
Abstract
Hygienic monitoring of laboratory rodents has focused more and more on the analysis of environmental sample material by quantitative polymerase chain reaction (qPCR) assays. This approach requires profound knowledge of specific genetic sequences of the agents to be monitored and the assays need to be permanently adapted to take the latest research into account. [Pasteurella] pneumotropica was recently reclassified into the new genus Rodentibacter, with Rodentibacter (R.) pneumotropicus and R. heylii as the most commonly detected species in laboratory mouse colonies. This study aimed at the development of a specific qPCR assay for the simultaneous detection of both agents.
A novel primer probe set, based on detection of the specific virulence factor‚ ‘inclusion body protein A’ gene (ibpA), was confirmed by testing the assay on currently described Rodentibacter type species and other Pasteurellaceae. Furthermore, it was validated within four different barrier units and results were compared with the cultural analysis of sentinel mice.
The assay was suitable to specifically detect R. pneumotropicus and R. heylii and discriminate them from other murine Rodentibacter spp. In addition, it revealed high sensitivity for the detection of both agents in environmental sampling material including exhaust air dust in individually ventilated cage systems. Altogether, higher pathogen prevalence was detected via qPCR of environmental samples compared with cultural diagnostics of sentinel mice.
This study describes a qPCR assay for the simultaneous detection of R. pneumotropicus and R. heylii. This assay was demonstrated to be beneficial during routine health monitoring, especially with regard to environmental sampling strategies.
Introduction
Hygienic monitoring of laboratory rodents is the fundamental basis for healthy animals and reliable data in biomedical research. As most experimental mouse colonies are housed in individually ventilated cage (IVC) systems, transmission of pathogens between adjacent animal groups is generally prevented. Consequently, sentinel animals are exposed to a preferably high infection risk within the colony unit in terms of representative testing, reflecting the hygienic status of the colony. To achieve greater sensitivity for the detection of specific agents and to reduce the number of laboratory animals used for monitoring, referring to the 3R principle, current research has focused on the analysis of environmental sample material by quantitative polymerase chain reaction (qPCR) assays.1–4 This approach requires profound knowledge of specific genetic sequences of the agents to be monitored and the assays need to be permanently adapted to take the latest research into account.
Rodentibacter (R.) pneumotropicus and R. heylii, formerly known as [Pasteurella] pneumotropica, are commonly found in laboratory mouse colonies. As facultative pathogenic agents both bacteria species are known to cause abscesses in predisposed hosts.5–8 While colonizing the mucous membranes of the upper respiratory tract and also the lower parts of the genital or gastrointestinal tract, infectious agents are mostly shed by the animal’s saliva, faeces or genital excretions.5,9 Infection is either transmitted by direct contact or used bedding; however there have been various reports where detection of Rodentibacter spp. in positive colonies failed due to insufficient infection of the sentinel animals.1,10–12 Consequently Miller et al. reported that qPCR-based testing of exhaust air dust may be superior to the use of sentinel animals, especially in terms of animal welfare and the 3R principle.3,10
Due to recent findings published by Adhikary et al. [Pasteurella] pneumotropica was reclassified into the new genus Rodentibacter, including the species R. pneumotropicus, R. heylii, R. ratti, R. trehalosifermentans, R. heidelbergensis, R. rarus, R. mrazii, R. myodis, R. genomospecies 1 and 2. 13 Since cultivation of some isolates is growth factor dependent13–15 and also phenotypic characteristics can differ within the species, detection of Rodentibacter sp. by commercial biochemical reaction kits could lead to misinterpreted results. Furthermore, the detection panels of published PCR assays are not validated for the currently reclassified Rodentibacter sp., leading to confusion about the documentation within health reports. 14 Therefore this study aims at the development of a specific qPCR assay for the simultaneous detection of R. pneumotropicus and R. heylii based on recently published gene sequence analyses of strain specific virulence factors. 16
To this end a novel primer probe set was developed, based on detection of the virulence factor ‘inclusion body protein A’ gene (ibpA) which was shown to be exclusively present in the genomes of R. pneumotropicus and R. heylii type strains. 16 The specificity and sensitivity was confirmed by analysis of bacterial spiked and also animal-derived swabs and by testing the assay on all currently described Rodentibacter type species as well as other Pasteurellaceae. Furthermore, it was validated within four different barrier units harbouring IVC systems and results were compared with the findings obtained by cultural analysis of sentinel animals.
Within the genus Rodentibacter, the designed primer probe set was shown to be suitable to detect R. pneumotropicus and R. heylii without cross-reactivity to other murine associated bacterial species indicating high pathogen specificity. In addition, this qPCR assay revealed high sensitivity for the detection of R. pneumotropicus and R. heylii in environmental sampling material including exhaust air dust in IVC systems. Altogether, higher pathogen prevalence was detected via qPCR analysis of environmental samples compared with cultural diagnostics of sentinel mice.
Materials and methods
Mice
Samples were taken from mice during routine health-monitoring procedures out of three experimental units (barrier 1–3) and one strict breeding barrier (barrier 4). In the experimental units, female F1 hybrids (Ztm:NMRI × C57BL/6JHanZtm) were used as sentinel mice and were obtained from the Central Animal Facility (Hannover Medical School, Hannover, Germany). Additionally, oral swabs of colony mice of various strains were used for microbiological diagnostics within the hygienic monitoring programme. Animals were housed in Type 22 or Type 25 Polysulfon IVC systems (BioZoneGlobal, UK) with positive pressure in groups of two to four mice per cage with ad libitum food (Altromin TPF-1314, Lage, Germany) and water (autoclaved, 134℃/10 min.) supply. Further environmental standardization was achieved with 21 ± 2℃ temperature, 50% ± 5% relative humidity, 10–14 × air change hourly and 10:14 hour dark:light cycle generated by a 400 lux light source. The cages were provided with bedding material (Lignocel, Altromin, Lage, Germany) supplemented by nesting material (Enviro-Dri®, LBS Biotechnologies, Limburgerhof, Germany) and ‘Nestlets’ (ANT Tierbedarf, Buxtehude, Germany) (all autoclaved). Barrier access was restricted to personnel and a limited number of researchers. Animal handling, cage-changing process and sampling procedure were performed under a laminar flow cage changing station (Hereaus, Hanau, Germany) wearing dedicated protective clothing. Routine microbiological monitoring according to the FELASA recommendations 17 did not reveal any evidence of infection with common murine pathogens except for [Pasteurella] pneumotropica, Staphylococcus aureus, Klebsiella oxytoca, Helicobacter sp., Pneumocystis sp., murine norovirus, Trichomonas sp. and apathogenic intestinal flagellates.
In the strict breeding barrier, access was granted to two dedicated animal care takers after wet shower and complete change of clothes (cleanroom garments). Mice were maintained in the same housing system as in the other units and environmental parameters were identical. All materials entering this area, including water, were autoclaved and food (ssniff Spezialdiäten GmbH, Soest, Germany) was treated by gamma irradiation (50 kGY). This barrier was originally founded using Altered Schaedler colonized mice and limited microbial colonization was confirmed by sequencing. 18 In addition, this area has been tested negative for any FELASA-listed agents 17 since establishment.
All procedures were in accordance with the German Animal Welfare Legislation, approved by the local Institutional Animal Care and Research Advisory Committee and covered by the permission of the local veterinary authorities (reference number 42500/1H).
Routine hygienic monitoring programme
Routine microbiological monitoring was performed according to the FELASA recommendations 17 using female F1 hybrids (Ztm:NMRI × C57BL/6JHanZtm) as sentinel animals. The sentinel mice were housed in groups of two animals and kept in the used cages (50–100% dirty bedding) of the monitored colony in a cage rotating system. Every three months one mouse was sacrificed for health monitoring and replaced by a new animal which was then exposed to dirty bedding together with the remaining sentinel mouse maximizing pathogen transmission by preserving contact infection. Bacteriological testing was performed by direct cultural analysis of swabs from the oral cavity and fresh faecal pellets. Samples were cultivated in thioglycolate broth, blood agar base No. 2 containing 5% sheep blood and MacConkey agar plates under aerobic conditions (24–48 hours, 37℃). Bacterial colonies were sub-cultured, purified and further identified by colony morphology, Gram staining, oxidase and catalase tests, followed by biochemical evaluation using API 20 NE (bioMérieux, Nurtingen, Germany).
Environmental sampling procedure
For subsequent qPCR analysis, different types of environmental sample material were investigated. Three to five dried cage faecal pellets, a pooled amount of bedding and nesting material as well as cage swabs (nylon flock fibre, Hain-Lifesciences, Nehren, Germany) were directly obtained from the home cage of the sentinel mice. Additionally, the exhaust air dust filters (EAD) of the IVC rack systems were sampled as currently described by Miller et al.10,19 Sampling procedure was performed using sterile forceps and the respective material was transferred to an autoclaved 2 ml reaction tube (Sarstedt, Nümbrecht, Germany) for further processing.
DNA isolation
For the molecular biology-based testing of R. pneumotropicus and R. heylii, bacterial DNA was isolated from mouse associated (oral swabs and fresh faeces) as well as environmental sampling material (cage faeces, pooled bedding and nesting material, cage swab, EAD). DNA isolation from faeces was carried out using the PSP Spin Stool DNA Kit (Stratec, Pforzheim, Germany). For all other sample material, the MasterPure™ DNA Purification Kit (Epicentre Technologies, Madison, USA) was utilized. Both kits were used according to the manufacturer’s instructions. In case of high volume of sample material (e.g. pooled bedding and nesting material) the amount of lysis buffer was increased to attain a full sample cover for optimizing the nucleic acid extraction procedure.
qPCR assay
The R. pneumotropicus and R. heylii specific gene ibpA (GenBank: AB919058.1 and LC056027.1) was detected by qPCR using the forward primer 5′-GATGTGGGTGTCTCTGTAG-3′ and the reverse primer 5′-CCATCCGRCTCGTTTCATC-3′ (Biomers, Ulm, Germany) in combination with a double labelled probe 5′-6-FAM-ACCAACAGGTAGCGTAACAGTGGGTTATGGTCAG-BHQ1-3′ (Biomers, Ulm, Germany). Assay design was based on conventional guidelines 20 and verified using the Primer Express Software (Applied Biosystems, Darmstadt, Germany). Using a positive control plasmid containing the relevant genetic sequence, the qPCR protocol was optimized performing a primer matrix assay with different primer, probe and temperature conditions. In our hands best possible amplification was achieved using 900 nM primer and 200 nM probe concentrations with 60℃ annealing temperature. The qPCR assay was performed using the StepOnePlus™ Real-Time PCR System (Applied Biosystems, Darmstadt, Germany) and TaqMan® Fast Advanced Master Mix (Applied Biosystems, Darmstadt, Germany). All samples were tested in triplicates. As thermocycling parameters an initial denaturation process of 95℃ for 20 s, followed by 40 cycles of denaturation (95℃ for 1 s) and subsequent annealing and elongation (60℃ for 20 s) was performed and results were analysed with the StepOne™ Software v2.3.
Sample preparation, assay performance and data evaluation were carried out by three independent investigators. Sample preparation and assay performance were performed in a blinded manner.
Plasmid generation
For further quantification of the PCR product a sufficient positive control was established by amplifying the 105 bp sequence with the qPCR primer pair, using the proof-reading OneTaq®Hot Start 2 x Master Mix with Standard Buffer (New England Biolabs, Ipswich, USA). The fresh PCR product was directly cloned using the StrataClone™ PCR Cloning Kit (Agilent Technologies, Waldbronn, Germany) into the TOPO-T/A cloning vector pSC-A amp/kan and was transformed in SoloPack Escherichia coli cells. Insert specificity and fidelity were proven by sequencing (Eurofins-Genomics, Ebersberg, Germany). For large scale production of the positive control, the plasmid was multiplied with the NucleoBond® Xtra Maxi Kit (Macherey-Nagel, Düren, Germany). For standard curve generation plasmids were amplified in 10-fold dilution series from 109 to 1 copies/µl during each run for subsequent quantification of the bacterial copy quantities present in the sample material. In terms of assessing the standard curve parameters a linear regression analysis was performed using Graph Pad Prism software version 6 (GraphPad Software, LA Jolla, CA, USA) with subsequent estimation of the coefficient of determination (R2) as well as the curve-specific slope. The assay’s efficiency was then evaluated as described by Kralik and Ricchi 2017. 21
Validation 1
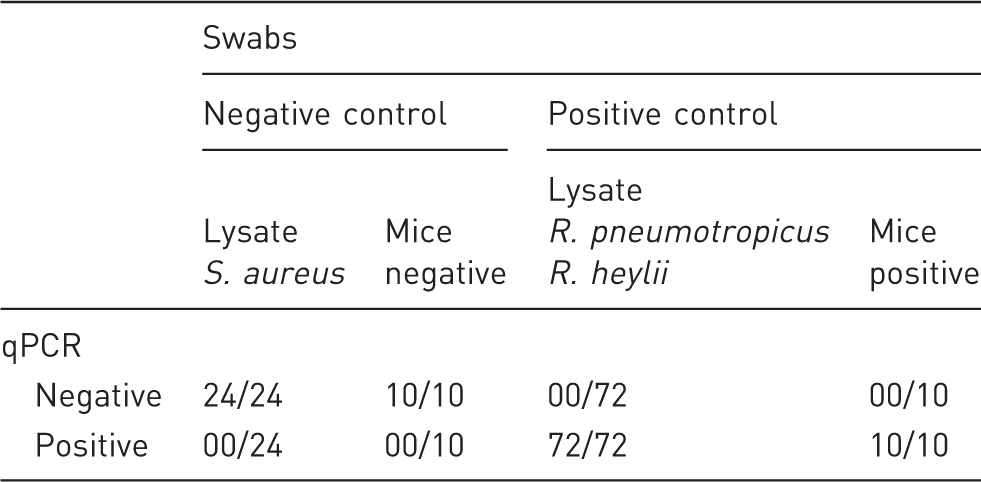
Initially, the qPCR assay was validated by testing the primer probe sensitivity and specificity by analysing different kinds of swabs. Therefore, bacterial lysates (OD600 0.5) were prepared in sterile double distilled water, which were then diluted in 10-fold dilution steps ranging from 100 to 10−10. Next, cotton wool swabs (Sarstedt, Nümbrecht, Germany) were spiked with 90 µl of each concentration, followed by subsequent DNA extraction and qPCR analysis. A 10−2 dilution was found to result in moderate copy quantities (∼CT 20, ∼105 copies/µl) and was therefore chosen for sensitivity and specificity estimation. For this approach 24 swabs were spiked either with bacterial lysates made of S. aureus (negative control), R. pneumotropicus, R. heylii or both (positive control). Additionally, oral swabs (nylon flock fibre, Hain-Lifesciences, Nehren, Germany) obtained from mice housed in the strict breeding barrier (negative controls) as well as from mice with culture proven colonization with [Pasteurella] pneumotropica (positive controls) were analysed to further test the assay under field conditions. False negative and false positive results were then used for calculation of the diagnostic sensitivity (= true positive/[true positive + false negative]) and specificity (= true negative/[true negative + false positive]).
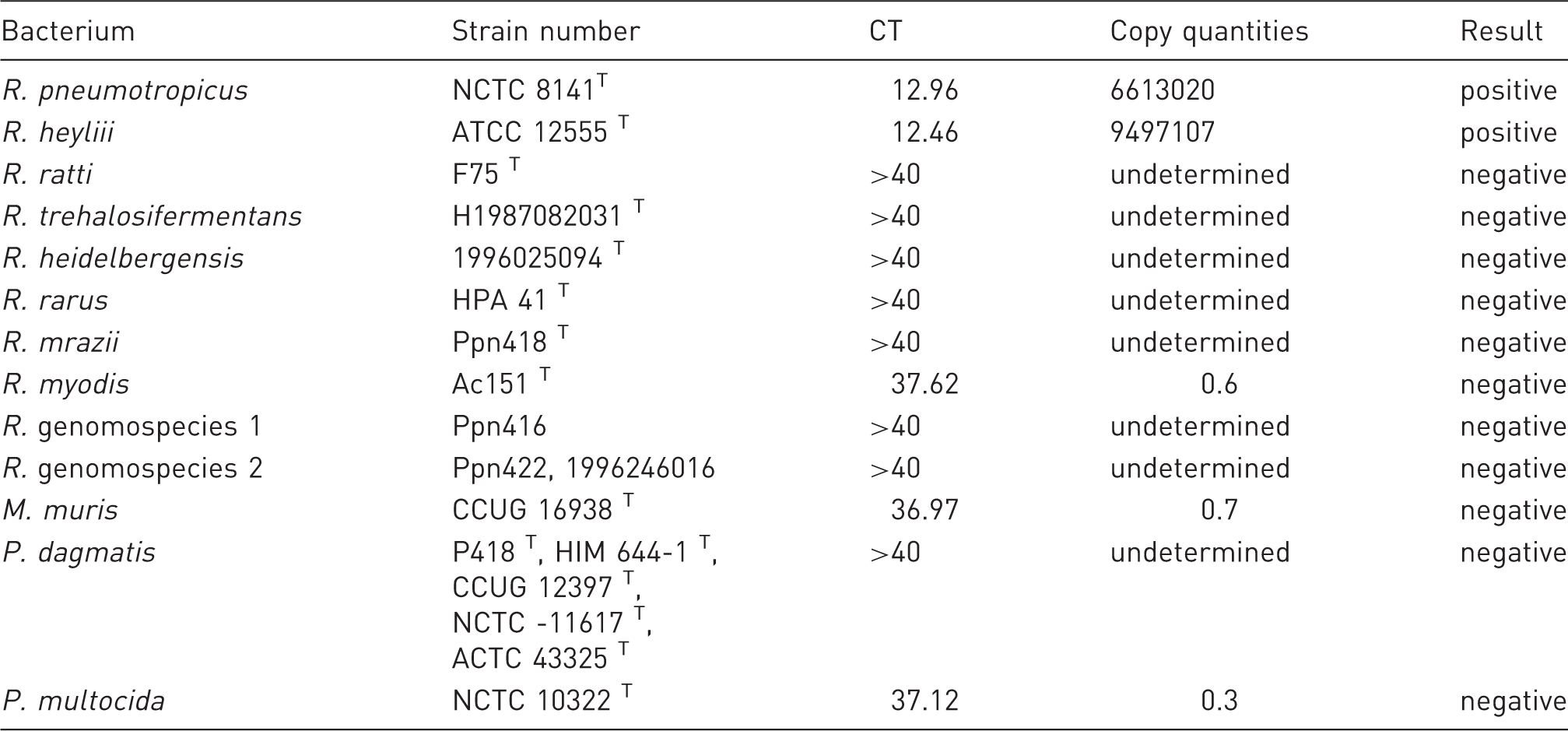
The assay’s specificity was further confirmed testing the currently described Rodentibacter type species (R. pneumotropicus, R. heylii, R. ratti, R. trehalosifermentans, R. heidelbergensis, R. rarus, R. maize, R. myodis, R. genomospecies 1 and 2), Muribacter muris, P. multocida and P. dagmatis (all provided by HC) with the herein described qPCR assay. For this approach bacterial lysates were prepared in sterile double distilled water followed by DNA isolation. For subsequent qPCR analysis bacterial nucleic acid concentrations were adjusted to 50 ng/µl by spectrophotometric measurement (NanoDrop® spectrophotometer, Peqlab Biotechnologie GmbH, Erlangen, Germany).
Validation 2
For the purpose of testing the assay during routine health monitoring, qPCR results were compared with those obtained by traditional diagnostics based on bacterial culture analysis of sentinel mice. For that reason, the assay was tested within the four barrier systems harbouring IVCs including mouse associated sampling (oral swabs, fresh faeces) as well as environmental sample strategies (cage faeces, pooled bedding and nesting material, cage swabs, EAD). Each rack is denominated as one subunit hereinafter. Furthermore, the bacterial amount found in the different sample types was quantified to further asses the most suitable sampling strategy for reliable pathogen detection.
Statistical analysis
Statistical analysis of standard curve parameters was performed by using the GraphPad Prism software version 6 (GraphPad Software, LA Jolla, CA, USA). For standard curve estimation, data were plotted in semilog (X)–line (Y) format and a subsequent linear regression was used for fitting the model. To compare the bacteria quantities found in different sample material, data were tested against the null-hypothesis of normality using the Shapiro-Wilk test. Significant differences between groups were determined using the Kruskal-Wallis test with Dunn’s test for multiple comparisons as a post hoc test.
Results
Standard curve
Beforehand, the novel designed qPCR assay was first tested by determination of standard curve parameters. The standard curve (Figure 1) was generated by analysing specific plasmids which were amplified in a 10-fold dilution series (109 to 1 copies/µl). Observed cycle threshold (CT) values of 26 independent qPCR runs were assigned to the respective copy quantities. Linear regression analysis was performed on semilog–linear plotted data, resulting in the following equation: CT = −3.37 × concentration + 36.58. The goodness of fit shows an R2 of 0.99 with 10 analysed points in total and eight degrees of freedom. The 95% confidence intervals for the slope range between [−3.48, −3.30] and for the intercept [36.10, 37.06]. The assay’s limit of detection was determined at CT 35.5.
Standard curve generated by analysing specific plasmids which were amplified in a 10-fold dilution series (109 to 1 copies/µl). Observed cycle threshold (CT) values of 26 independent quantitative polymerase chain reaction runs were assigned to the respective copy quantities. Linear regression analysis was performed on semilog–linear plotted data.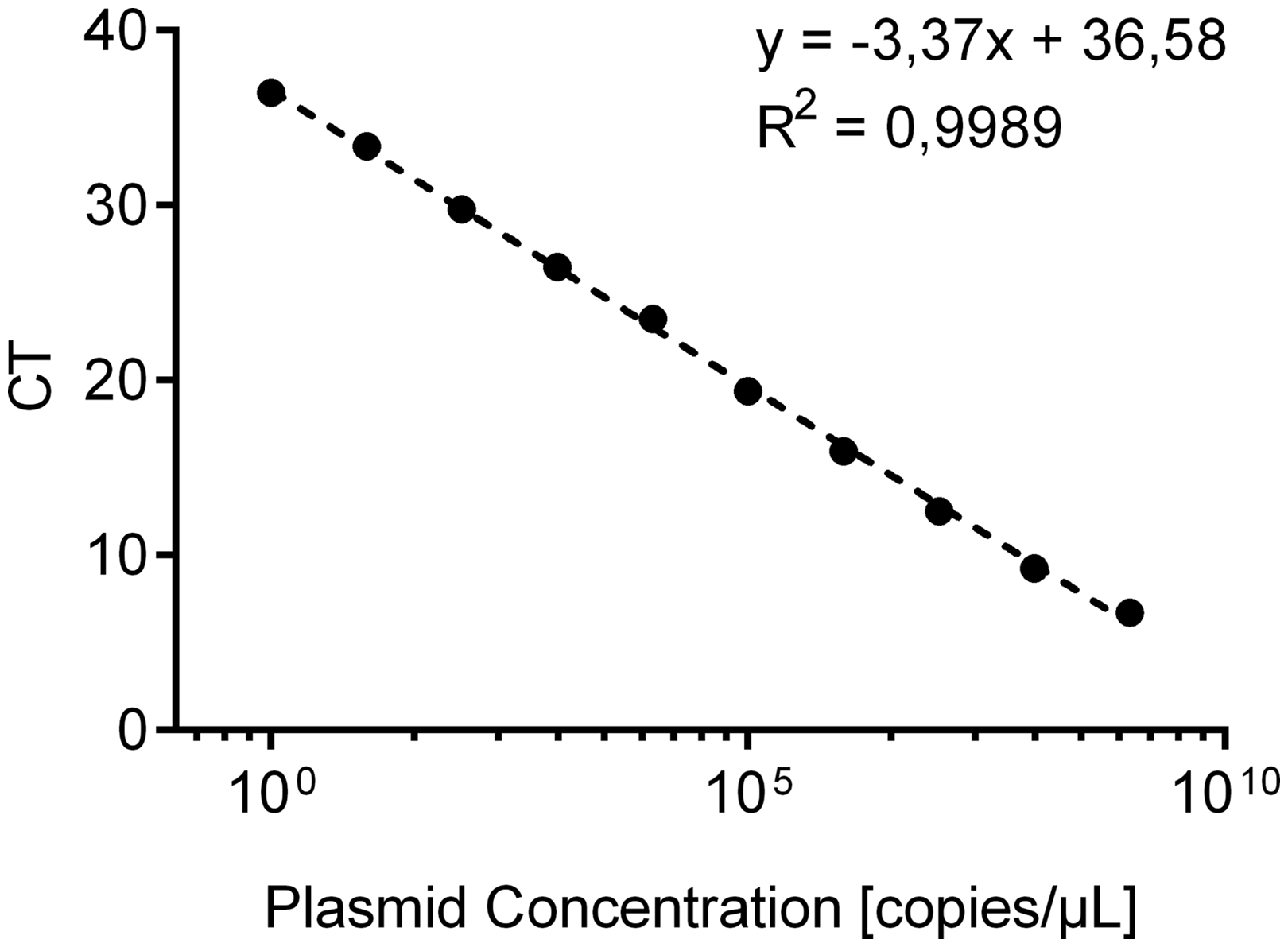
Validation 1
Quantitative polymerase chain reaction validation (qPCR) by evaluation of defined swabs. Diagnostic sensitivity and specificity were estimated based on false negative and false positive results.
Quantitative polymerase chain reaction (qPCR) analysis of the currently described Rodentibacter type species and other Pasteurellaceae. The table shows generated cycle threshold (CT) values as well as the calculated bacterial copy quantities and subsequent categorization in positive or negative results.
Validation 2
For the purpose of further testing the herein described qPCR assay during routine health monitoring, validation 2 was performed using sentinel mice, comparing the qPCR results with those obtained by traditional diagnostics based on bacterial culture analysis.
Comparison of R. pneumotropicus and R. heylii detection during routine health monitoring by bacterial culture and quantitative polymerase chain reaction (qPCR) analysis. The table shows the results for oral swabs (OS) and fresh faeces (FF) as well as qPCR testing of environmental sample material including dry cage faeces (CF), pooled bedding and nesting material (PBN), cage swabs (CS) and exhaust air dust filter material (EAD). Results are indicated as – (negative) or + (positive).
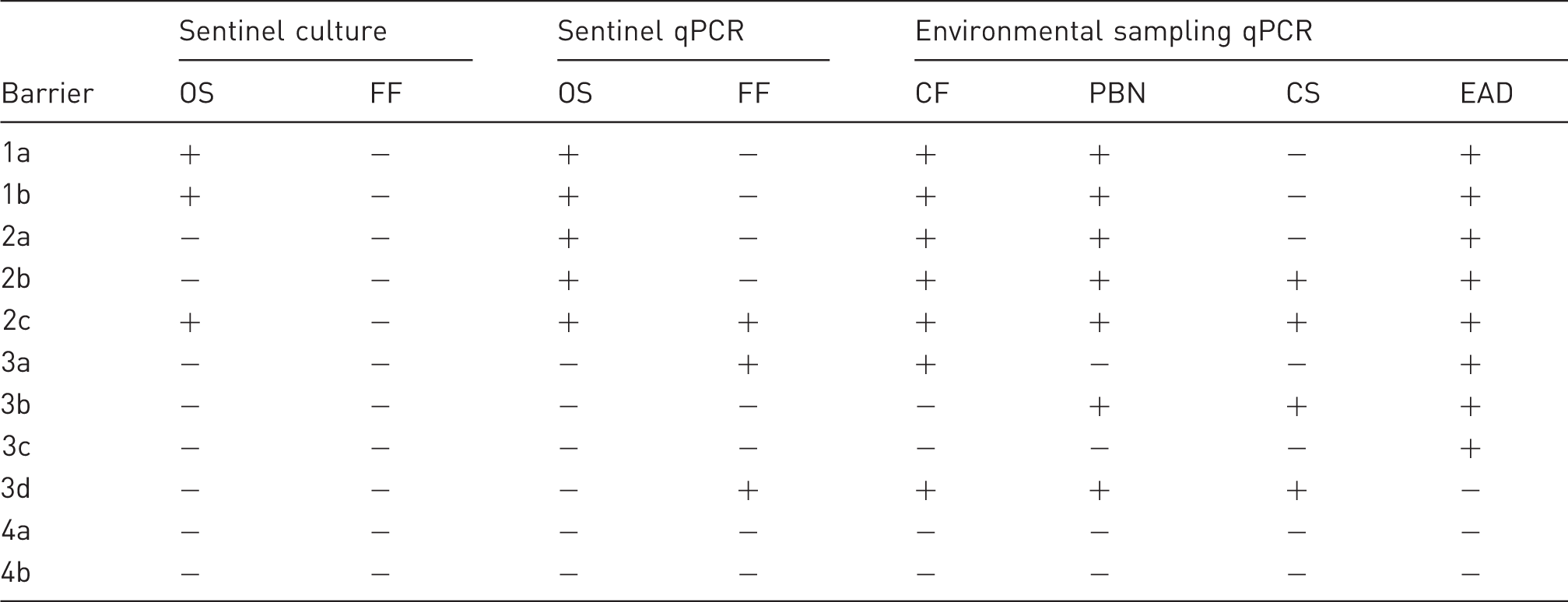
In barrier 1, oral swabs of all sentinel mice were tested positive by both, cultural analysis and qPCR, supported by positive qPCR results in cage faeces, pooled bedding and nesting material and EAD sample material. On the contrary, in barrier 2, two of the three sentinels were tested negative by bacterial culture (oral swabs, fresh faeces), whereas showing positive results in qPCR analysis of oral swabs and nearly all environmental sample types tested by qPCR, indicating higher diagnostic sensitivity of the latter method. Most interestingly, qPCR analysis in barrier 3 revealed positive results in different environmental sample material and also fresh faeces of sentinel mice, without showing any positive results during cultural bacteriology. Thus, molecular biological-based testing of barrier 3 was shown to be beneficial during routine health monitoring of laboratory mouse colonies, especially with regard to environmental sampling strategies. In barrier 4, no positive results were obtained. Overall, in the three affected units, culture revealed 3/9 and qPCR 7/9 positive animals using the mouse-based sampling approach while environment-based sampling revealed positive results in all nine subunits.
To verify the qPCR results of environmental sampling in barrier 3, oral swabs from the colony mice were analysed on single cage level to further evaluate the prevalence of R. pneumotropicus and R. heylii in subunit 3a (positive EAD result) and 3d (negative EAD result). These swabs revealed a pathogen prevalence of 43% (29/68) (3a) and 24% (18/75) (3d), possibly pointing out that bacterial transmission to sentinel animals was insufficient during the respective health monitoring period, whereas environmental sampling was suitable to reflect the real hygienic status of the monitored colony. It has to be noted that EAD sampling in this unit missed the positive finding in subunit 3d. Therefore, one negative EAD sampling alone may not be sufficient to estimate the hygienic status of the colony and may at best be supported by other sampling methods, if possible.
Comparison of sample materials
During the second validation process, R. pneumotropicus and R. heylii could be detected in a broad spectrum of mouse-associated and also environmental sample material including oral swabs, fresh faeces, cage faeces, pooled bedding and nesting material, cage swabs and EAD material of IVC systems. Figure 2 summarizes the bacterial quantities found in different sample types. Taken together, all environmental sample types revealed higher bacterial copy quantities than the animal-associated samples. Within the environmental sampling panel, cage faeces, pooled bedding and nesting material and EAD material were shown to be harbouring the highest bacterial concentrations and were so proven to be most suitable for reliable pathogen detection.
Comparison of the bacterial quantities found in different sampling materials, including oral swabs (OS) and fresh faeces (FF) obtained directly from the investigated animals, and also dry cage faeces (CF), pooled bedding and nesting material (PBN), cage swabs (CS) and exhaust air dust filter material (EAD) serving as an environmental sampling panel. Bacterial quantities are indicated by copies/µl. Data were tested against the null-hypothesis of normality using the Shapiro-Wilk test. In case of a rejected null-hypothesis, as shown here, non-parametric data are shown in a box-and-whiskers plot with 5–95% percentiles (n = 15). Significant differences between groups were determined using the Kruskal-Wallis test with Dunn’s test for multiple comparisons as post hoc test. Levels of significant differences are indicated as p ≤ 0.05, p ≤ 0.01 or p ≤ 0.001.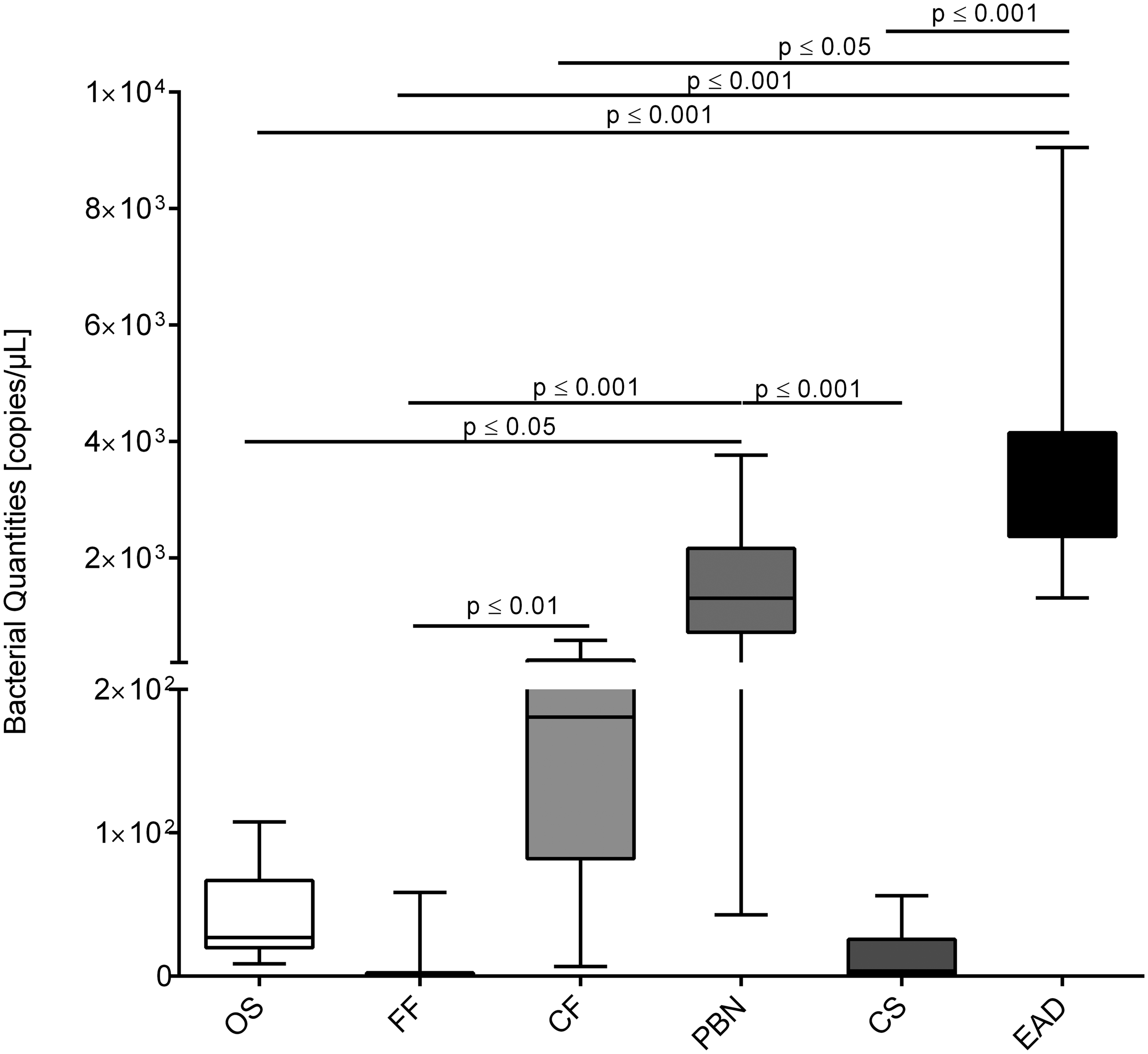
Discussion
Hygienic monitoring is an essential basis for standardized animal-based biomedical research. Yet detection of some infectious agents in sentinel animals remains challenging due to insufficient transmission or uneven shedding of the respective pathogens.22–24
A qPCR-based approach aiming for pathogen detection in environmental sampling material (e.g. EAD) was shown to be beneficial during routine health monitoring,2,3 which is why the development of useful primer/probe combinations for specific antigen detection became increasingly important.
Due to recent findings published by Adhikary et al. [Pasteurella] pneumotropica was reclassified into the new genus Rodentibacter sp., including R. pneumotropicus, R. heylii, R. ratti, R. trehalosifermentans, R. heidelbergensis, R. rarus, R. mrazii, R. myodis, and R. genomospecies 1 and 2. 13 This alteration of the former nomenclature implies adjustment of documentation within health reports as well as methods used for pathogen detection. 14
In this study we aimed at the development of a novel qPCR assay for the simultaneous detection of R. pneumotropicus and R. heylii based on a specific virulence factor, which was predicted by genome sequence analysis. 16 In terms of achieving the acquired pathogen specificity, the virulence factor ‘inclusion body protein A’ (IbpA) was chosen for subsequent assay design, as its gene ibpA was shown to be exclusively present in the genomes of R. pneumotropicus and R. heylii type strains, according to current results.5,16
Assay development based on virulence factor expression was chosen here as a novel approach for primer/probe design and hitherto appears to be a useful alternative to more traditional ways, which are mostly based on 16S rRNA sequence, internal transcribed spacer, or rpoB gene sequences analysis.5,25–27 However, virulence factors possibly vary between strains and might not be present in low-virulent strains. Nonetheless, in our hands the herein described assay revealed excellent species specificity and reliably detected not only the respective bacterial type strains but also worked on a broad spectrum of swab material, obtained from mice infected with Rodentibacter sp. within our facility. For a more complete validation of the assay’s sensitivity, further analysis of defined field strains of both bacterial species would be necessary, especially since ibpA gene sequences are only publicly available for their type strains. Future work should focus on filling this gap to achieve extensive assay validation. Furthermore, the assay design should be re-evaluated on a regular basis by taking novel sequencing data and latest findings into account.
During the initial validation process, the assay’s specificity and sensitivity were evaluated by serial dilution and subsequent analysis of bacteria-spiked swabs as well as of swabs obtained from mice with confirmed hygienic status. The assay revealed neither false positive nor false negative results, indicating a diagnostic sensitivity and specificity of 100% within those experiments.
Subsequently, the assay was tested on the currently described Rodentibacter type species and other Pasteurellaceae in laboratory mouse colonies.
In our hands positive results were obtained analysing bacterial DNA of R. pneumotropicus and R. heylii, whereas all other Rodentibacter sp. and other tested Pasteurellaceae remained negative. It has to be noted that a very slight fluorescent signal could be observed in the case of R. myodis (average CT 37.6), M. muris (average CT 37.0) and Pasteurella multocida (average CT 37.1), resulting in calculated copy quantities clearly below 1 copy/µl. In our understanding those results should be interpreted as negative on the basis of the respective standard curve, having CT 35.5 as a limit of detection. Those results indicate high specificity for double detection of R. pneumotropicus and R. heylii within this assay.
It has to be pointed out that the herein described assay is not suitable to detect all possible mouse-related Rodentibacter sp., as for example R. ratti is also able to infect laboratory mice as an alternative host. If the health monitoring of an animal facility aims to scan for all relevant murine species, a common primer probe combination detecting all members of the family Pasteurellaceae should be used instead.28,29
In terms of further validation, the assay was successively tested during routine health monitoring within three experimental research barrier units (1, 2, 3) and one strict breeding barrier unit (barrier 4) harbouring IVCs, and findings were compared with the results obtained by cultural diagnostics of sentinel mice. For this, animal associated samples (oral swabs, fresh faeces) as well as environmental sample material (cage faeces, pooled nesting and bedding material, cage swabs and EAD) were analysed by qPCR while oral swabs and faeces of sentinel mice were diagnosed by culture for the presence of R. pneumotropicus and R. heylii.
Historical results revealed a moderate prevalence of [Pasteurella] pneumotropica in barrier 1 and also a low prevalence in barrier 2, whereas barrier 3 and 4 were tested negative within the study period. As expected, presence of R. pneumotropicus and R. heylii could be confirmed by qPCR analysis of sentinel mice in barrier 1 and 2. Furthermore, the assay showed high bacterial numbers in the environmental sample material including EAD material of IVC racks. Surprisingly barrier 3 was tested positive for R. pneumotropicus and R. heylii by qPCR in environmental samples, although negative results were obtained by cultural diagnostics as well as by qPCR of oral swabs from sentinel mice. This is in line with others who could also show that qPCR of sentinel animals failed to show positive results for [Pasteurella] pneumotropica while environmental testing was superior in terms of pathogen detection. 10
To further asses the prevalence of R. pneumotropicus and R. heylii in barrier 3, oral swabs of the colony mice were tested by qPCR on the single cage level. As the sentinel mice were again negative, 44% (in the case of barrier 3a) and 26% (in the case of barrier 3d) of the colony mice were tested positive by qPCR. Positive findings were subsequently confirmed by PCR analysis in a commercially working laboratory as a reference method (data not shown). Those results indicate that the designed qPCR assay comprehensively revealed pathogen presence in barrier 3, whereas former hygienic monitoring, performed by cultural diagnostics of sentinel animals, historically failed to detect R. pneumotropicus and R. heylii. It has to be noted that bacterial culture performed on sheep blood agar under aerobic conditions with subsequent bacterial differentiation using the API system is not a very reliable method for Pasteurellaceae identification,13–15 as the system will probably fail to identify all members of this bacterial family correctly. Therefore, it is in the end not unexpected that positive results first occurred using a molecular biology-based approach.
Finally, bacterial copy quantities were evaluated in the different sample material used in this study to assess which sampling strategy may be the most suitable one for reliable pathogen detection. To our knowledge, this is the first study directly comparing the bacterial quantities of Rodentibacter sp. found in different sample materials. As we expected, higher copy quantities were found in the environmental sample material compared with animal-related sample types. Most interestingly, very low bacterial copy quantities could be found in the fresh faecal samples, whereas the dried faeces obtained from the animal’s home cage revealed very high pathogen concentrations. Notably, analysis of EAD material of the IVC systems revealed the highest copy quantities. This corroborates findings of others who found that qPCR analysis of EAD material was a suitable and highly sensitive approach for hygienic monitoring in laboratory mouse colonies.2,3,10 Yet it has to be mentioned, that a PCR-based test method cannot discriminate between dead and living bacteria, which is why positive results do not automatically imply the presence of infectious agents. During routine health monitoring it might be challenging, therefore, to decide whether DNA detection in the animal’s environment indicates infection of the respective colony or is it just the result of residual DNA, possibly still present even after autoclaving the cage system. Cage-level testing certainly adds additional value in this respect as cages are thoroughly cleaned on a regular basis. Future work should focus on these aspects and may also address further questions regarding decontamination of IVC systems, especially in terms of practicability within complex breeding barriers.
This study describes a novel qPCR assay based on the expression of a specific virulence factor for the simultaneous detection of R. pneumotropicus and R. heylii. This assay was demonstrated to be beneficial during routine health monitoring of laboratory mouse colonies, especially with regard to environmental sampling strategies. Thus, this study not only describes a novel approach for qPCR assay design, possibly improving Pasteurellaceae diagnostics, but also contributes to the 3R principle.
Footnotes
Acknowledgements
We thank Ulrike Freischmidt, Susann Roesel and Birthe Heinemann for their excellent technical assistance.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.