
Abstract
Objective
Sepsis, a systemic inflammatory response triggered by infection, is characterized by organ dysfunction. NETosis, a form of cell death involving the release of neutrophil extracellular traps (NETs), plays a crucial antimicrobial role during sepsis. This study aimed to explore the relationship between NETosis-related genes (NRGs) and 28-day mortality in patients with sepsis.
Methods
This retrospective observational study utilized the Medical Information Mart for Intensive Care IV (MIMIC-IV) database. Univariate and multivariate logistic regression analyses were conducted to identify independent prognostic factors. A nomogram was then constructed to assess the potential of these factors in predicting 28-day mortality in patients with sepsis. Additionally, a Mendelian randomization (MR) study was performed to identify NRGs with causal associations to 28-day mortality in sepsis. Expression validation was carried out using the GSE65682 dataset from a public database, followed by identification of key genes. Enrichment analysis was performed to uncover the molecular mechanisms associated with these key genes in sepsis.
Results
A total of 909 patients with sepsis (706 survivors and 203 non-survivors) were identified from the MIMIC-IV database. Seven independent prognostic factors, including absolute neutrophil counts, were identified. The nomogram developed proved to be a reliable tool for predicting 28-day mortality in patients with sepsis. The MR study identified 12 NRGs with a unidirectional causal relationship to 28-day mortality, with AKT1 and CXCR2 emerging as key genes. Both genes are predominantly involved in immune-related pathways.
Conclusion
Analysis of the MIMIC-IV database highlighted neutrophil_abs as a significant prognostic factor for 28-day mortality in sepsis. Transcriptomic analysis identified AKT1 and CXCR2 as critical genes associated with 28-day mortality, providing insights into potential therapeutic strategies for sepsis.
Introduction
Sepsis is a host inflammatory response triggered by severe, life-threatening infections, often resulting in organ dysfunction. 1 It is a leading cause of mortality in intensive care units (ICUs), accounting for over 250,000 deaths annually in the United States. 2 While the incidence and mortality rates of sepsis have declined, it remains a significant burden, particularly in developing countries. 2 According to the Global Burden of Disease report, 48.9 million cases of sepsis were diagnosed globally in 2017, with a mortality rate of 22.5%, contributing to nearly 20% of all global deaths. 2
Neutrophils represent the first line of defense in the innate immune response to infections. They eliminate pathogens through degranulation, phagocytosis, and the formation of neutrophil extracellular traps (NETs), which capture and kill microorganisms. 3 NET formation, or NETosis, can occur via two major pathways: the classical NETosis pathway, which involves nuclear lobulation, disintegration of the nuclear membrane, loss of cellular polarization, chromatin decondensation, and plasma membrane rupture; and nonlytic NETosis, where chromatin is expelled and granular proteins released without cell death. 3 These NET components, once released, form an extracellular matrix that enhances antimicrobial defense, leaving behind anucleate neutrophils capable of chemotaxis and pathogen phagocytosis. In sepsis, NETosis serves as a crucial immune response to control pathogen spread by trapping microorganisms and delivering antimicrobial molecules directly to them. However, excessive NET formation due to persistent inflammatory stimuli can exacerbate tissue damage and contribute to harmful inflammation. 4
Several observational studies have implicated NETosis-driving factors in the development and progression of sepsis.4,5 However, such studies are often limited by confounding factors and reverse causality, leading to potential biases in the conclusions. Mendelian randomization (MR) overcomes these limitations by using genetic variants as instrumental variables to assess causal relationships between risk factors and disease outcomes. 6 Genetic variation, inherited randomly from parents, is unaffected by common confounders, making MR a powerful tool for establishing causal inference. For MR to yield reliable results, three key assumptions must hold: (i) the genetic variant must be reliably associated with the exposure of interest (relevance assumption); (ii) the genetic variant should not be associated with known or unknown confounders (independence assumption); and (iii) the variant should influence the outcome solely through the exposure, not through a direct causal pathway (exclusion restriction assumption). 6 MR analysis has been widely applied to explore causal links between exposures and diseases.6,7
This study utilized the MIMIC-IV database to analyze the relationship between clinical characteristics and 28-day mortality risk in patients with sepsis. Independent prognostic variables were identified, and a nomogram was constructed to assess their predictive accuracy for 28-day mortality in this cohort. Additionally, a series of bioinformatics analyses were employed to investigate the causal relationships between NETosis-related genes (NRGs) and sepsis mortality, uncovering the molecular mechanisms underlying these genes and providing insights into potential therapeutic targets for sepsis management.
Materials and methods
Data acquisition
The GSE65682 dataset (platform: GPL13667) was obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), which includes 28-day survival data for septic individuals. This dataset comprises blood samples from 760 patients with sepsis (479 with 28-day survival data) and 42 healthy controls. Additionally, sepsis-related data on 28-day mortality (ieu-b-5086) were acquired from the Integrative Epidemiology Unit Open Genome-Wide Association Study (IEU OpenGWAS) database (https://gwas.mrcieu.ac.uk/). This dataset includes 12,243,487 single-nucleotide polymorphisms (SNPs) from 486,484 European individuals (cases: controls = 1896: 484,588). Furthermore, 69 NRGs (Table S1) were sourced from the literature. 8 Data on quantitative trait loci (eQTLs) for NRGs were retrieved from the IEU OpenGWAS database and treated as exposure variables.
Study population
The MIMIC-IV database (https://mimic.mit.edu/) is a publicly available repository containing over 50,000 ICU admissions from Beth Israel Deaconess Medical Center in Boston, Massachusetts, covering the years 2008–2019. 9 This database employs a retrospective observational study design with longitudinal patient follow-up. The study population was selected based on predefined inclusion and exclusion criteria. Eligible patients were those aged ≥18 years, meeting the Sepsis 3.0 diagnostic criteria, having infections, and a Sequential Organ Failure Assessment (SOFA) score ≥2. A total of 479 patients with sepsis had complete 28-day survival follow-up data. The healthy control group was matched with the septic cohort for age and gender and had no acute or chronic infectious diseases. Septic cases were identified using the International Classification of Diseases, 9th Revision (ICD-9) diagnostic codes: 99591, 99592, 67024, 67022, and from the 10th Revision (ICD-10): R6521, A4181, R6520, T8144XA, A419, A408, A4102, A4159, A4189, and B377. Patients were excluded if they had multiple ICU admissions, no ICU admission, or an ICU stay of less than 48 hours, or if their clinical information was incomplete. Of the 25,591 patients with sepsis screened from 2008 to 2019, 15,072 were excluded based on the criteria, and an additional 9610 were excluded due to missing data. Ultimately, 909 patients were enrolled, as illustrated in the recruitment flowchart (Fig. S1).
This study employed a retrospective observational analysis using de-identified public data from the MIMIC-IV database, GSE65682 (GEO), and ieu-b-5086 (IEU OpenGWAS). As the data were sourced from historical medical records and public databases, prospective patient follow-up was not involved. All patient-specific information (e.g., names, medical record numbers, contact details) was de-identified to ensure that individual identities could not be identified. The study adhered to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines. 10
Variable extraction and creation of baseline characteristics
Clinical data for patients with sepsis were collected, and the following variables were extracted for analysis: (1) Demographic characteristics: age, sex, and weight; (2) Comorbidities: chronic pulmonary disease, severe liver disease, renal disease, myocardial infarction, diabetes, congestive heart failure, and cancer; (3) Severity scores upon ICU admission: simplified acute physiology score II (SAPS II) and SOFA score; (4) Vital signs: temperature, pulse oximetry saturation (SpO2), respiratory rate, mean blood pressure (MBP), and heart rate; (5) Laboratory data: white blood cell (WBC) count, hemoglobin, platelet count, lactate, creatinine, absolute neutrophil counts (neutrophils_abs), absolute lymphocyte counts (lymphocytes_abs), and blood urea nitrogen (BUN); (6) Treatment and interventions: use of dopamine, epinephrine, phenylephrine, norepinephrine, vasopressin, milrinone, dobutamine, and mechanical ventilation. The analysis focused on septic participants from the MIMIC-IV database, dividing them into two groups: the death group (those who died within 28 days of ICU admission) and the survival group (those who survived). The baseline characteristics were summarized in a table using the tableone package (v 0.13.2).
In this study, categorical variables describing baseline characteristics were expressed as percentages. The weighted chi-square test was applied to assess differences between groups. Variables with P < .05 in univariate logistic regression, along with clinically significant confounding factors, were selected as covariates for inclusion in multivariate logistic regression analysis.
Multivariate logistic regression analyses
Multivariate logistic regression (OR ≠ 1, P < .05) was performed using the Stats package (v 4.2.2), incorporating the clinical variables that showed significant differences between the survival and death groups in prior analyses. The clinical variables with significant differences (P < .05) were identified as independent prognostic factors.
To account for potential collinearity among the independent prognostic variables, collinearity diagnostics were conducted. Variance inflation factor (VIF) values were calculated using the vif() function from the car package, with one variable treated as the dependent variable and the others as independent variables. A VIF value of less than 5 for each variable indicated an acceptable level of collinearity.
Development of the nomogram and machine learning algorithm
A nomogram was developed using the rms package (v 6.5.0) to estimate the 28-day mortality risk among patients with sepsis based on independent prognostic factors. Each independent prognostic factor was assigned a score, and the total score was derived by aggregating these individual scores. A higher total score indicated an increased probability of 28-day mortality in sepsis. Receiver operating characteristic (ROC) curves were plotted using the pROC package (v 1.18.0) to assess the predictive accuracy of the nomogram, with the area under the curve (AUC) being calculated. An AUC value greater than 0.7 was considered indicative of good predictive ability. Decision curve analysis (DCA) was performed using the rmda package (v 1.6) to evaluate the clinical utility of the nomogram. Additionally, an eXtreme Gradient Boosting (XGBoost) machine learning model was built using the xgboost package (v 1.7.3.1) to assess the predictive performance and importance of the independent prognostic factors in predicting 28-day mortality in patients with sepsis.
Screening instrumental variables (IVs)
For the MR analysis, the sepsis (28-day mortality)-related dataset (ieu-b-5086) served as the outcome variable, while eQTL data for NRGs were used as the exposure factor. Exposure factors were initially processed, and IVs were identified using the extract_instruments function from the TwoSampleMR package (v 0.5.6). The screening criterion 11 was set to P < 5 × 10–6, which identified IVs significantly associated with the exposure factors. To mitigate bias from linkage disequilibrium (LD), IVs exhibiting LD were excluded using the parameters: “clump = TRUE, r2 = 0.001, and clumping distance = 10 kb.” F-statistic tests were then applied to the IVs, with those having F-statistics below 10 being excluded. Additionally, exposure factors with fewer than three SNPs were excluded. The harmonize_data function from the TwoSampleMR package (v 0.5.6) was used to standardize the effect alleles and sizes, ensuring proper alignment of exposure factors, IVs, and outcomes.
MR analysis
The causal relationships between NRGs and sepsis-related 28-day mortality were examined using the mr function from the TwoSampleMR package (v 0.5.6) along with five algorithms: MR Egger, weighted median, inverse variance weighted (IVW), simple mode, and weighted mode. IVW was the primary method for result assessment. A statistically significant causal relationship between NRGs and 28-day sepsis mortality was inferred if the IVW analysis yielded a P-value < .05. NRGs with odds ratios (ORs) greater than 1 were identified as risk factors for sepsis, while those with ORs less than 1 were considered protective factors. Forest and scatter plots were generated to visualize MR results, and funnel plots were used to assess the validity of Mendel's second law. To ensure the reliability of the findings, a sensitivity analysis was conducted. A horizontal pleiotropy test (P > .05) was performed using the mr_pleiotropy_test function from the TwoSampleMR package (v 0.5.6) and the mr_presso function from the MRPRESSO package (v 1.0). Heterogeneity was assessed using the mr_heterogeneity function from the TwoSampleMR package (v 0.5.6). If heterogeneity was detected (Cochran's Q test; P < .05), IVW with random effects (IVW-random) was employed. If no heterogeneity was found (Cochran's Q test; P > .05), IVW with fixed effects (IVW-fixed) was used. Additionally, a leave-one-out (LOO) sensitivity analysis was performed to evaluate whether any single SNP influenced the overall effect of all SNPs in the IVW analysis. Finally, the Steiger directionality test was conducted using the directionality_test function from the TwoSampleMR package (v 0.5.6) to assess the causality of NRGs in patients with sepsis who died within 28 days. A “correct causal direction = TRUE and P < .05” indicated a unidirectional causal relationship between NRGs and sepsis mortality. NRGs exhibiting a significant causal association with 28-day mortality in patients with sepsis, as determined through MR analysis, were considered candidate genes for further investigation.
Discerning key genes by expression validation combined with MR analysis
The Wilcoxon test (P < .05) was performed to compare candidate gene expression levels between patients who died within 28 days (death group) and those who survived (survival group) in GSE65682. Genes exhibiting significant differences (P < .05) between the groups were identified as potential key genes. Subsequently, genes with ORs greater than 1 and high expression in the death group, as well as those with ORs less than 1 and high expression in the survival group, were considered key genes.
Kaplan–Meier (K–M) survival analysis and analysis of clinical characteristics
To explore the relationship between key gene expression levels and survival in patients with sepsis, the 479 patients with complete survival data from GSE65682 were categorized into high-expression or low-expression groups based on the optimal threshold for each key gene. K–M survival curves were generated using the survminer package (v 0.4.9) to assess differences in overall survival (OS) between the high-expression and low-expression groups (P < .05). The same cohort of 479 patients was further classified based on clinical characteristics such as age, sex, diabetes status, endotype class, ICU-acquired infection (ICUA), and pneumonia diagnosis. The Wilcoxon test (P < .05) was then applied to compare key gene expression differences across these subgroups.
Nomogram construction and evaluation based on key genes
To predict patient incidence based on key gene expression, a nomogram model incorporating key genes was constructed using the rms package (v 6.5.0) in the training set. This model used key gene expression levels as predictors, with disease status as the outcome. DCA was employed to evaluate the clinical utility of the model by plotting decision curves based on the key genes. Additionally, a calibration curve was constructed using the same nomogram prediction model in the rms package (v 6.5.0) to assess the consistency between predicted and actual probabilities.
Gene set enrichment analysis (GSEA)
GSEA was conducted to explore the functional roles of the identified key genes in sepsis. The previously identified key genes from the high-expression and low-expression groups in the K–M survival analysis were subjected to differential analysis using the limma package (v 3.54.0). Log2 fold-change (FC) values were calculated and sorted in descending order. The gene set “h.all.v2023.2.Hs.symbols.gmt” from the Molecular Signatures Database (MSigDB) (https://www.gsea-msigdb.org/gsea/msigdb) was used as the reference. GSEA was performed for each key gene using the clusterProfiler package (v 4.7.1.003), with a normalized enrichment score (NES) threshold of |NES| > 1 and P < .05.
Construction of regulatory networks and drug prediction
Molecular regulatory networks offer valuable insights into the core mechanisms of gene regulation in disease-related processes. To explore the regulation of key genes by microRNAs (miRNAs), the miRNA target prediction database (miRDB) (https://mirdb.org/) was utilized to predict miRNAs that target the key genes. The upstream long noncoding RNAs (lncRNAs) of the miRNAs were then estimated using the StarBase database (https://starbase.sysu.edu.cn/index.phpStarBase) (clipExpNum > 4). Transcription factors (TFs) regulating the key genes were predicted using the JASPAR plugin from the NetworkAnalyst database (https://www.networkanalyst.ca/). Potential drugs targeting key genes were identified through the Drug-Gene Interaction Database (DGIdb) (https://dgidb.org/). The resulting networks—lncRNA-miRNA-key genes, TF-key genes, and drug-key gene interactions—were visualized using Cytoscape software (v 3.10.2).
Statistical analysis
All statistical analyses were conducted using R (v 4.2.2). After confirming data normality through the Kolmogorov-Smirnov test, normally distributed variables were presented as means (SDs), while non-normally distributed variables were presented as medians (IQRs). Continuous variables were analyzed using the Mann–Whitney U test and Student's t-test, while categorical variables were assessed via Fisher's exact test or Chi-squared tests. Group comparisons were performed using the chi-square test or independent samples t-test. Results were considered statistically significant at P < .05 (two-sided). To ensure the statistical power of the study, a power analysis for two-sample t-tests was performed using the “pwr” package in R (v 1.3-0). 12 For the MIMIC dataset, under a significance level of α = .05, with a large effect size (Cohen's d = 0.8) and sample sizes of 706 (group 1) and 203 (group 2), the calculated statistical power for detecting intergroup differences reached 100% (power = 1). For the transcriptome dataset, with the same significance level (α = .05) and effect size (Cohen's d = 0.8), and sample sizes of 479 (group 1) and 42 (group 2), the statistical power was 99.9% (power = 0.9986591). These results indicate that the sample sizes in both datasets provided sufficient statistical power to detect the expected effects, ensuring the reliability of the statistical significance of the findings.
Ethical statement
This study was conducted in accordance with the Helsinki Declaration of 1975 as revised in 2024. This study utilized retrospective data and was exempt from institutional review board approval. Given the study design and data characteristics, direct patient participation was not involved. Thus, written informed consent was not required.
Results
Comparison of baseline characteristics between deceased and surviving patients with sepsis
Using the MIMIC-IV database, 203 patients with sepsis who died within 28 days of ICU admission (death group) and 706 patients who survived the same period (survival group) were identified. Analysis of the baseline characteristics revealed significant differences between the two groups (Table 1). Notably, 20 clinical variables showed significant discrepancies (P < .05), including age, weight, myocardial infarction, congestive heart failure, cancer, WBC count, SAPS II score, and SOFA score. Among these, neutrophil_abs demonstrated extremely significant differences (P < .001) between the groups.
Comparison of baseline characteristics between the survival and deceased groups.

SD: standard deviation; Diabetes without_cc: Diabetes without chronic complications; SAPS: simplified acute physiology score; SOFA: Sequential Organ Failure Assessment; MBp: mean blood pressure; WBC: white blood cell counts; abs: absolute counts. *Bold values represent P < .05, indicating a significant difference between survival and decaesed patients, and entered into the multivariate logistic regression model.
Identifying independent prognostic variables
These 20 variables were subsequently included in a multivariate logistic regression analysis (OR ≠ 1, P < .05), which identified seven independent prognostic factors: weight, SAPS II score, neutrophil_abs, BUN, cancer, and the use of vasopressin and norepinephrine (Table 2). Of these, weight was found to be a protective factor against sepsis (OR < 1), while neutrophil_abs and the remaining five variables were identified as risk factors (OR > 1). To assess potential multicollinearity among the seven factors, VIFs were calculated, with each VIF being less than 5, indicating an acceptable level of collinearity. Visualization using the ggplot function further confirmed the absence of strong multicollinearity among these variables (Fig. S2).
Multivariate logistic regression analysis of in patients with sepsis.

OR: odd ratio; CI: confidence interval. *Bold values represent P < .05.
Neutrophil_abs: an important prognostic variable for patients with sepsis
As shown in Fig. 1A, a nomogram was developed to estimate sepsis-related mortality based on the seven independent prognostic variables. The nomogram demonstrated that a higher total score correlated with a decreased survival probability for patients with sepsis. The ROC curve for the nomogram yielded an AUC of 0.787 (Fig. 1B), indicating good accuracy in predicting 28-day mortality. DCA demonstrated that the nomogram outperformed individual prognostic variables in terms of net benefit (Fig. 1C), highlighting its potential for clinical application. Additionally, in the XGBoost model, neutrophil_abs was identified as the second most important factor in predicting 28-day mortality in patients with sepsis (Fig. 1D).
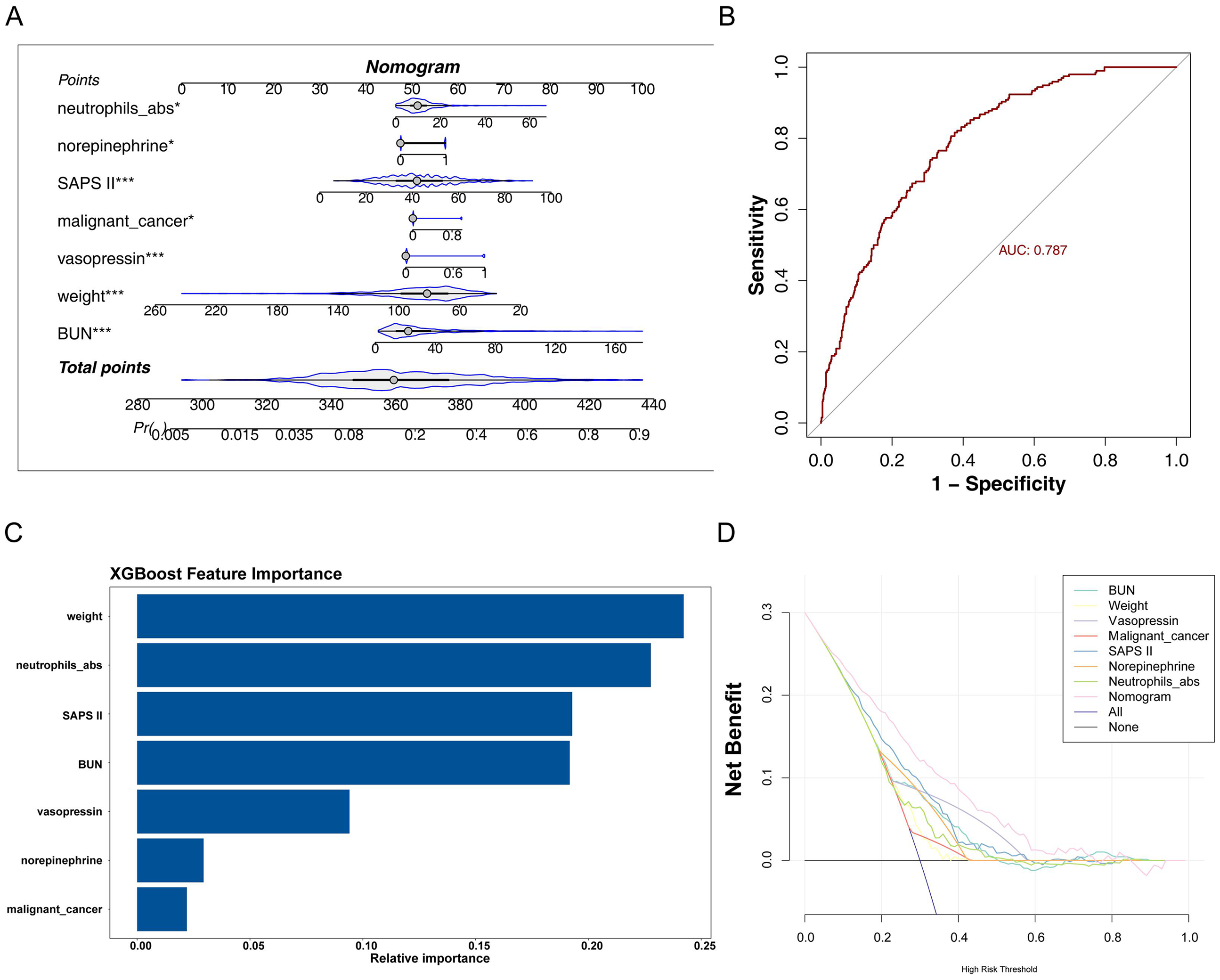
Construction and validation of the nomogram model. (A) A nomogram was developed to predict the risk of sepsis-related mortality, based on seven independent prognostic variables. Each variable corresponds to a score, and the total score predicts the probability of death. (B) The ROC curve was constructed to assess the predictive efficiency of the nomogram model. (C) DCA demonstrated that the nomogram model provided greater net benefit compared to individual prognostic variables. (D) The importance of the seven prognostic factors included in the nomogram was evaluated using the XGBoost model.
Acquisition of 12 candidate genes through MR analysis
The MIMIC-IV database study identified neutrophil_abs as a significant prognostic variable for sepsis development, justifying the use of 69 NRGs as exposure factors and 28-day mortality from sepsis as the outcome for investigating causal genes. After screening the IVs, 59 genes were identified (Table S2). The IVW method revealed that 20 genes were significantly associated with 28-day mortality in patients with sepsis (P < .05) (Table S3). Among these, 12 genes (e.g., AKT1, CXCR2, and CTSG) were identified as protective factors for sepsis (OR < 1), while 8 genes (e.g., MAPK3 and CRISPLD2) were associated with increased risk (OR > 1). Scatter plots demonstrated a strong correlation between these 20 genes and sepsis-related 28-day mortality, with intercepts near 0, indicating minimal confounding effects (Fig. S3A). In these plots, positive slopes represented risk factors, while negative slopes indicated protective factors, according to the IVW method. Forest plots revealed that 12 genes (such as AKT1, CXCR2, and CTSG) had effect sizes less than 0, signifying their protective role (Fig. S4). Conversely, the effect sizes of 8 genes (e.g., MAPK3 and CRISPLD2) exceeded 0, highlighting their role as risk factors. Funnel plots indicated that the SNP distribution for all 20 genes was symmetric and evenly dispersed, suggesting adherence to Mendel's second law in the MR analysis (Fig. S5).
The sensitivity analysis further confirmed the reliability of the MR study. The horizontal pleiotropy test results showed that 15 genes, including AKT1 and CXCR2, were unaffected by confounding factors (P > .05) (Table S4). The heterogeneity test revealed that 14 genes displayed no heterogeneity (Cochran's Q test; P > .05), confirming that the IVW-random method was appropriate for MR analysis (Table S5). However, heterogeneity was detected for CRISPLD2 (Cochran's Q test; P < .05), so IVW-fixed analysis was applied. LOO analysis demonstrated that excluding any single SNP had a negligible impact on the effect of the remaining SNPs, reinforcing the robustness of the MR results (Fig. S6). The results of the Steiger directionality test confirmed that 12 genes had a unidirectional causal relationship with 28-day mortality in patients with sepsis (correct_causal_direction = TRUE and P < .05) (Table S6). Based on these findings, the following 12 genes were identified as candidate genes associated with sepsis-related 28-day mortality: CTSG, CRISPLD2, BST1, PIK3CA, G0S2, TLR2, AKT1, PADI4, CXCR1, CXCR2, CYP4F3, and ATG7.
Identification and analysis of key genes AKT1 and CXCR2
The expression of the 12 candidate genes was further examined in the death and survival groups of GSE65682. CTSG expression significantly increased in the death group, while AKT1 and CXCR2 expressions were notably higher in the survival group (P < .05) (Fig. 2A). The MR analysis indicated that CTSG, AKT1, and CXCR2 were protective factors against sepsis. These results suggest that the elevated expression of AKT1 and CXCR2 in the survivor group identifies these genes as key factors in sepsis prognosis.

Analysis of candidate gene expression levels and survival analysis of key genes. (A) Boxplots validating the expression of 12 candidate genes. Note: Different colors represent different groups, with 1 indicating death within 28 days and 0 indicating survival within 28 days. (B) Kaplan–Meier survival curves showed that patients with sepsis exhibiting high AKT1 expression had a higher survival probability. (C) Kaplan–Meier survival curves revealed that patients with sepsis exhibiting high CXCR2 expression had a higher survival probability. (D) Expression of AKT1 in different clinical subtypes. (E) Expression of CXCR2 in different clinical subtypes.
Further analyses assessed the correlation between the expression levels of key genes and OS in patients with sepsis. K–M curve analysis demonstrated that patients with high expression of AKT1 and CXCR2 had a significantly higher survival probability (Fig. 2B–C) (P < .01). Both AKT1 and CXCR2 showed differential expression across different endotype classes (Fig. 2D–E). The endotype classes included Mars1, Mars2, Mars3, and Mars4. Significant differences in AKT1 expression were observed between the two groups in all endotype class comparisons (P < .05), except for Mars2 and Mars4, where no significant difference was detected. Similarly, CXCR2 expression differed significantly between the two groups in all comparisons, except for Mars3 and Mars4, where no significant differences were found. Additionally, CXCR2 expression was higher in males than in females.
Construction and evaluation of nomograms for key genes AKT1 and CXCR2
As shown in Fig. 3A, a nomogram model was constructed based on the expression levels of AKT1 and CXCR2, with disease status as the outcome event. This model calculated a total score by summing the scale values of both genes, where a higher total score indicated a higher risk of disease onset. The total score was directly correlated with gene expression levels (e.g., a total score of 37.4 corresponds to a 22% incidence rate). DCA was also performed using the key genes, and the curves for all models were above the gray solid line, suggesting that the models provided better clinical benefits following intervention (Fig. 3B). Furthermore, as shown in Fig. 3C, the predicted probabilities from the nomogram closely aligned with the reference line, indicating high prediction accuracy.

Construction and evaluation of the nomogram for key genes AKT1 and CXCR2. (A) Nomogram for the key genes AKT1 and CXCR2. Note: The variable names in the prediction model are AKT1 and CXCR2. Each variable is represented by a corresponding line segment marked with scales, indicating the range of the variable, with the length of the line segment reflecting the contribution of that factor to the outcome. The “Points” represent the individual score corresponding to each variable under different values, and the “Total Points” represent the sum of the individual scores across all variables. “Pr” stands for the probability of being diseased. (B) Decision curves of the nomogram. (C) Calibration curve of the nomogram. Note: The abscissa represents the predicted event rate, and the ordinate represents the observed actual event rate, both ranging from 0 to 1.
Enrichment analysis of key genes
The functions of AKT1 and CXCR2 were further explored through GSEA, which revealed that AKT1 and CXCR2 were linked to 49 and 44 pathways, respectively (Tables S7-8). The top five pathways associated with AKT1 include “intestinal immune network for IgA production,” “autoimmune thyroid disease,” “allograft rejection,” and “viral myocarditis” (Fig. 4A). Similarly, the top five pathways for CXCR2 include “natural killer cell-mediated cytotoxicity,” “Leishmania infection,” and the “chemokine signaling pathway” (Fig. 4B). These results suggest that both AKT1 and CXCR2 are involved in immune processes and may play key roles in the pathogenic mechanisms of sepsis.

Enrichment analysis of key genes. (A) GSEA plot for the AKT1 gene. (B) GSEA plot for the CXCR2 gene. The GSEA plots consist of two parts. The first part: the line represents the Enrichment Score (ES) of each gene. Different colors correspond to distinct pathways, and the ordinate shows the corresponding running ES. A peak in the line indicates the enrichment score of the gene set, with the genes preceding the peak being considered core genes within that set. The second part: the barcode-like section represents hits, where genes are sorted by fold change (from high to low), and each vertical line corresponds to a gene within the set.
Prediction of AKT1-associated and CXCR2-associated regulators and drugs
Molecular regulatory networks were further examined to gain insights into the regulatory factors influencing AKT1 and CXCR2. miRDB predictions indicated that AKT1 is regulated by hsa-miR-656-3p and CXCR2 by hsa-miR-3163 (Fig. 5A). Following this, 26 lncRNAs were predicted to target hsa-miR-656-3p and hsa-miR-3163. Among these, XIST, NEAT1, NORAD, and HCG11 were identified as shared upstream lncRNAs for both miRNAs. The NetworkAnalyst database identified seven transcription factors (TFs), including TEAD1, regulating AKT1, and six TFs, including STAT3, regulating CXCR2 (Fig. 5B). These results indicate that AKT1 and CXCR2 are regulated by multiple factors. Furthermore, drug prediction analysis identified 71 potential drugs, including desipramine, targeting AKT1 (Fig. 5C), and 18 potential drugs, including genistein, targeting CXCR2.
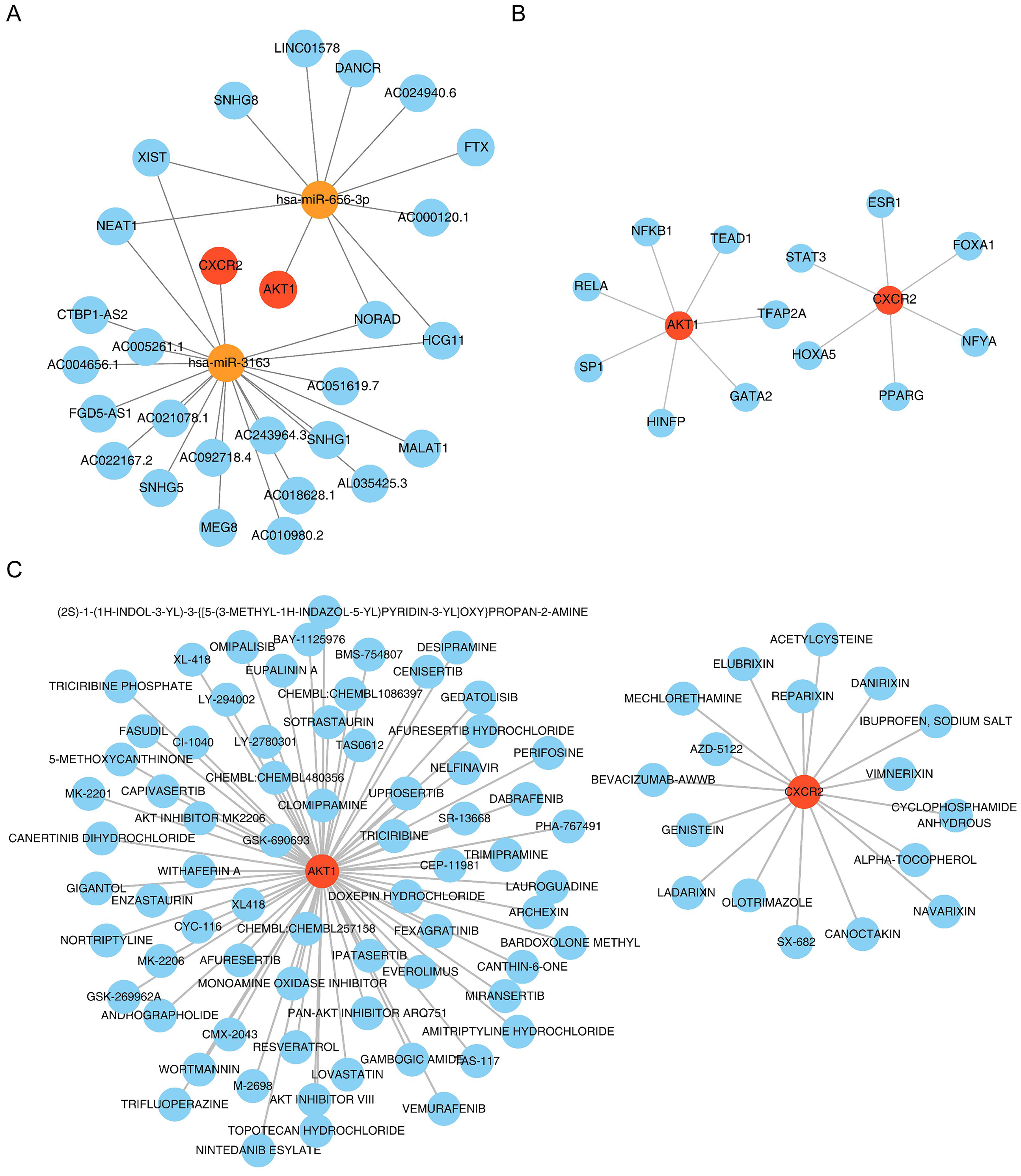
Prediction of AKT1-associated and CXCR2-associated regulators and drugs. (A) Construction of the lncRNA-miRNA key gene interaction network. Note: Red circles represent prognostic genes, yellow circles represent predicted miRNAs, and green circles represent lncRNAs. (B) TF-mRNA regulatory network. Note: Red circles represent prognostic genes, and the surrounding blue dots represent targeted TFs. (C) Key gene-targeted drug prediction network. In this diagram, red circles represent key genes, and blue circles represent drugs.
Discussion
This comprehensive genetic and observational study explores the relationships between neutrophils, NRGs, and 28-day mortality in patients with sepsis. Our findings demonstrate that neutrophil_abs independently predicts the risk of death in patients with sepsis. Genetic analysis further revealed that AKT1 and CXCR2 are key genes associated with reduced 28-day mortality in sepsis, potentially linked to sepsis immunopathogenesis.
Neutrophils play a critical role as the first line of defense in innate immunity, responding to infectious agents.3,13 Severe microbial infections stimulate granulocyte production and the release of both mature and immature neutrophils from the bone marrow into peripheral blood. Elevated levels of immature granulocytes in the blood during sepsis are associated with disease progression. 14 Immature neutrophils in patients with sepsis exhibit reduced functionality, including compromised respiratory bursts and phagocytosis. These cells tend to produce and release more NETs. 14 Persistent NETs in tissues and the vasculature, resulting from excessive production or inadequate clearance, can lead to hypercoagulation and endothelial damage. Additionally, during emergency granulopoiesis, immature myeloid cells migrate to the blood in response to infection and may differentiate into myeloid-derived suppressor cells (MDSCs), which have potent immunosuppressive effects on both innate and adaptive immunity. 14 Although neutrophils produce relatively few cytokines, they can secrete large amounts of the immunosuppressive factor IL-10 during sepsis. Mature CD16 hiCD62Llow neutrophils, which suppress T-cell proliferation, have been detected in an endotoxemia model where healthy individuals were administered low-dose endotoxin. 15 Uhel et al. further highlighted the connection between increased neutrophilic MDSC numbers and nosocomial infections following sepsis. 16 These cells are believed to sustain long-term immunosuppressive effects in individuals who develop chronic critical diseases.
In this study, functional analyses indicated that AKT1 and CXCR2 may play a pivotal role in the pathogenesis of sepsis by regulating immune responses. The downstream target gene of AKT, PI3K, is activated upon the stimulation of the mTOR/S6K/4EBP1 pathway, promoting protein production by enhancing elongation and translation. 17 Activated AKT inhibits apoptosis through the phosphorylation of Bad, a member of the Bcl-2 family, thus disrupting the apoptotic signaling pathway and promoting cell survival. 18 Several sepsis-related studies have demonstrated that activation of the AKT pathway can suppress apoptotic cell death, reduce oxidative stress and inflammation, and mitigate organ damage during sepsis. 19 Among the three isoforms of AKT—AKT1, AKT2, and AKT3—AKT1 is most strongly associated with apoptosis regulation. Activation of the PI3K-AKT-mTOR pathway has been shown to reduce apoptosis in human umbilical vein endothelial cells (HUVECs). For instance, Compound 7460–0250, a highly selective AKT1 activator, inhibited HUVEC apoptosis, enhanced inflammatory responses, and prevented immune cell leakage into the interstitial space, thereby preserving endothelial barrier function, as evidenced by in vitro and in vivo experiments. 20 Moreover, overexpression of AKT1 suppressed lymphocyte apoptosis in sepsis, increased the production of the Th1 cytokine IFN-ɤ, promoted the translocation of phospho-forkhead to the cytosol, and improved survival outcomes during sepsis. 21 In contrast, inhibition of PI3K activity with wortmannin in a murine polymicrobial sepsis model led to increased plasma factor levels and reduced survival. 22 Furthermore, inhibiting the PI3K-AKT pathway in endotoxemic mice enhanced LPS-induced activation of the p38MAPK pathway, which resulted in elevated levels of proinflammatory cytokines (e.g., TNF-α, IL-6, and MCP-1) and procoagulant molecules. 23 Additionally, Samsum ant venom has been shown to alleviate LPS-induced tissue injury and oxidative damage by upregulating AKT1 in rats, supporting the notion that AKT1 could serve as a potential therapeutic target in sepsis. 24
C-X-C motif chemokine receptor 2 (CXCR2), a chemokine receptor predominantly expressed on leukocytes, plays a pivotal role in the pathogenesis of various inflammatory conditions. 25 It is essential for the regulation of neutrophil recruitment, particularly during infections, and its expression is downregulated in neutrophils from patients with sepsis. Circulating CXCR2 ligands, such as IL-8, are associated with NETosis in these patients. 26 NETs, primarily observed in the lungs during experimental sepsis, contribute to lung injury and fibrin deposition. 27 In septic mice, inhibition of CXCR2 by reparixin reduced NET generation, mitigated multiorgan injury, and improved survival, without impairing bacterial clearance. 28 CXCR2 surface expression decreases due to the internalization of the receptor triggered by circulating chemokines. 29 Moreover, CXCR2 surface receptor deficiency has been linked to neutrophilia in septic mouse models, along with severe neutrophil hyperplasia in the bone marrow. A reduction in CXCR2 surface expression is also associated with sepsis, even in the presence of infections. 30 In patients with septic shock, CXCR2 surface expression is significantly lower after disease onset compared to individuals with infections. 31 Regression models suggest that septic shock status can predict CXCR2 surface expression in early sepsis, with severely affected patients showing markedly lower expression compared to those with milder forms of the condition. 32 In contrast, CXCR2 surface expression remains high in infected patients, resembling the pattern observed in healthy individuals. These findings suggest that reduced CXCR2 surface expression correlates with peak disease activity and reflects the body's ability to revert to a normal state, demonstrating a dose-response relationship with clinical outcomes. 33 As patients transition into the less dynamic phase of the disease, CXCR2 surface expression increases, and no significant difference in expression is observed between septic and infected patients within one week. 34 Disrupted neutrophil migration due to reduced CXCR2 surface receptor expression serves as an immunopathological marker, distinguishing septic from infected patients post-onset. This reduction impairs neutrophil chemotaxis, leading to improper extravasation across various organs, contributing to tissue injury and potentially resulting in multiorgan failure in sepsis. 30
To thoroughly evaluate the therapeutic implications of these findings, drugs targeting the two key genes were identified. Desipramine, an antidepressant, exhibits significant anti-inflammatory effects in septic shock animal models. Preventive treatment with desipramine markedly reduced TNF-α production and mortality in an LPS-induced septic shock model. 35 Similar to the glucocorticoid prednisolone, desipramine offers protective effects against sepsis-related death. 36 Reparixin, a noncompetitive allosteric inhibitor of the IL-8 receptors CXCR1 and CXCR2, emerged as an effective treatment for severe COVID-19-related acute respiratory distress syndrome (ARDS). 37 Its use is based on the hypothesis that the autocrine and systemic IL-8–CXCR1/2 axis plays a central role in neutrophil-driven immunopathology during sepsis. 38 A meta-analysis of 406 severe cases, including lung and kidney transplant recipients and patients with critical COVID-19, found that the reparixin group had significantly lower all-cause mortality compared to the control group [5/220 (2.3%) vs. 12/186 (6.5%)]. 39 These results suggest that reparixin could serve as a novel treatment for ARDS, modulating the inflammatory response and improving clinical outcomes across various etiologies.
Limitations
This study has several limitations. First, the MIMIC-IV and IEU OpenGWAS datasets predominantly consist of European populations, limiting the generalizability of the findings to non-European ethnic groups. Future studies in more diverse populations are essential to validate these results. Second, MR analysis is constrained by the heritability of IVs and sample size, which may reduce the power to detect weak causal associations. Future research should incorporate multi-omics data and implement stricter screening criteria to select higher-quality IVs. Third, sepsis is a dynamic disease with varying definitions across studies, which can lead to sample heterogeneity. To address this, a multi-center collaborative database should be established to systematically collect clinical data from patients with sepsis, minimizing bias from single-center studies and enhancing the external validity of the results. Fourth, discrepancies in data sources between MR and observational analyses prevent consistent adjustment for confounding factors, potentially introducing residual bias in causal inference. These limitations should be considered when interpreting our results, as they may hinder the direct application of AKT1/CXCR2 as therapeutic targets unless further validated. To mitigate this, future studies should conduct both observational and MR analyses within the same cohort to ensure consistent data sources and proper control for confounding factors. Fifth, due to the absence of independent sepsis datasets that include both survival data and blood samples, validation of the nomogram's performance in external cohorts was not possible. This limits the evidence supporting the model's universal applicability across different clinical settings. Future multi-center studies with standardized data collection are needed to address this gap.
Conclusions
In conclusion, this comprehensive analysis suggests that neutrophil_abs may independently predict the 28-day mortality risk in patients with sepsis based on the MIMIC-IV database. Additionally, a nomogram incorporating seven variables was developed to assess the survival probability of patients with sepsis. Two NRGs, AKT1 and CXCR2, were identified as key genes associated with sepsis prognosis, potentially involved in the immunopathogenesis of sepsis. These findings offer insights into potential treatment strategies, but further confirmation through larger prospective studies is required. In vitro and in vivo research is necessary to explore the mechanistic roles of these NRGs in sepsis progression.
Supplemental Material
sj-pdf-1-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-pdf-1-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-pdf-2-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-pdf-2-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-pdf-3-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-pdf-3-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-pdf-4-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-pdf-4-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-pdf-5-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-pdf-5-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-pdf-6-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-pdf-6-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-pdf-7-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-pdf-7-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-pdf-8-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-pdf-8-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-docx-9-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-docx-9-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-docx-10-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-docx-10-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-xlsx-11-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-xlsx-11-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-xlsx-12-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-xlsx-12-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-xlsx-13-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-xlsx-13-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-xlsx-14-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-xlsx-14-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-xlsx-15-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-xlsx-15-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-xlsx-16-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-xlsx-16-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-xlsx-17-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-xlsx-17-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Supplemental Material
sj-xlsx-18-sci-10.1177_00368504251374929 - Supplemental material for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis
Supplemental material, sj-xlsx-18-sci-10.1177_00368504251374929 for Associations between NETosis-related genes and 28-day mortality in patients with sepsis: An observational retrospective study and bioinformatic analysis by Liu Yang and Liang Chen in Science Progress
Footnotes
Acknowledgements
Authors’ contributions
YL: conception, design, data collection, data analysis, manuscript writing, manuscript revision. LC: conception, design, data collection, data analysis, manuscript writing, manuscript revision, fund acquisition, and supervision.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Nanjing Medical Science and Technology Development Fund (grant number YKK22239).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Availability of data and materials
Publicly available datasets were analyzed in this study, including the MIMIC-IV database (https://mimic.mit.edu/), GSE65682 (https://www.ncbi.nlm.nih.gov/geo/), and ieu-b-5086 (![]() ). The original research data generated in this study are available from the corresponding author upon reasonable request.
). The original research data generated in this study are available from the corresponding author upon reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.