study, low expression of CYP3A4 and CYP2E1 may be associated with carcinogen down-metabolism, which leads to increased exposure or accumulation of carcinogens. The above findings could contribute to the discovery of bio-targets for HCC treatment.
Conclusion
In summary, this study analyzed the global gene expression signature and identified potential diagnostic or prognostic HCC biomarkers by integrating bioinformatics datasets from the TCGA and GEO platforms. The results will provide some evidence for a better understanding of tumorigenesis and progression of HCC, and help to explore the candidate targets for disease diagnosis and treatment. The shortcoming of the research mainly lies in the lack of a more in-depth mechanism exploration that will guide our efforts in future studies.
Supplemental Material
sj-pdf-1-sci-10.1177_00368504211029429 – Supplemental material for Global analysis of gene expression signature and diagnostic/prognostic biomarker identification of hepatocellular carcinoma
Supplemental material, sj-pdf-1-sci-10.1177_00368504211029429 for Global analysis of gene expression signature and diagnostic/prognostic biomarker identification of hepatocellular carcinoma by Jihan Wang, Yangyang Wang, Jing Xu, Qiying Song, Jingbo Shangguan, Mengju Xue, Hanghui Wang, Jingyi Gan and Wenjie Gao in Science Progress
Footnotes
Authors’ note
This study was not required to obtain approval as the study was based on deidentified retrospective patient data published at public domains.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (No. 81702067), Fundamental Research Funds for the Central Universities (No. 20ykpy94), Yat-sen Scholarship for Young Scientist for Wenjie Gao, General Financial Grant from China.
Supplemental material
Supplemental material for this article is available online.
Author biographies
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.