
Abstract
Diabetic wounds, a prevalent and severe complication of diabetes, pose a significant burden on the global healthcare system. This burden arises from their high incidence, high disability rates, and substantial treatment costs. Research has shown that excessive oxidative stress damage plays a crucial role in diabetic wounds. This oxidative stress not only complicates the healing process but also triggers various forms of programmed cell death, such as apoptosis, pyroptosis, necrosis, and ferroptosis. These programmed cell deaths significantly hinder wound recovery. In this narrative review, we comprehensively review the impact of oxidative stress on diabetic wounds and its intricate interplay with programmed cell death. With ongoing intensive research into oxidative stress in diabetic wounds, we are confident that innovative therapies will emerge in the near future.
Introduction
As the global population ages, diabetes prevalence is soaring, placing immense strain on healthcare systems. By 2030, it is projected that 195.2 million individuals aged 65 to 99 will have diabetes, with Asia, notably China and India, facing severe challenges.1,2 Type 2 diabetes primarily results from insulin resistance in muscle and adipose tissue, leading to excessive liver glucose production, combined with impaired beta-cell function that restricts insulin secretion, disrupting glucose homeostasis. 3 This results in persistent hyperglycemia, damaging multiple organ systems and disrupting metabolic pathways, ultimately causing organ impairment and exacerbating diabetes-related morbidity, mortality, and economic burdens.4,5
Diabetic ulcers, a severe complication of diabetes, significantly elevate the risk of sepsis and other severe sequelae due to their chronic nature, sometimes necessitating amputation. These ulcers, characterized by full-thickness skin damage below the ankle that penetrates through the dermal tissue, often arise from a complex interplay between neuropathy (loss of sensation in the feet) and ischemia (reduced blood flow to the feet).6–8 These ulcers arise from neuropathy and ischemia, classified as neuropathic, ischemic, or neuroischemic. Neuropathic ulcers are painless, circular, often surrounded by calluses, while ischemic/neuroischemic ulcers are irregularly shaped, prone to gangrene, and frequently accompanied by complications like cellulitis or osteomyelitis.9–11
Wound healing involves hemostasis, inflammation, proliferation, and maturation phases. During the proliferation phase, angiogenesis—the formation and remodeling of new blood vessels—is crucial for wound repair. 12 In a healthy state, the microvascular network maintains a delicate balance, ensuring adequate nutrient and oxygen supply to tissues while effectively removing metabolic byproducts and waste. 13 However, diabetes, a chronic metabolic disorder, disrupts this balance upon tissue injury. This disruption is caused by multiple intricate biochemical pathways that contribute to the accumulation of non-enzymatic glycation and excessive activation of oxidative stress at diabetic wound sites. 14 Oxidative stress, as a central driving force in the pathological progression of diabetes, profoundly influences cell fate by modulating various programmed cell death pathways. These reactions significantly hinder neovascularization, impeding the efficient transportation of nutrients and immune cells to the wound site. Consequently, this not only escalates the risk and severity of infections but also accelerates the formation and progression of diabetic ulcers. 15
Current therapeutic approaches have seen researchers extensively evaluating innovative strategies, including the utilization of growth factors, cytokine modulators, anti-inflammatory agents, matrix metalloproteinase inhibitors, angiogenesis stimulators, extracellular matrix activators, stem cells, and various natural products. 16 While these treatments have demonstrated some potential in preliminary studies, their clinical efficacy remains limited, and there is a lack of significantly effective prevention and treatment protocols. The pathophysiological mechanisms underlying diabetic wounds involve excessively activated oxidative stress and subsequent programmed cell death. Thus, the key to preventing and treating diabetic wounds lies in promptly halting the excessive activation of oxidative stress, preventing programmed cell death in the injured area, promoting angiogenesis, and restoring blood flow supply. In summary, this narrative review will summarize current research on the roles of oxidative stress and programmed cell death in diabetic wounds, aiming to provide direction for future therapeutic research. This review is guided by the Scale for the Assessment of narrative review articles (SANRA). 17
Oxidative stress
The concept of “oxidative stress,” first introduced in the inaugural chapter of the book “Oxidative Stress” in 1985, has undergone 39 years of extensive research and development. It has emerged as a vital link between redox biology and medicine. 18 At its essence, oxidative stress occurs when the oxidative capacity within an organism exceeds the balancing capabilities of its antioxidant defense system. This imbalance typically arises due to an increase in the production of reactive oxygen species (ROS), a decline in antioxidant capacity, or both. ROS, which are highly reactive molecules derived from oxygen, can be categorized into two main types based on their chemical properties: non-radical and radical species. Radical ROS, distinguished by their unpaired electrons, are particularly unstable and highly reactive. 19
The role of oxidative stress in biological and pathological processes
Maintaining low or moderate concentrations of free radicals is crucial for various biological processes. However, excessive levels of free radicals and oxidants can be harmful, causing negative effects on several cellular structures.
Biological processes
Cells contain various endogenous antioxidant enzymes crucial for maintaining redox homeostasis by neutralizing harmful ROS.20,21 Glutathione (GSH), the most abundant intracellular antioxidant, is a tripeptide composed of glutamate, glycine, and cysteine, which suppresses ROS and preserves protein thiol status. 22
Other key enzymes include superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPX), peroxiredoxin (PRX), and thioredoxin (Trx). SOD exists in three forms and catalyzes the conversion of superoxide into hydrogen peroxide and oxygen.19,23 CAT and GPX collaborate to decompose hydrogen peroxide into water and oxygen, mitigating oxidative stress. Together, these enzymes form a robust defense system.
At low or moderate concentrations, free radicals play vital roles in biological processes: Cellular Structure and Defense: 1.
The role of oxidative stress in pathological processes
Research shows that diabetes can elevate oxidative stress in obese individuals.29,30 Oxidative stress occurs when excessive free radicals and oxidants harm cellular structures such as membranes, lipids, proteins, lipoproteins, and DNA. 24 This harmful process contributes to disease through two major mechanisms.
The first mechanism involves direct oxidation of macromolecules. Reactive species like •OH, ONOO-, and HOCl- are produced during oxidative stress and target membrane lipids, structural proteins, enzymes, and nucleic acids. This damage leads to aberrant cell function and potentially cell death, causing significant disruption at the fundamental level of cellular functioning. 31
The second mechanism relates to aberrant redox signaling. Normally, oxidants like H2O2 serve as essential second messengers for cellular communication. However, during oxidative stress, non-physiological levels of H2O2 disrupt this balance, causing redox signaling to malfunction. 32 This disruption exacerbates cellular damage and promotes disease progression.
In diabetes, both oxidative stress mechanisms often coexist and contribute to the development of diabetic complications. Advanced glycation products accumulate, and stress signaling pathways are aberrantly activated. Additionally, oxidative stress triggers increased H2O2 production and iron release from proteins by reactive species like O2•− and ONOO−, leading to the generation of lipid peroxidation products such as 4-hydroxy-2-nonenal (4HNE).33,34 These products exacerbate aberrant signaling and further complicate the disease process.
Excessive and uncontrolled oxidative stress also exacerbates the diabetic condition and deregulates inflammatory processes, which are crucial in the pathogenesis of chronic non-healing wounds in diabetic patients. 35 Elevated and prolonged presence of pro-inflammatory macrophages in diabetic wounds produces excessive amounts of ROS. 36 This ROS production fuels the inflammatory response and hinders the healing process, leading to the persistence of chronic wounds in diabetic patients.
Generation pathways of reactive oxygen species
ROS, which function as essential intracellular signaling molecules and potential sources of damage, are produced through a variety of pathways (Figure 1). Broadly, these pathways can be divided into two main categories: endogenous cellular generation and exogenous induction.
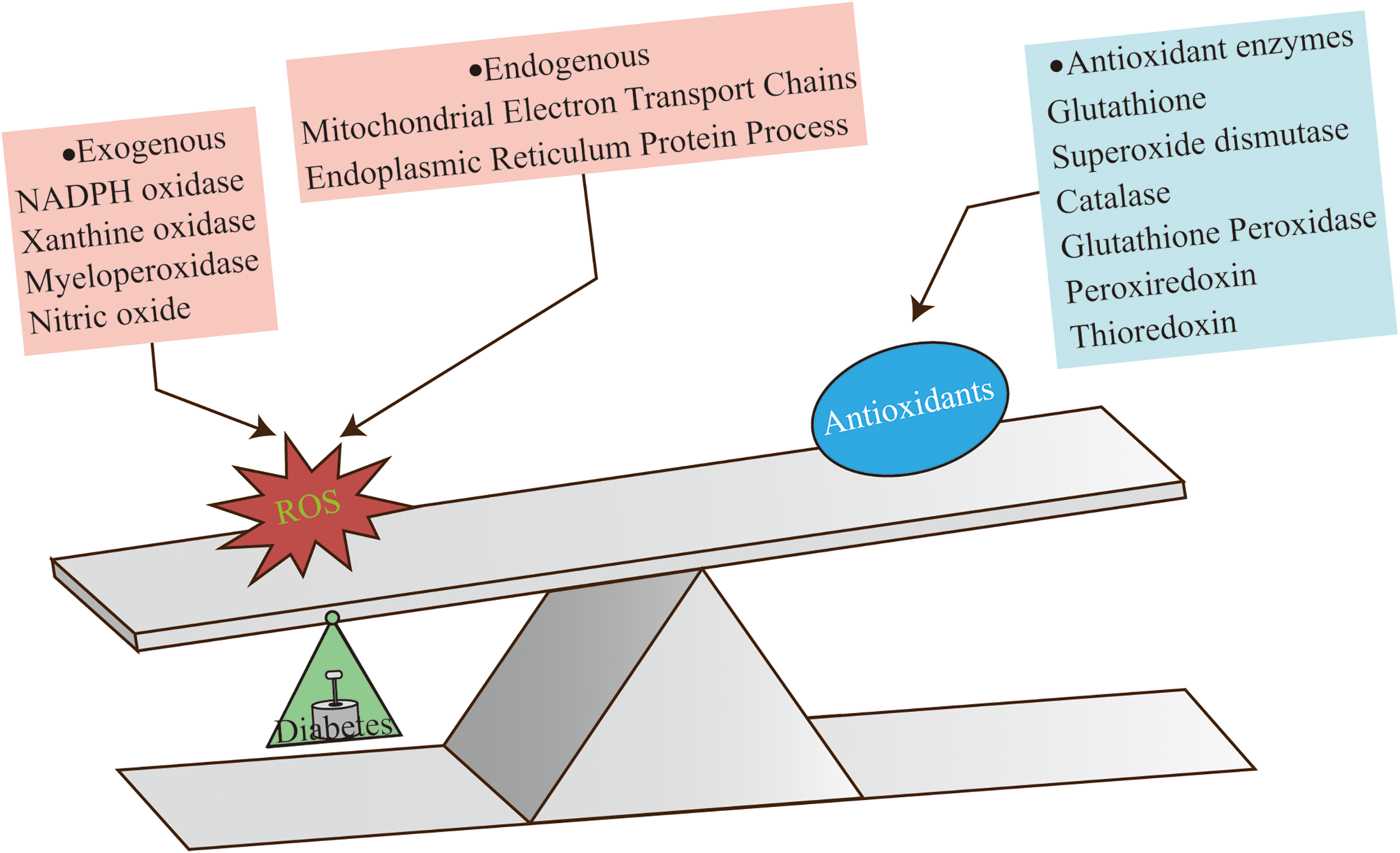
Redox imbalance in diabetic wounds.
Endogenous cellular generation of ROS: The primary mechanism of endogenous ROS generation relies on oxidative phosphorylation within mitochondria. This process not only provides cells with energy but also serves as a major source of ROS production. Additionally, cellular metabolic activities, such as the formation of protein disulfide bonds, also contribute to the generation of ROS. 37
Exogenous induction of ROS: External factors, including foreign substances, microbial invasions, and the influence of cytokines, can significantly induce ROS generation. These external stimuli activate intracellular signal transduction pathways, which in turn modulate the activities of enzymes involved in ROS production, ultimately increasing ROS levels. The enzymes involved in ROS generation encompass a range of crucial proteins, including xanthine oxidase, cyclooxygenase, lipoxygenase, nitric oxide synthase (NOS), heme oxygenase, peroxidases, hemoproteins, and NADPH oxidase, among others. 38 These enzymes are widely distributed within cells and collaborate to form a complex network of ROS generation by catalyzing distinct chemical reaction pathways.
Mitochondria
Mitochondria, known as the cell's energy factories, have a unique structure consisting of both an outer and an inner membrane. Within the inner membrane, there are five essential complexes (I-V) that together form the electron transport chain (ETC). 39 This complex system uses redox reactions to produce ATP, the cell's primary energy source. 40
The ETC process begins with the oxidation of NADH and succinate in complexes I and II, respectively. 41 These reactions convert ubiquinone to ubiquinol, which is then oxidized in complex III, transferring electrons to cytochrome c. 42 Cytochrome c then undergoes oxidation in complex IV, where electrons are ultimately transferred to oxygen molecules, resulting in the formation of water. 43 This series of reactions creates an H + gradient across the inner mitochondrial membrane, which drives the synthesis of ATP.
However, during electron transfer, some electrons may leak and reduce oxygen molecules, generating superoxide, a ROS. To counteract these ROS, mitochondria have developed mechanisms such as SOD1 and SOD2, which are located in the intermembrane space and mitochondrial matrix respectively. These enzymes convert superoxide to oxygen and H2O2. 44 Additionally, enzymes like GPX and PRX further reduce H2O2 to water, maintaining a redox balance within the mitochondria.
When the production of ROS exceeds the antioxidant capacity of the mitochondria due to conditions such as diabetes or disease, the delicate redox balance is disrupted, leading to oxidative stress. 24 This imbalance can have adverse effects on cellular function and health. Hyperglycemia has several detrimental effects on cellular processes. Firstly, it boosts ROS production by altering mitochondria and oxygen use, which leads to reduced ATP formation and increased membrane potential. 45 Additionally, hyperglycemia raises the NADH/FADH2 ratio, causes mitochondrial fission, and results in the accumulation of fragmented mitochondria. 46 Secondly, hyperglycemia increases nuclear DNA mutation rates due to exposure to oxygen radicals. 47 ROS-induced breaks in DNA strands may be linked to the release of superoxide and hydroxyl radicals. 48 Consequently, these DNA fragments activate PARP, a repair enzyme, which in turn disrupts glycolysis. Therefore, the delicate interplay between ROS generation and antioxidant defense mechanisms in mitochondria is essential for maintaining cellular homeostasis.
Endoplasmic reticulum
Within the endoplasmic reticulum (ER), H2O2 is primarily generated by two enzymes: ER oxidoreductin 1 (ERO1) during oxidative protein folding, which involves disulfide bond formation,, 49 and ER-ROS. 50 Peroxisomes, however, maintain oxidative balance in the cell by harboring antioxidant enzymes such as CAT, superoxide dismutase 1 (SOD1), and PRX, which regulate ROS levels. 50
Under physiological conditions, glutathione (GSH) constitutes approximately 98% of the total glutathione pool and plays a crucial role in cellular antioxidant defense, especially during protein folding processes. 51 When GSH is oxidized to glutathione disulfide, glutathione reductase recycles it back to its active form, ensuring a continuous supply for antioxidant defense . Additionally, the ER is safeguarded from oxidative insults by other antioxidants such as glutathione peroxidase, peroxiredoxin 4, and ascorbate peroxidase, which work together to maintain the oxidative balance within the ER and protect it from damage caused by ROS. 52
However, under pathological conditions such as diabetes, there is an increased demand for protein production, leading to excessive consumption of GSH and the formation of non-native disulfide bridges. 53
This depletion of GSH reserves exacerbates oxidative stress within cells, 54 while ROS generated due to hyperglycemia inhibits disulfide isomerase, causing protein misfolding and unfolding . These misfolded and unfolded proteins contribute to ATP degradation, prompting increased glucose utilization and mitochondrial oxidative phosphorylation to boost ATP production. Unfortunately, this process further enhances ROS generation, creating a vicious cycle.
Moreover, the accumulation of misfolded proteins in the ER lumen triggers Ca2+ leakage into the cytosol, elevating its concentration and further stimulating ROS production from mitochondria. This vicious cycle of ER stress, Ca2+ leakage, and ROS generation exacerbates oxidative stress within the cell. Additionally, the depletion of Ca2+ from the ER lumen contributes to even more ER stress, as Ca2+ is essential for various ER functions, including protein folding and quality control. Thus, the depletion of Ca2+ and the accumulation of misfolded proteins reinforce each other, perpetuating a cycle of stress and damage to the cell. 51
NADPH oxidases
The NADPH oxidase (NOX) family encompasses seven homologues: NOX1 to NOX5, as well as DUOX1 and DUOX2. Among these, NOX2 and NOX4 are especially significant due to their high expression levels in diabetes.55–57
NOX2, a six-transmembrane protein, activates via complex interactions, while NOX4 localizes broadly, including plasma membrane and intracellular compartments. 58 Hyperglycemia, a key characteristic of diabetes, intensifies the assembly of NOX enzymes, leading to increased production of ROS. 59 Additionally, the accumulation of advanced glycation end product (AGE) further promotes ROS generation. 60 Under hyperglycemic conditions, a harmful cellular environment emerges, characterized by oxidative stress, acidosis, and hypercoagulability, ultimately fostering progressive ischemic necrosis.
Xanthine oxidase
Xanthine oxidase (XO), also known as xanthine oxidoreductase (XOR), belongs to the molybdoflavoenzyme family found in both prokaryotes and eukaryotes. 61 In mammals, XO is widely distributed, especially in the liver, vascular endothelial cells, and mammary gland, and is crucial for nucleotide catabolism, maintaining cellular homeostasis.62,63
A key feature of XO in mammals is its ability to undergo post-translational modification, converting it into xanthine dehydrogenase, which accepts NAD + as an electron acceptor, or remaining as XO. Circulating XO can bind to endothelial glycosaminoglycans, initiating a reaction that generates ROS such as O2- and H2O2. 64
In diabetic wounds, XO expression and activity are significantly upregulated compared to wild-type or acute wounds, leading to overproduction of free radicals and prolonged wound closure.65,66 In vitro studies show that XO-derived ROS can mediate the release of interleukin-1β (IL-1β), a proinflammatory cytokine elevated in chronic venous leg ulcer wound fluids and tissues. 67
Additionally, uric acid, the end product of XO activity, is elevated in wound fluids from chronic venous leg ulcers. High concentrations of uric acid can crystallize into monosodium urate crystals, stimulating the production of proinflammatory cytokines and further perpetuating inflammation. 67 Thus, XO plays a significant role in the inflammatory response observed in diabetic wounds.
Myeloperoxidase
Myeloperoxidase (MPO) is a heme-containing peroxidase that is synthesized during myeloid differentiation and stored in azurophil granules of leukocytes. It plays a crucial role in phagocyte function by generating HOCl- and other oxidants through the oxidation of halide ions by H2O2. These oxidants effectively eliminate pathogens 68 and contribute to the formation of neutrophil extracellular traps, enhancing the capture and killing of bacteria. 69
However, MPO activity can also have detrimental effects. During diabetic disease states, MPO is released extracellularly through degranulation, resulting in the oxidation of not only halide ions but also other substrates. This can mediate tissue damage, as reported in several studies.70,71 Furthermore, MPO is implicated in the oxidative modification of low-density lipoprotein, catalyzing lipid peroxidation linked to coronary artery disease. Evidence suggests a correlation between circulating MPO concentrations, MPO-derived oxidized molecules, and the disease.72,73 Of particular interest, research has shown that MPO levels are elevated during acute inflammation and can impair diabetic wound healing.74–76
NOS
Nitric oxide (NO) and related nitrogen species are vital in physiological processes, especially skin, cardiovascular, and metabolic functions.77,78 The body produces NO mainly through the l-arginine-dependent NOS pathway and, when NOS activity is compromised, via the nitrate–nitrite–NO pathway enhanced by diet. 78
Abnormal NO production characterizes diabetes, impairing wound healing. 79 In healthy wounds, cells express iNOS for healing, but this is suppressed in diabetes, reducing NO.80,81 Inducible nitric oxide synthase (iNOS) is a well-characterized mediator of diabetic complications. In hyperglycemic conditions, sustained iNOS upregulation generates excessive NO, which reacts with superoxide to form peroxynitrite, a potent oxidant that damages cellular components through protein nitration and lipid peroxidation. This oxidative-nitrative stress cascade impairs macrophage function and exacerbates inflammation. 82 The decreased NO levels in diabetic wounds have several detrimental effects. Firstly, the diminished recruiting effect of NO results in the macrophage phenotype shifts from a pro-repair to a pro-inflammatory state. 83 Secondly, inhibited NO production adversely affects keratinocyte function during re-epithelization, reducing their number, antagonizing proliferation and differentiation, and ultimately delaying wound closure.84,85 Thirdly, fibroblasts in diabetic chronic wounds exhibit reduced iNOS and eNOS expression, undergo senescence, and fail to produce collagen or form the extracellular matrix, further compromising wound healing. 86 Consequently, decreased NO levels in extracellular wound fluid correlate with reduced collagen content and weaker wound breaking strength.86,87
Oxidative stress induced programmed cell death in diabetic wound
Cell death is caused by damage, either from inside or outside the body. 88 Initially, it was divided into necrosis (passive cell death) and apoptosis (active, gene-controlled cell death). 89 Subsequent research has identified additional regulated PCD pathways beyond apoptosis, including pyroptosis and ferroptosis,90,91 which exhibit distinct molecular mechanisms while maintaining the orderly, genetically controlled characteristics of PCD.
PCD can be divided into two types based on how cells die. The first type is apoptosis, where cells don't break open or release their contents. Instead, they form apoptotic bodies that are eaten by other cells. This “silent” PCD doesn't cause inflammation.
The second type includes pyroptosis and necroptosis, where cells eventually break open and release inflammatory factors. 92 Pyroptosis happens when certain sensors, like NLRs, AIM2, and the pyrin receptor, are activated. Necroptosis occurs when the apoptotic pathway is blocked. 93
Recently, two new types of PCD have been discovered: ferroptosis and cuproptosis. Ferroptosis involves cell death caused by lipid peroxidation due to iron and a lack of antioxidants like GSH and GPX4.
Apoptosis
Apoptosis is a well-studied type of controlled cell death. For many years, it was thought to be the only regulated form of cell death. 93
The intrinsic pathway of apoptosis starts when cells are damaged by toxins or DNA damage. This leads to the release of cytochrome c from mitochondria, forming apoptosomes and activating Caspase-3. Caspase-3 activation marks the point of no return in cell death. 94
Cytochrome c release is helped by proapoptotic proteins like BAX, BAK, and PUMA. These proteins can be activators (BIM, BID, and PUMA) 95 or sensitizers (BAD, NOXA, and BIK). 96 Activators bind to BAX and BAK, causing them to change shape and form pores in the mitochondrial membrane.97,98
These pores allow cytochrome c to enter the cytosol, starting the formation of apoptosomes. Apoptosomes activate caspase-9, which then activates caspase-3 and caspase-7. These caspases cleave other caspases, creating a cascade that ensures apoptosis is completed.99–102
The extrinsic pathway of apoptosis starts when cell surface death receptors like TNFR1/2, Fas, and TRAIL receptors are activated. This leads to the formation of the DISC, which can either form a prosurvival or proapoptotic complex, ultimately activating effector caspases and feeding back into the intrinsic pathway.103–109
Oxidative stress occurs when the balance between ROS and antioxidants is disrupted. ROS activate ASK1, which is controlled by Trx. When Trx is inactivated by ROS, ASK1 oligomerizes and autophosphorylates, leading to the activation of JNK.110,111
Apoptosis plays a key role in diabetes and wound healing. Abnormalities in apoptosis affect various cells, leading to uncontrolled inflammation, hindered angiogenesis, and impaired re-epithelialization. 92
AGEs induce oxidative stress and inflammation in fibroblasts, linked to apoptosis. TIMP-1 reduces apoptosis in fibroblasts treated with AGEs. GDF11 promotes CTGF expression and improves wound healing by regulating YAP and Smad2/3.112–114
Given the central role of oxidative stress in diabetic wound pathology, recent therapeutic strategies have focused on targeting these pathways. Among promising candidates, natural compounds like xanthohumol, astragaloside IV, and Berberine have emerged as particularly relevant due to their dual modulation of oxidative stress and apoptosis pathways in diabetic wounds.
In high glucose, Nrf2 inhibition decreases HO-1 expression, leading to increased keratinocyte (KC) apoptosis and impaired wound healing. Xanthohumol reduces KC apoptosis by increasing Nrf2 expression and activating AMPKα and modifying Keap1. A Keap1/Nrf2 loop increases oxidative stress-mediated EC apoptosis in diabetes.115–117 Astragaloside IV weakens KC apoptosis by activating the TGF-β/Smad pathway. MMP9 increases FasL expression and promotes KC apoptosis through the FasL/Fas pathway, delaying wound healing. Berberine down-regulates MMP9, accelerating healing. Cx31.1 is associated with KC apoptosis in diabetic wounds.118–121 Melatonin promotes diabetic wound healing by mediating mitochondrial function and apoptosis in endothelial cells through the AMPK/SIRT1/HIF-1α pathway. 122
Necroptosis
Necroptosis, a form of cell death distinct from apoptosis, can be initiated by various receptors such as death receptors (Fas/FasL), 123 Toll-like receptors (TLR4 and TLR3), 124 and cytosolic nucleic acid sensors (RIG-I and STING).125–127 These receptors often trigger NFκB-dependent proinflammatory signals but can also lead to necroptosis when Caspase-8 is inhibited. Active RIPK1 forms a complex with FADD, caspase-8, and caspase-10, and in the absence of caspase-8 activity, RIPK1 phosphorylates RIPK3 to form the ripoptosome.128,129
The RIPK1/RIPK3 complex recruits and phosphorylates MLKL, forming the necrosome. This complex disrupts cellular integrity by facilitating ion influx and forming pores in the plasma membrane. 130 MLKL oligomerization and membrane translocation depend on specific inositol phosphate interactions, and a mutant MLKL that alters its structure leads to constitutive killing activity and lethal inflammation in mice. MLKL oligomerization also controls the kinetics and threshold of necroptotic cell death.131–133
Necroptosis interacts with ROS in a positive feedback loop, and TNF uses ROS as secondary messengers to induce cell death. ROS modifies RIP1, promoting its autophosphorylation and triggering TNF-induced necroptosis.134–136 RIP3 kinase activity may also be linked to mitochondrial bioenergetics, and RIPK1 regulates various signaling pathways including NF-κB, caspase 8-mediated cell death, and the MAP kinase cascade.137,138
Necroptosis is prevalent in diabetic wounds, significantly impacting healing. 139 Decreased SIRT3 expression in diabetic wounds leads to prolonged healing due to compromised mitochondrial function, heightened oxidative stress, and enhanced necroptosis. 140 In contrast, hydrogen sulfide can modulate macrophage polarization and necroptosis, accelerating wound healing in diabetic skin. 141 The brief figure of initial programmed cell death is shown in Figure 2.

Mechanisms of programmed cell death in diabetic wound healing. (A) Intrinsic and extrinsic apoptotic pathways interacting with ROS. (B) Necrosome formation and ROS-dependent necroptosis pathways.
Pyroptosis
Pyroptosis is a programmed cell death induced by various stimuli, featuring inflammasome activation and membrane pore formation. Inflammasome activation involves assembly and activation in response to PAMPs or DAMPs, leading to pro-caspase-1 cleavage and generating active caspase-1. Active caspase-1 triggers two downstream events: formation of mature N-GSDMD by cleaving porin GSDMD, and maturation and release of cytokines like IL-1β and IL-18 by cleaving pro-inflammatory cytokines. 142
Based on caspase-1 activation, pyroptosis is divided into classical and non-canonical pathways.. 143 In the classical pathway, inflammasome assembly and activation are crucial. In the non-canonical pathway, caspase-11 (in humans) or caspase-4/5 (in mice) is directly activated by bacterial components, cleaving GSDMD to form membrane pores that mediate pyroptosis.144,145
The NLRP3 inflammasome-mediated pyroptosis is a well-studied pathway consisting of NLRP3, ASC, and pro-caspase-1. 142 Multiple signals, including calcium flux, ER stress, mitochondrial dysfunction, ROS, and lysosomal disruption, activate the NLRP3 inflammasome.146–149
ROS play a crucial role in this activation through two stages: initiation and activation.150,151 During initiation, ROS upregulate NLRP3, pro-caspase-1, and pro-IL-1β. In the activation stage, ROS promote NLRP3 assembly via TXNIP, which dissociates from Trx under high ROS levels to bind NLRP3.152,153
GSDMD is essential for pyroptosis and has three domains: PFD (N-GSDMD) for pore formation, RD (C-GSDMD) for inhibition, and a linker for activation. 154
During pyroptosis, caspase cleavage of the linker dissociates C-GSDMD, allowing N-GSDMD to oligomerize and form pores in the cell membrane.155,156 These pores disrupt membrane integrity, causing cell swelling, rupture, and release of DAMPs like HMGB1 and cytokines IL-1β and IL-18 into the extracellular space.157–159
Diabetic wounds show prolonged inflammation due to hyperglycemia and hyperlipidemia, delaying healing. Studies have focused on the NLRP3 inflammasome and related pathways in this process. Yang et al. found excessive NLRP3/Caspase-1/GSDMD activation in diabetic mouse wounds, while GSDMD knockout mice showed improved healing. 160 Bitto et al. also observed NLRP3 activation impairing wound healing. 161
Pyroptosis plays a role in diabetic wound healing. Ao et al. showed that lavender oil accelerating healing by inhibiting caspase-11-mediated macrophage pyroptosis. 162 Ou ZL et al. indicated NOD1, NOD2, and GSDMD-N-mediated pyroptosis hinder healing. 163 Chen et al. proved classical pyroptosis and excessive inflammation delay healing in fibroblasts. 164
MicroRNA and long non-coding RNA regulate pyroptosis in diabetic wounds. Song Y et al. proved that mir-374a-5p upregulation and MALAT1 downregulation activate pyroptosis and the NLRP3/Caspase-1 pathway, respectively.165–169
Liu D et al. observed that neutrophil extracellular traps (NETs) also play a role, activating the NLRP3 inflammasome and triggering inflammation. 170 Sustained NLRP3 activation in macrophages adversely affects healing. Kambara et al. found neutrophil elastase cleaves GSDMD, mediating NET formation. 171
Hyperglycemia increases NLRP3 and protein kinase R expression in diabetic wounds, activating the Caspase-1/NLRP3 axis and elevating cytokines, exacerbating hyperglycemia, AGEs, and ROS formation. ROS production induces endothelial progenitor cell senescence, disrupting tissue repair and angiogenesis.172–175 Kailun Zhang found that bioglass promotes wound healing by inhibiting endothelial cell pyroptosis through regulation of the connexin 43/ROS signaling pathway. 176
Ferroptosis
Ferroptosis, coined by the Stockwell Lab in 2012, refers to a unique form of cell death in human fibrosarcoma cells induced by erastin. 177 This iron-dependent process differs from apoptosis, necrosis, autophagy-dependent cell death, and pyroptosis. Ferroptotic cells exhibit intact membranes, swelling, reduced or absent mitochondrial cristae, and no nuclear condensation. 178
Biochemically, ferroptosis is marked by low GSH levels and inactivated GPX4, leading to lipid peroxide accumulation and cell death. The exact mechanism remains unclear, but hypotheses include the formation of lipid pores due to oxidized phospholipids, increasing membrane permeability and promoting the production of toxic aldehydes like 4-HNEs and malondialdehydes, which inactivate cellular proteins and enhance ferroptosis.156,179
During ferroptosis, several key regulators and signaling pathways are crucial. One important regulator is GPX4, which converts toxic lipid hydroperoxides to non-toxic forms using GSH, an important antioxidant. Lipid hydroperoxides can decompose into reactive compounds that act as “oxidative stress second messengers.” 180
Accumulated intracellular iron also plays a key role in ferroptosis.181,182 Iron is imported into the cell, reduced, and transported into the labile iron pool. Excess iron generates hydroxyl radicals through the Fenton reaction, leading to phospholipid hydroperoxide accumulation and a lipid radical chain reaction.179,183
Lipoxygenases (LOX) catalyze the peroxidation of polyunsaturated fatty acids (PUFAs) to generate lipid peroxyl radicals, further contributing to ferroptosis.184,185 Ferroptosis suppressor protein 1 (FSP1) is another key regulator, although its mechanisms are not fully understood here.
GPX4's activity is vital for maintaining membrane lipid bilayer balance and cellular health. 186 A decline in GPX4 activity increases cells’ vulnerability to ferroptosis. 187 GPX4 functionality is linked to GSH levels, regulated by the cystine/glutamate antiporter SLC7A11.188,189
ROS under oxidative stress can deplete GSH levels, reduce GPX4 expression, and activate p53, inhibiting SLC7A11 transcription and further impacting GSH synthesis and GPX4 activity.190–193
Oxidative stress triggers the formation and opening of the mitochondrial permeability transition pore (mPTP), initiating a vicious cycle known as “ROS-induced ROS release.”194,195 This cycle exacerbates ROS production, which reacts with PUFAs on the mitochondrial membrane, leading to lipid peroxidation and mitochondrial DNA (mtDNA) damage. Specifically, damaged mtDNA that affects ETC complex-coding subunits can result in metabolic dysfunction. 194
Oxidative stress triggers the opening of the mPTP, initiating a cycle of ROS-induced ROS release, exacerbating ROS production, and leading to lipid peroxidation, mitochondrial DNA damage, and metabolic dysfunction.196–198
In diabetic wounds, an overly active ferroptosis response delays healing. Amini et al. noted that diabetic skin wound patients had lower levels of selenium, vitamin E, and antioxidants, potentially impairing GPX4 function and leading to ferroptosis and ulcer development.199–201
Wu et al. found that hyperglycemia causes endothelial oxidative stress and inflammation, which iron overload exacerbates. 202 Chaudhary et al. demonstrated that iron overload enhances oxidative stress, activates NLRP3 inflammasome signaling, and triggers inflammatory responses and renal dysfunction in rats. 203
Chen et al. observed that high iron levels in diabetic rats led to AGEs accumulation in the liver and increased AGE-RAGE axis expression in the testes. AGEs disrupt wound healing by promoting glycated collagen formation and oxidative stress. 204 They also bind to neutrophil scavenger receptors, inhibiting neutrophil migration and bactericidal capacity. Consequently, neutrophils fail to reach the wound site promptly, leading to inflammatory regions and chronic wounds due to excessive cytokine release.205,206 The brief figure of other two types of programmed cell death is shown in Figure 3.
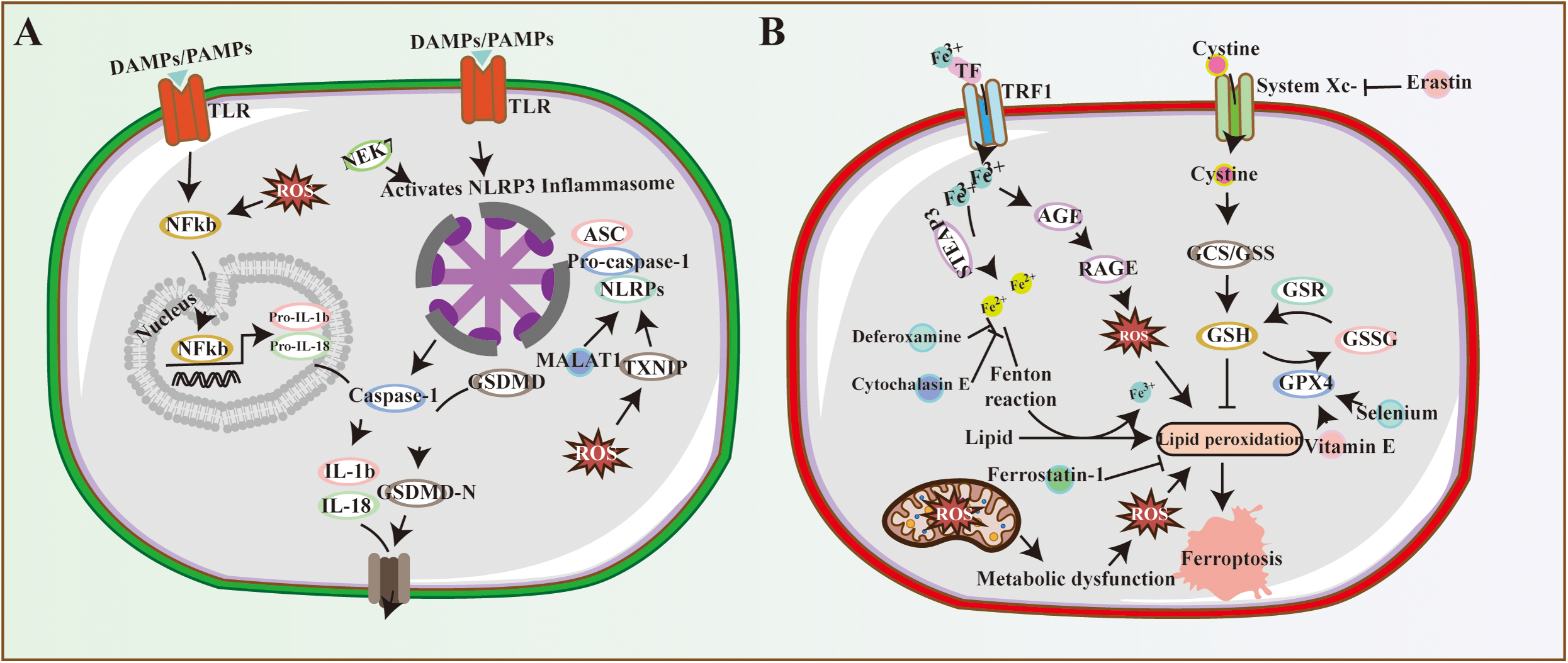
Non-classical programmed cell death in diabetic wounds. (A) Pyroptosis mechanisms: NLRP3 inflammasome activation, caspase-1/11-mediated pathways, and GSDMD pore formation. (B) Ferroptosis regulators: GPX4, FSP1, and SLC7A11-mediated iron-dependent lipid peroxidation.
Conclusion
The healing process of diabetic wounds is a highly complex and multifaceted biological challenge, influenced by numerous intricate interactions and factors. Key barriers to effective wound healing in diabetics include the persistence of hyperglycemia, neuropathy, inadequate blood supply, disrupted matrix metabolism, impaired wound contraction capabilities, and microbial imbalance.
Oxidative stress plays a pivotal role at the core of this physiological and pathological cascade. As a critical regulatory factor, excessive activation of oxidative stress triggers programmed cell death through multiple signaling pathways, thereby stalling the healing process of diabetic wounds.
In recognition of the significant contribution of excessive oxidative stress to delayed wound healing in diabetes, current research and therapeutic strategies are increasingly focused on controlling ROS levels and mitigating their tissue damage. This approach holds profound theoretical significance and showcases immense potential for clinical application, promising to drive advancements in the treatment of diabetic wound healing.
Footnotes
Acknowledgements
We express our gratitude for the technical support provided by the libraries at Zhejiang Chinese medical university and Zhejiang University.
Author contributions
Y.P. Fan conceived and designed the review framework; M.F. Li conducted extensive literature search and analysis; Y.P. Fan and L. Hong wrote the manuscript. All authors read and approved the final paper.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Availability of data and materials
All data and materials used in this review paper are publicly available and can be accessed through the Literature Sources.