
Abstract
Health monitoring of laboratory rodents not only improves animal health but also enhances the validity of animal experiments. In particular, infections of laboratory animals with murine parvoviruses influence biomedical research data. Despite strict barrier housing, prevalence remains high in animal facilities, leading to increased risk of parvovirus introduction after the import of contaminated mice. Unfortunately, hygienic rederivation can be challenging, since gametes often contain residual virus material. Consequently, the process has to be closely monitored with highly sensitive diagnostic methods to verify parvovirus decontamination of the rederived progeny. However, diagnostic sensitivity of traditional methods is often low and requires testing of large animal cohorts. Therefore, we aimed to develop a powerful quantitative real-time polymerase chain reaction (qPCR) assay for the fast and reliable detection of murine parvoviruses in different sample materials. We validated the assay within an infection experiment and systematically analysed various animal-derived and environmental sample materials. We further developed a strategic risk assessment procedure for parvovirus monitoring after embryo transfer. Our novel qPCR assay reliably detected parvovirus DNA in a broad variety of sample materials, with environmental samples dominating in the acute phase of infection, whereas animal-derived samples were more suitable to detect low virus loads in the chronic phase. Here, the assay served as a highly sensitive screening method for parvovirus contamination in mouse colonies, requiring significantly lower sample sizes than traditional methods like conventional PCR and serology. Thus, the use of our novel qPCR assay substantially improves parvovirus diagnostics, enhancing research validity according to the 6Rs.
Introduction
Systematic health monitoring is a key element in the husbandry of laboratory rodents as it not only ensures animal health but also enhances the validity of biomedical research.1,2 Among all relevant pathogens, murine parvoviruses are certainly one of the most troubling agents, since maintaining virus-free mouse colonies is exceptionally challenging.3–6 Up to 2018, in mice two parvoviruses have been identified, namely mouse parvovirus (MPV) and minute virus of mice (MVM), which can be further subdivided into distinctly related strains (MVMp, MVMi, MVMm, MVMc, MPV1, MPV2, MPV3, MPV4, MPV5).4,7–15 Furthermore, a novel virus was recently discovered that causes inclusion body nephropathy in immunodeficient mouse strains and was therefore named mouse kidney parvovirus (MKPV). 16
As characteristic members of the Protoparvovirus genus, both MVM and MPV are extremely small (18–26 nm), non-enveloped, single-stranded DNA viruses consisting of approximately 5000 nucleotides. Typically requiring actively dividing or differentiating cells, murine parvoviruses predominantly infect endothelial cells, lymphocytes and haematopoietic precursors. 4 In contrast to other animal species, gastrointestinal lesions are not observed in infected mice and natural infection is in most cases subclinical with occasional congenital abnormalities in the offspring of infected dams. Despite minimal overt clinical manifestations, infection with murine parvoviruses affects cell proliferation, leading to myelo- and oncosupression, and thereby significantly impacting biomedical research, especially in the fields of immunology, oncology and haematology. 17 Therefore, comprehensive parvovirus diagnostics of mouse colonies is crucial to ensure research validity. Deeper knowledge about the hygienic status of laboratory animals directly contributes to the concept of the 6Rs, which besides animal welfare also take into account the scientific value of studies.2,18
While viral contaminants such as Sendai virus or lymphocytic choriomeningitis virus are nowadays rare in mouse colonies due to broad rederivation strategies and strict barrier housing, parvoviruses still remain prevalent in animal facilities all over the world,3,19–25 leading to an increased risk of introducing parvovirus into a facility after unknowingly importing contaminated mouse stocks. 26 The underlying reasons for this relatively stable epidemiological situation include: i) environmental tenacity of infectious parvovirus, ii) relative difficulty to detect ongoing infections due to mouse strain- and virus-dose dependent seroconversion rates, iii) intermittent viral shedding and iv) the ability of parvoviruses to cause persistent infections despite the onset of immunity.4,14,27–35 Consequently, the simultaneous use of indirect (antibody detection via serology, e.g. indirect immunofluorescence assay (IFA)) and direct (antigen detection via molecular methods, e.g. polymerase chain reaction (PCR)) methods are required for successful parvovirus diagnostics.34,36,37 Although generally detectable via traditional sentinel systems (such as direct contact or soiled bedding sentinel mice) 38 diagnostic success rates are often low within a given colony. Therefore, the analysis of very large sample sizes is required to reliably confirm the parvovirus status of a hygienic unit,1,4,39,40 which in the case of invasive blood sampling procedures can be stressful for the tested animals. Environmental sampling strategies, as already established for other pathogens,41–44 could be beneficial in this context but have not yet been convincingly employed for parvovirus diagnostics.45,46
The most successful approach for decontamination of parvovirus-positive colonies is the hygienic rederivation via embryo transfer,47,48 which involves the collection of embryos from infected mouse stocks and subsequent transfer into virus-free surrogate dams. 49 Due to their extremely small size, parvoviruses tend to firmly adhere to the zona pellucida of the embryos. 50 Consequently, prior to transfer, virus particles have to be carefully removed from the embryos through several washing steps in sterile media (at least 10 times is recommended) or routine removal of the zona pellucida.48,51 Despite all precautionary measures, there is always a remaining risk that embryo transfer might fail, especially when viral contamination occurred from the female donor. In this context Mahabir et al. reported that transfer of embryos, containing residual virus material, into naïve recipients led to seropositive progeny. 51 Consequently, virus eradication has to be closely monitored throughout the procedure with highly sensitive diagnostic methods.
To address this issue the aim of this study was to establish a powerful quantitative real-time PCR (qPCR) assay for the fast and reliable detection of murine parvoviruses in different sample materials for strategic risk assessment procedures during routine health monitoring programmes.
Altogether, our novel qPCR assay proved to be a powerful diagnostic tool for fast and highly sensitive screening of parvovirus contamination in mouse colonies. A risk-based sampling strategy allowed for the reliable detection of parvovirus DNA in a broad variety of animal-derived as well as environmental materials, requiring significantly lower sample sizes than traditional methods such as conventional PCR and serology. Thus this novel qPCR system substantially improves parvovirus diagnostics, contributing not only to animal welfare by enabling reliable environmental sampling strategies, but also to enhancing the validity of biomedical research according to the 6Rs.
Materials and methods
Study outline and ethical statement
The first part of the study (Part A) describes the validation of a novel qPCR assay for the simultaneous detection of murine Parvoviridae, with the exception of MKPV.
The second part of the study (Part B) was used to establish a risk assessment procedure for parvovirus diagnostics in routine health monitoring. We used our novel qPCR assay in combination with serological testing during the hygienic rederivation process of a historically parvovirus-positive mouse colony. Different sample materials were analysed to find the optimal risk-based sampling strategy for future monitoring procedures.
All experiments were performed in strict accordance with the German Animal Welfare Legislation, approved by the local Institutional Animal Care and Research Advisory Committee and covered by the permission of the local governmental authorities (reference numbers 42500/1H, 13A404, 18A300, 18A311 and 18A315).
Animal husbandry and experiments
MVMi infection of mice and collection of animal-derived and environmental samples
In Part A of our study an infection experiment with MVMi was performed to generate positive material for qPCR assay validation. In total, four female C57BL/6J (B6/J) mice (Central Animal Facility of the Hannover Medical School, Hannover, Germany) and four Crl:CD1 (ICR) females (Charles River Laboratories, Wilmington, USA) were used. Mice were housed in pairs of the same strain background, but differing in age – an 8 week old was paired with a 22 week old. After two weeks of acclimatization, the 10 week old mouse of each cage was infected oronasally with a suspension of MVMi-positive cell culture cells, while the other, 24 week old, animal was used as a contact sentinel. All mice were housed in individually ventilated polysulfone biocontainment units (BCUs) (Allentown, NJ, USA) with negative pressure and ad libitum food (Altromin TPF-1324, Lage, Germany) and water supply (autoclaved, 30 min, 105°C). Standardized environmental conditions included the following parameters: temperature 21 ± 2°C, relative humidity 55 ± 5%, artificial light (14:10 h light:dark), autoclaved softwood granulate (poplar wood, AB 368P, AsBe-wood GmbH, Buxtehude, Germany), nesting material (Enviro-Dri®, LBS Biotechnologies, Limburgerhof, Germany) and cotton rolls (autoclaved, ANT Tierhaltungsbedarf, Buxtehude, Germany). All human–animal interactions such as handling, sampling or cage changing took place under a laminar flow utilizing sterile tools. Access to the barrier was restricted to a limited number of staff. The animal room was entered through an air shower after a complete change of clothing (shoes, bonnets, surgical face masks and gloves). Animal-derived and environmental samples were collected daily during the first week post infection and then twice a week until the end of the experiment. Four weeks post infection, blood and necropsy samples were taken during a final dissection. Mice were sacrificed by CO2 overdose (25% fill rate) and subsequent desanguination via cardiac puncture.
Hygienic rederivation of a parvovirus-positive mouse colony
Cryopreserved two-cell embryos derived from parvovirus-positive B6;129S2-Stra6tm1Nbg/Lzt and B6;129S2-Stra6™ 2 .1Nbg /Lzt donor mice 52 were imported from a collaborating research facility (Part B). As a means of hygienic rederivation, 8–12-week-old CD2F1 surrogate dams (BALB/cJHanZtm × DBA/2JHanZtm) obtained from the Central Animal Facility were used for embryo transfer, as described in detail by Dorsch et al. 49 Morphologically intact embryos were washed 11 times with a sterile phosphate buffer before being transferred into the oviducts of anaesthetized surrogate dams. For further information on the procedure see the Material and methods section of the Supplemental material online.
As the first precautionary measure in this study’s risk assessment procedure, the wash media as well as non-transferred discarded embryos were collected and examined for parvoviral DNA via conventional PCR and qPCR.
After surgery, surrogate dams were kept in autoclaved static microisolators (Han-gnotocages). Environmental sample material was collected out of the gnotocages and analysed for the presence of parvovirus DNA as a means of further risk assessment. For detailed housing descriptions see the Supplemental Material and Methods.
Shortly before giving birth, the surrogate dams were separated, relocated to BCU cages and housed similarly to the infection experiment animals but received a breeding diet (Altromin TPF-1314, Lage, Germany) ad libitum. Again, environmental samples were taken directly out of the surrogate dams’ home cages. After weaning of their litter, dams were sacrificed for necropsy sample collection. The offspring of the surrogate dams (Fx + 1, Fx + 2 and Fx + 3 generations) were kept under the exact same conditions as the infection experiment animals. For further risk assessment, blood and environmental samples were routinely collected and dissections were performed on a regular basis.
In vivo infectivity test
To discriminate between infectious virus and residual DNA found in the surrogate dams’ offspring, an in vivo infectivity test was performed. For this purpose spleens, mesenteric lymph nodes and fresh faeces were obtained from the Fx + 2 generation of mice. Samples that tested positive for murine parvoviruses via qPCR were homogenized in sterile PBS (PBS Dulbecco, Merck Biochrom GmbH, Berlin, Germany) and administered intraperitoneally and/or orally to 5–6-week-old recipient mice after a two week acclimatization period. The recipient cohort consisted of 12 animals, six of which were female FVB/NHanZtm and the other six male B6/J mice. Two recipients of each strain received suspensions of donor spleens (100 µl orally + 200 µl intraperitoneally), mesenteric lymph nodes (100 µl orally + 100 µl intraperitoneally) or fresh faecal pellets (200 µl orally). During the course of the in vivo infectivity test environmental sample material was also frequently collected from the animals’ home cages and blood samples were taken on a weekly basis. Animals were sacrificed after two or 12 weeks for necropsy sample collection.
Sampling procedures are depicted in Figure 1.
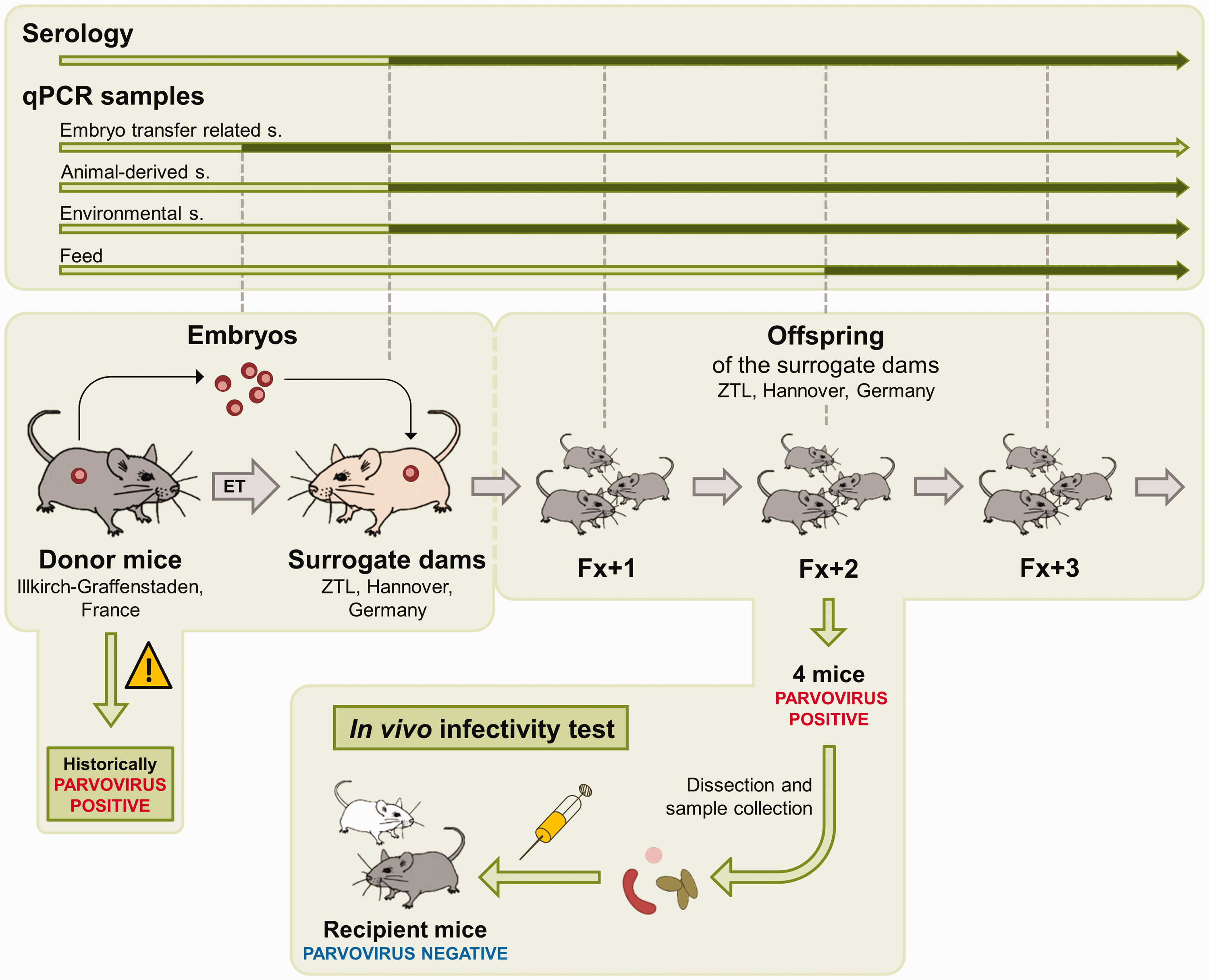
Study Part B: schematic overview of the risk assessment procedures for the hygienic rederivation of a parvovirus-positive mouse colony.
Routine microbiological monitoring was performed according to FELASA recommendations 1 and did not reveal any evidence of infection with specified opportunistic bacteria or common murine pathogens. Prior to the start of each experiment, all experimental mice were examined serologically and were additionally analysed for the presence of parvoviral DNA via qPCR of faecal samples.
Sampling procedures
If designated for DNA extraction, samples were collected in autoclaved 1.5 ml reaction tubes (Sarstedt, Nümbrecht, Germany) and stored at –20°C until further processing. Samples intended for RNA isolation were transferred to sterile 2 ml cryo tubes (Cryo.s, Greiner Bio-One, Frickenhausen, Germany) that were immediately plunged into liquid nitrogen immediately and stored at –80°C until use.
Embryo transfer related samples
In the course of the embryo transfers both the discarded embryos and the phosphate buffered wash media of the first and last washing step were collected.
Animal-derived samples
During dissections, approximately 20 mg of spleen tissue, 0.5 cm of small intestine, colon or caecum and one to two mesenteric lymph nodes were taken. From one to two (live animals) or three to five (dissection animals) fresh faecal pellets were obtained during stimulated defecation (live animals) or directly from the distal intestinal tract (dissected animals). Blood for serological analysis was either collected via facial vein phlebotomy (live animals) or by terminal cardiac puncture (dissected animals). After blood samples were allowed to clot at room temperature for 1 h, serum was collected by centrifugation at 7500 rev/min for 5 min.
Environmental samples
Dirty bedding material, 10–15 dried faecal pellets and flocked cage swabs (FLOQSwabs, Copan, Brescia, Italy) were obtained from the BCU homecages of animals to be investigated. In addition, cage-level exhaust air dust (EAD) pre-filter material was also collected.
RNA and DNA isolation
Viral nucleic acids (both RNA and DNA) were extracted from all sample types using the Invisorb Spin Virus RNA Mini Kit (INVITEC Molecular, Berlin, Germany) according to the manufacturer’s instructions. After the isolation process, DNA samples were stored at –20°C while all RNA samples were stored at –80°C until use.
Detection of parvovirus
Conventional PCR
Conventional PCR was performed using a generic PCR assay that detects both autonomous murine members of the family Parvoviridae.
RT-PCR
To identify replicating virus, mRNA was isolated, transcribed into cDNA and detected via reverse transcription PCR (RT-PCR) by utilizing intron spanning primers to distinguish cDNA (221 bp product) from genomic DNA (1292 bp product). 31 Further details are summarized in the Supplemental Material and methods.
qPCR
For the simultaneous detection of MVM and MPV, a specific primer and probe combination was designed to amplify an 80 bp fragment of the parvovirus specific non-structural protein 1 (NS1) gene (Table 1).
Primers and probe used in this study.

aDual labelled fluorescent probe: 5’-reporter (fluorophore):6-FAM (6-carboxyfluorescein) 3’-quencher: BHQ-1 (black hole quencher 1).
PCR: polymerase chain reaction; qPCR: quantitative real-time PCR; RT-PCR: reverse transcription PCR
qPCR assays were performed using the StepOnePlus™ and the QuantStudio™ 6 Flex Real-Time PCR Systems (Applied Biosystems, Darmstadt, Germany).
As a positive control and for quantification of viral particles in positive samples, specific plasmids were produced and amplified in a 10-fold dilution series. From a plasmid stock produced through TOPO TA cloning a 10-fold serial dilution ranging from 109 to 100 copies/µl was generated for standard curve analysis.
All samples and controls were tested in triplicate.
Optimal results were achieved using an annealing temperature of 60°C and oligonucleotide concentrations of 900 nM (primers) and 200 nM (probe).
Serological parvovirus detection
Indirect IFA
For detection of parvovirus specific antibodies, sera were tested in an indirect IFA using MVMi-infected newborn kidney cells. More details can be found in the Supplemental Material and methods.
Positive samples from in-house IFA and qPCR runs were sent to another laboratory for third-party testing (WK and Charles River Laboratories, Wilmington, USA) as a means of verification.
Enzyme linked immunosorbent assay (ELISA)
Some serum samples were re-tested by an external laboratory (GVG Diagnostics GmbH, Leipzig, Germany) with an ELISA as a reference method.
Statistical analysis
Animals were allocated to experimental groups by use of an online random number generator (https://www.graphpad.com/quickcalcs/randomize1/). Researchers were at all stages aware of group allocations. No animals, experimental units or data points were excluded from statistical analysis.
Standard curve parameters, as well as copy number data, were analysed using Prism software version 8 (GraphPad Software, San Diego, CA, USA).
For juxtaposition of viral quantities found in different sample materials, copy numbers were tested against the null-hypothesis of a Gaussian distribution via Shapiro–Wilk test. Kruskal–Wallis test with Dunn’s test for multiple comparisons as a post hoc test was used to determine significant differences between groups.
Results
(Part A) Validation of qPCR assay
Standard curve
Specific plasmids were amplified in a 10-fold dilution series (109–100 copies/ µl) to generate a standard curve (Figure 2). By plotting CT values obtained from 30 independent qPCR runs against their respective copy quantities, a semi-logarithmic regression graph (log [X] – lin [Y]) with the following characteristics was created: y = –3.54x + 39.32.
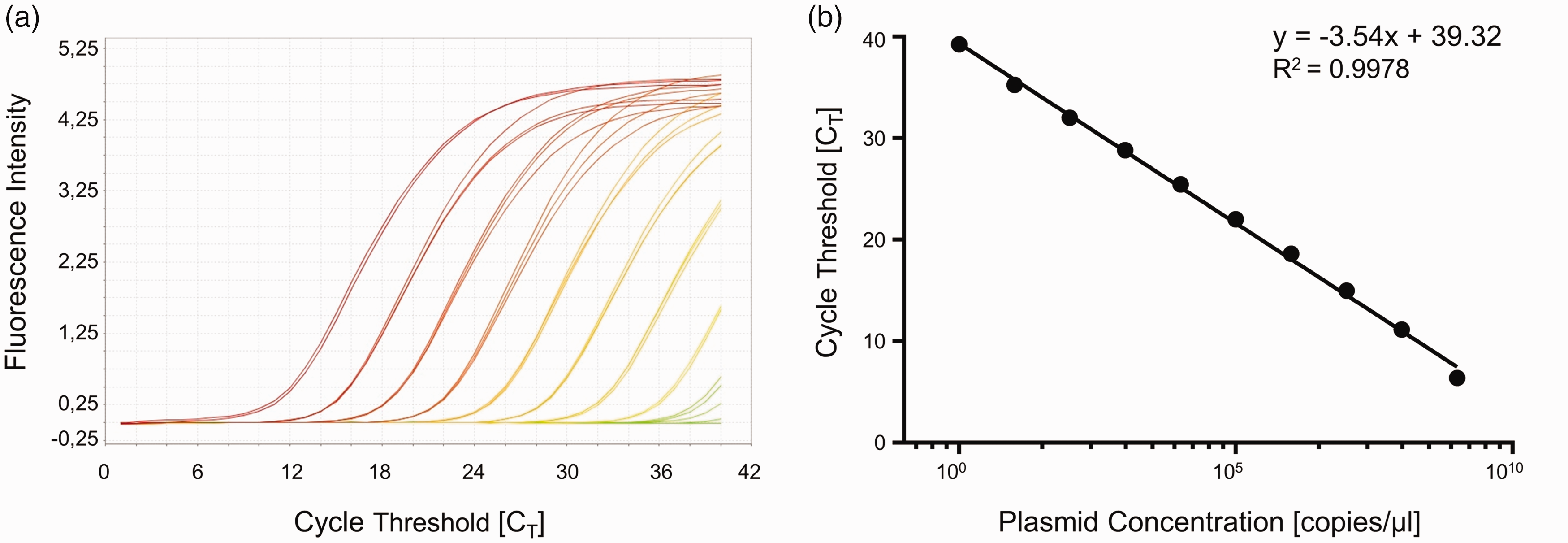
Validation of the qPCR assay.
At eight degrees of freedom the goodness of fit shows an R2 of 0.9978, while the 95% confidence intervals range between 38.60 and 40.03 for the Y-intercept and between –3.673 and –3.405 for the slope. The limit of detection was determined to be 10 copies/µl (CT 35.26).
Diagnostic sensitivity and specificity
For evaluation of the assay’s diagnostic sensitivity and specificity, either faecal pellets spiked with virus suspensions or sampled from mice with confirmed parvovirus-free hygienic status (negative controls) or animals used in the MVMi infection experiment (positive controls) were analysed via qPCR as described previously. 42 Positive and negative results were compared with expected numbers of positive and negative samples (Table 2), whereupon false negative and false positive results were used for calculation of diagnostic sensitivity (→ true positive/[true positive + false negative]) and specificity (→ true negative/[true negative + false positive]). In this study both the diagnostic specificity and sensitivity were found to be 100%.
Confusion matrix: qPCR assay validation via evaluation of defined faecal pellets. Faecal pellets were spiked with either Mouse Hepatitis Virus- (MHV) or MVMi-positive virus suspensions or collected from mice with confirmed hygienic status, including animals from the infection experiment. False positive and false negative results were used for calculation of diagnostic specificity and sensitivity.
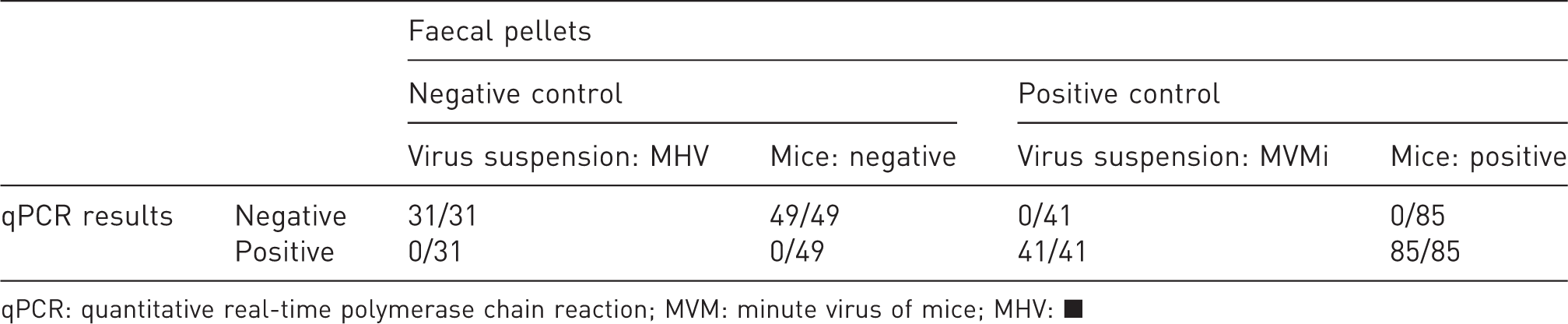
qPCR: quantitative real-time polymerase chain reaction; MVM: minute virus of mice; MHV: ▪
To further evaluate the novel qPCR assay for potential cross-reactivity, various sample materials containing defined common murine pathogens (including viruses, bacteria, fungi and parasites) were examined. Viral quantities less than 1 copy/µl were considered negative, samples with copy numbers between 1 and 10 were labelled uncertain until at least one re-testing confirmed the positive result, and values above 10 copies/µl were generally classified as positive. As expected, positive results were obtained for the targeted murine viruses MVM and MPV. Additionally, a rat-specific virus (Toolan’s H-1 Virus (H-1)) tested positive, indicating cross-reactivity with closely related rat parvoviruses. No further off-target amplification could be detected, proving high pathogen specificity. Detailed results are summarized in Table 4 of the Supplemental Results.
Field validation of qPCR-based parvovirus diagnostics in an infection experiment: comparison of sample materials
An infection experiment was performed to further validate the novel qPCR assay under field conditions. Accordingly, two mouse strains (B6/J and ICR) were oronasally infected with MVMi and subsequently housed in pairs comprising one inoculated animal and one contact sentinel mouse. To find the optimal sample type for successful parvovirus diagnostics in future health monitoring strategies, a large panel of samples was systematically collected including environmental as well as animal-derived sample materials.
qPCR analysis revealed parvoviral DNA in all examined sample types of both inoculated as well as contact sentinel mice. Of all sample types, the two kinds of faecal samples (cage faeces + fresh faeces) contained the highest viral concentrations and thus proved most suitable for reliable pathogen detection. Bedding material and EAD filter material were also highly positive for parvoviral DNA (Figure 3). All in all, environmental samples showed superior performances in the acute infection period (∼ first week post infection), whereas fresh faeces dominated in the chronic phases. Viral quantities ranged from 0 (cage swab) to 4.1 million (cage faeces) copies/µl in the acute phase and decreased to very low (i.e. 15 copies/µl in caecum) concentrations in the chronic phase of infection, underlining the fact that animals became latently infected with MVMi during the experiment.
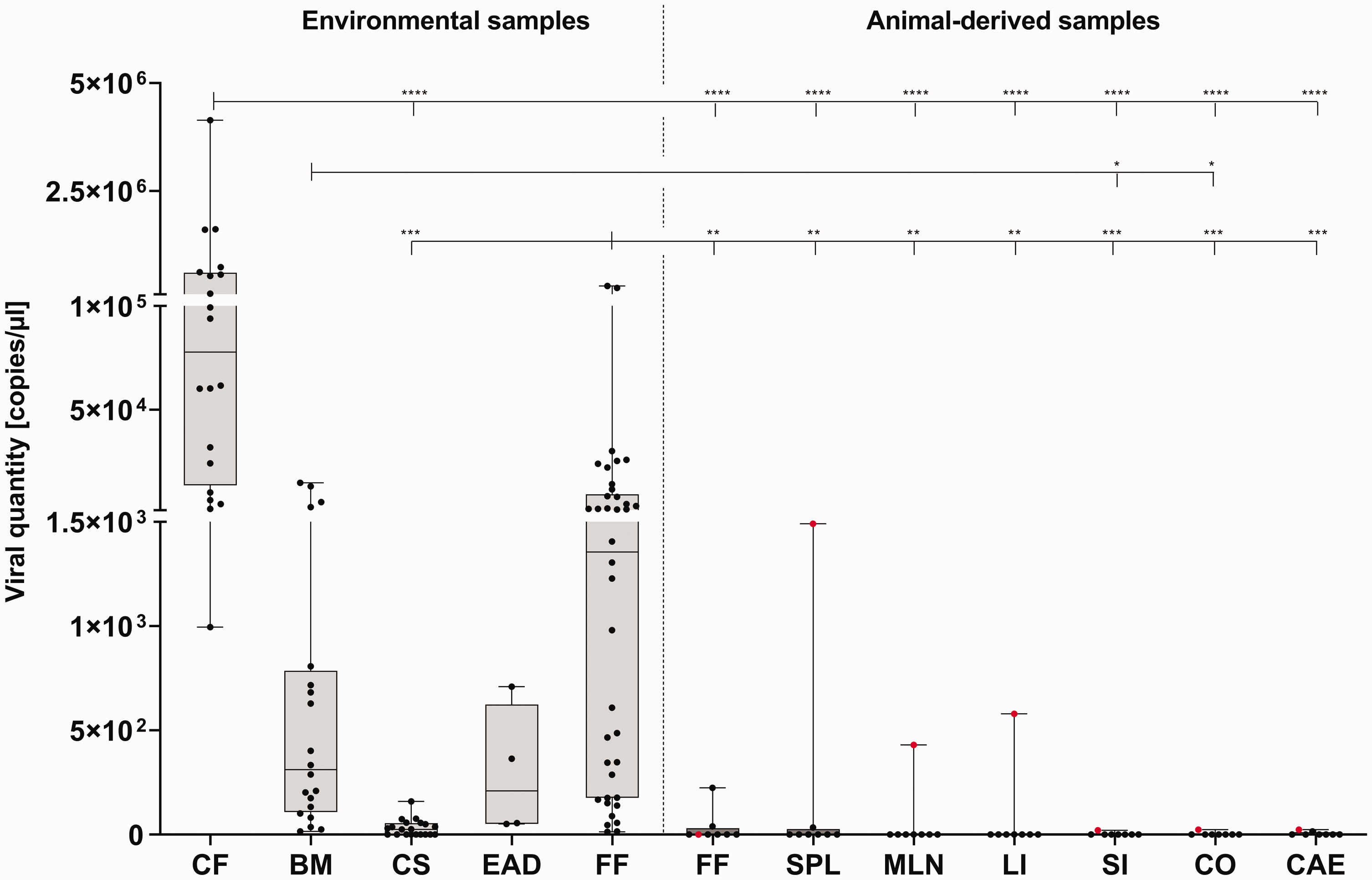
Quantitative real-time polymerase chain reaction (qPCR) analysis of samples obtained in the parvovirus infection experiment performed in C57BL/6J and Crl:CD1 mice.
In general, all eight mice tested positive for parvoviral DNA via qPCR, whereas only one of them seroconverted as indicated by a positive reaction in the IFA.
These results demonstrate that direct virus detection via qPCR is superior to serological diagnostics in terms of diagnostic sensitivity and that our qPCR assay is suitable to detect even very small traces of viral DNA.
Altogether, results from Part A demonstrated that the herein described novel qPCR assay not only shows very high diagnostic sensitivity and specificity but is also high performing during health monitoring procedures as it reliably detects viral DNA in very low quantities, especially in environmental sample material.
(Part B) Risk assessment and rederivation
Embryo transfer
For hygienic rederivation of a historically parvovirus-positive mouse colony, imported embryos were transferred into surrogate dams. As a first precautionary measure, discarded embryos and wash media of the first and last washing steps were examined for the presence of murine parvoviruses by conventional PCR as well as this study’s novel qPCR assay. Both sample types tested positive by conventional PCR and qPCR, with the embryos containing higher concentrations of parvoviral DNA (130 copies/µl) than the wash media (30 copies/µl), proving that washing steps did not reliably remove parvoviral DNA from the transferred embryos (Figure 4(b) and (c)).
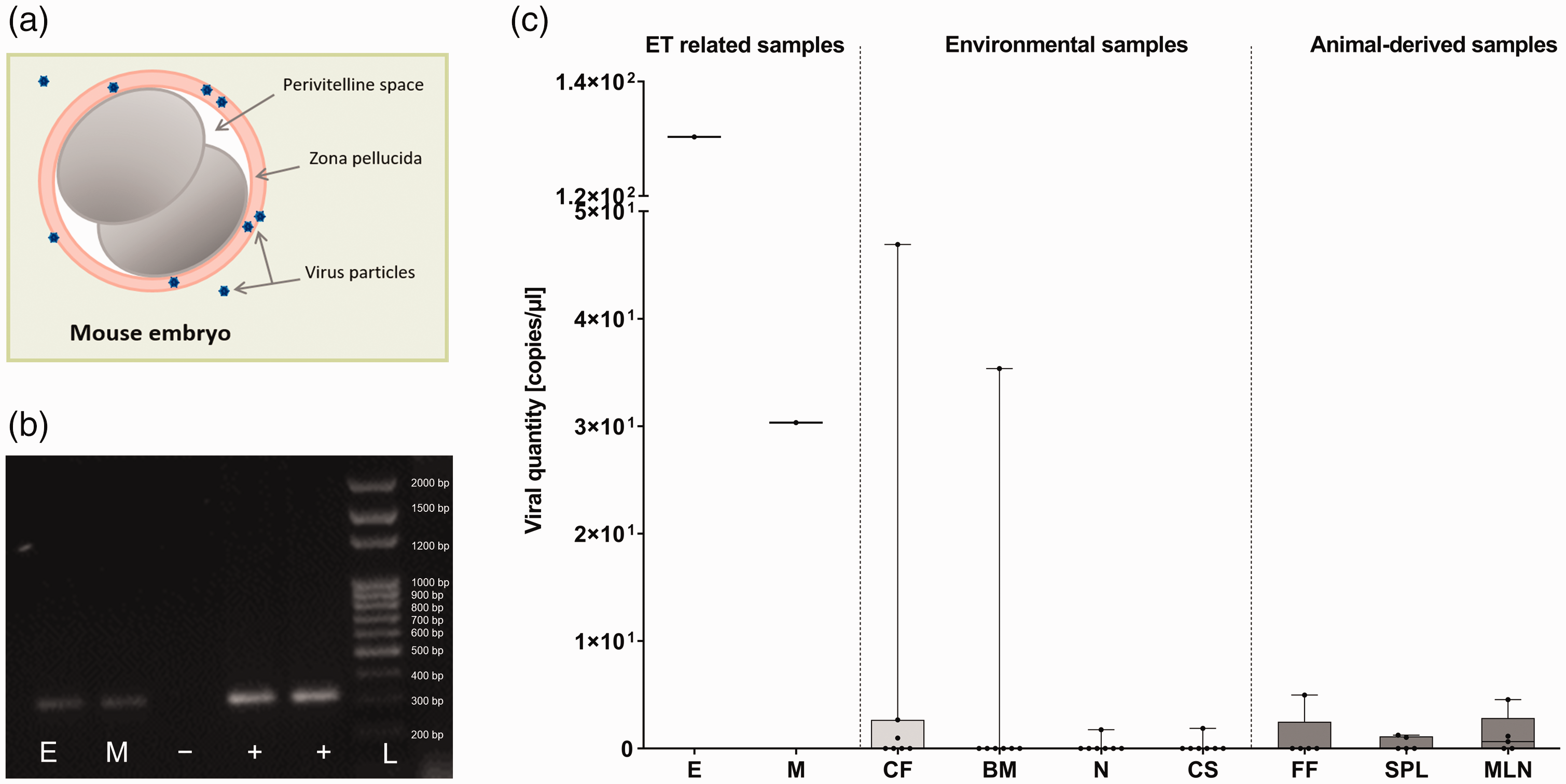
Risk-oriented molecular analysis of embryo transfer (ET) as well as surrogate dam-related samples for parvovirus contaminations. (a) Parvovirus particles adhere to the zona pellucida of a mouse embryo. Size ratios do not match reality. (b) Results of conventional polymerase chain reaction (PCR) analysis after gel electrophoresis. Discarded embryos (E) and wash media (M) were analysed using a generic parvovirus conventional PCR assay. Negative and positive controls are marked with – and + respectively. Size of the PCR product was measured by a low range DNA ladder (L). (c) Quantitative real-time PCR analysis of samples directly and indirectly connected to the ET. Depicted are copies of parvoviral DNA per microlitre found in discarded embryos (E) and wash media (M) as well as environmental (cage faeces (CF), bedding (BM) and nesting material (N), cage swabs (CS)) and necropsy sample material (fresh faeces (FF), spleen (SPL), mesenteric lymph nodes (MLN)) obtained from CD2F1 surrogate dams and their homecages. Copy numbers were tested against the null-hypothesis of a Gaussian distribution with the Shapiro–Wilk test. Since the null-hypothesis was rejected, non-parametric data are displayed in box-and-whiskers plots from minimum to maximum with mean.
Surrogate dams
As positive qPCR results of the embryo transfer-related sample material indicated a high risk of parvovirus introduction into the surrogate dams through embryo transfer, the potentially infected animals were housed in gnotocages instead of directly being transferred to the breeding barrier. To further estimate the risk of a parvovirus infection, environmental samples were collected out of the gnotocages and tested for parvoviral DNA. While conventional PCR was consistently negative (data not shown), four out of 10 environmental samples proved positive in the herein described qPCR.
These results strongly indicated that surrogate dams became infected through embryo transfer involving contaminated embryos, so they were relocated to a quarantine unit and housed in BCU cages for giving birth.
After weaning of their litter, all surrogate dams were sacrificed for routine health monitoring. During the dissection further environmental samples out of the animals’ homecages as well as animal-derived samples were collected and subsequently analysed by qPCR. Additionally, serum samples of all surrogate dams were tested for parvovirus specific antibodies via IFA.
All in all, positive qPCR results were obtained from 35 out of 50 tested samples, with the highest copy numbers found in cage faeces.
Generally, consistent with data from the infection experiment (Part A), environmental samples proved to be higher yielding compared with animal-derived samples (Figure 4(c)). Remarkably, specific antibodies could not be detected in any of the animals via IFA, suggesting that surrogate dams became parvovirus carriers without seroconverting.
Screening of following generations
Although the surrogate dams tested negative for parvovirus by serology, qPCR-based testing revealed parvovirus DNA in different sample material, even if the viral copy quantities were found to be relatively low. As these data were consistent with those obtained in the infection experiment of Part A, further risk assessment steps were implemented to prevent the risk of potential pathogen spread within the breeding units and to increase monitoring frequency of the potentially infected colony. Therefore, as another precautionary measure, the surrogate dams’ offspring were further kept under strict quarantine and underwent a three-fold screening process consisting of environmental sampling, blood collection and dissections to follow up any potentially remaining parvovirus infection in the rederived progeny. Regular sample collection of first, second and third filial generation mice revealed parvovirus DNA in numerous sample materials. Twelve of 159 animal-derived and 30 of 239 environmental samples tested positive for parvovirus by qPCR. It should be pointed out that in only two of the 110 sera tested were specific antibodies detected, proving that those animals became infected. Consistent with results from the infection experiment and the surrogate dams’ testing, most of the examined animals became carriers without showing seroconversion, as indicated by long term shedding of low viral loads. Detailed results of the screening procedure are summarized in Table 3.
Summary of test results of surrogate dams and offspring generations Fx+1 to Fx+3 and in vivo infectivity test mice.
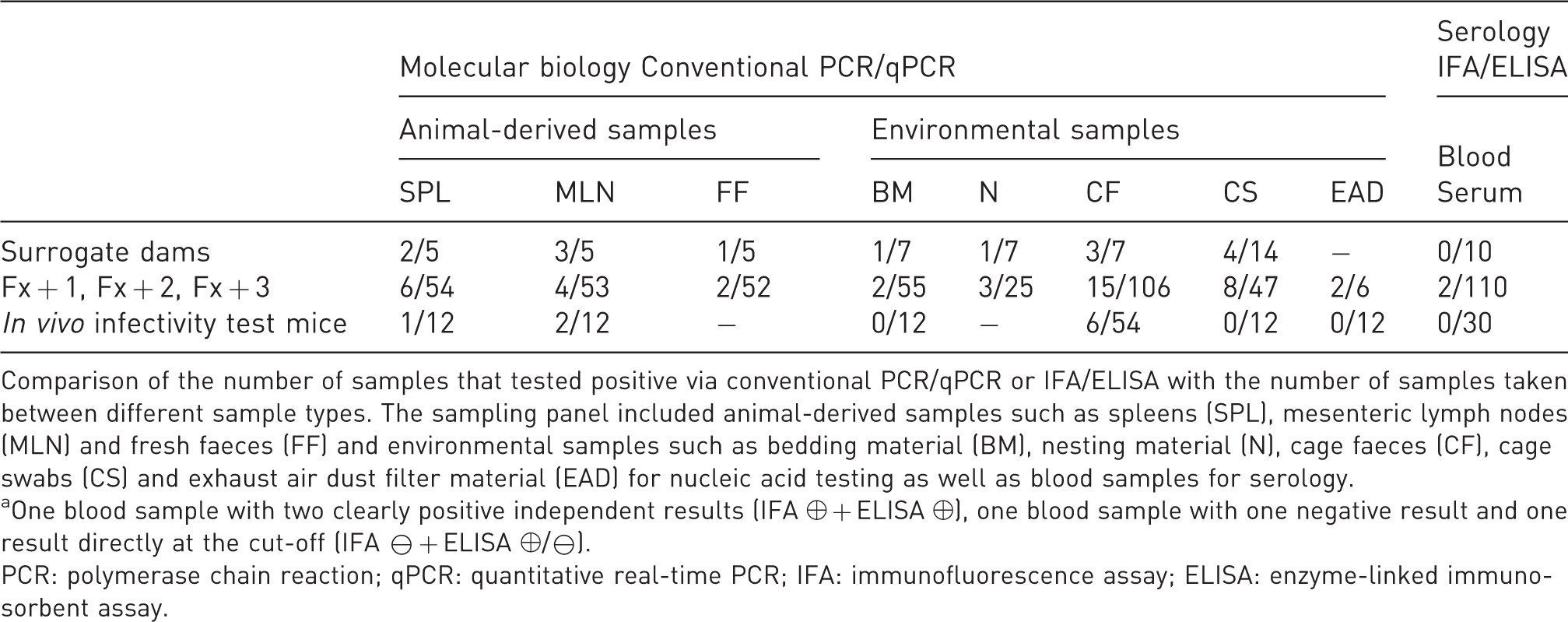
Comparison of the number of samples that tested positive via conventional PCR/qPCR or IFA/ELISA with the number of samples taken between different sample types. The sampling panel included animal-derived samples such as spleens (SPL), mesenteric lymph nodes (MLN) and fresh faeces (FF) and environmental samples such as bedding material (BM), nesting material (N), cage faeces (CF), cage swabs (CS) and exhaust air dust filter material (EAD) for nucleic acid testing as well as blood samples for serology.
aOne blood sample with two clearly positive independent results (IFA ⊕ + ELISA ⊕), one blood sample with one negative result and one result directly at the cut-off (IFA ⊖ + ELISA ⊕/⊖).
PCR: polymerase chain reaction; qPCR: quantitative real-time PCR; IFA: immunofluorescence assay; ELISA: enzyme-linked immunosorbent assay.
Although several samples turned out to be parvovirus positive by qPCR and two animals even seroconverted, there was no evidence that the detected virus materials were still infectious. Therefore, as a final step, the infectious relevance of detected parvoviral DNA was investigated by use of an in vivo infectivity test.
In vivo infectivity test
To distinguish between residual DNA and actually infectious parvovirus particles, parvovirus-positive samples derived from the Fx + 2 generation of mice were homogenized in sterile PBS and administered orally as well as intraperitoneally to parvovirus-negative recipient mice. Four qPCR-positive mice were selected due to their close relationship with the Fx + 1 generation animals with the highest parvoviral DNA copy numbers as well as the most positive results in earlier qPCR testing.
Seroconversion was found in none of the inoculated mice, whereas environmental (cage faeces) as well as necropsy sample materials (spleens, mesenteric lymph nodes) of seven of the 12 animals proved positive by qPCR. No parvoviral DNA was detected in bedding material, cage swabs and EAD filter material (Table 3).
In addition to positive qPCR results, enlarged mesenteric lymph nodes were observed during necropsy of parvovirus-positive mice, possibly indicating an immunological response.
As another way of determining virus transcription and thus infectivity of detected virus material, mRNA was isolated from qPCR parvovirus-positive spleens and mesenteric lymph nodes of the Fx + 2 generation of mice and in vivo infectivity test animals. No parvovirus-specific mRNA could be found in any of the tested samples (data not shown), suggesting a viral load too low for mRNA detection via RT-PCR.
Altogether, our results show compelling evidence that parvovirus monitoring during routine health monitoring should never exclusively rely on serological analyses, as long as very high sample numbers are not included. The large-scale use of molecular testing using a highly sensitive qPCR assay was proven to be a superior approach to successfully screen for potential virus carriers, especially by using an environmental sampling strategy. In our set-up, faeces was deemed the most suitable sample type, whereas EAD filter material contained only very low viral quantities.
In conclusion, the combination of molecular testing and targeted, risk-orientated serology of low shedding animals was suitable to detect latent parvovirus infections in contaminated mouse colonies.
Discussion
Murine parvoviruses are common contaminants of laboratory mouse colonies and, despite strict barrier housing, are still prevalent in animal facilities all around the globe.3,19–25 Hence, there is a permanent risk of accidentally introducing parvoviruses to the husbandry units by the import of infected animals. 26 Furthermore, some authors reported parvovirus outbreaks due to contaminated chow. 53 If undiagnosed, infectious agents may uncontrollably spread within a hygienic unit, leading to virus persistence in contaminated colonies.
Latent parvovirus infections of laboratory mice have a huge impact on biomedical research data, which is why health monitoring strategies should involve comprehensive parvovirus diagnostics to reliably confirm the hygienic status of the colony.2,4–6,17 Since infections with murine parvoviruses lead to inconsistent seroconversion rates, depending on the infected mouse strain and virus dose, the use of indirect virus detection methods requires very large sample sizes to successfully find specific antibodies in analysed blood sample material.4,5 Furthermore, shedding in persistently infected animals often involves very low viral concentrations, which is why sensitivity of direct detection methods (usually PCR) has to be exceptionally high. For the same reasons environmental sampling strategies, as already established for other pathogens,41–44 have not yet been convincingly employed for the routine parvovirus monitoring.45,46 These diagnostic difficulties demand complex health monitoring procedures to reliably detect ongoing parvovirus infection within a mouse colony.36,40
Therefore, the aim of this study was to develop a powerful qPCR assay for the fast and highly sensitive detection of murine parvoviruses in different sample materials and to establish a strategic risk-assessment procedure for routine health monitoring programmes. We designed a specific primer and probe combination based on the highly conserved parvovirus NS1 gene, aiming at detecting all mouse relevant parvoviruses, with the exception of MKPV.
Initial assay validation was performed by standard curve analysis where the regression analysis showed an excellent goodness of fit (R2 = 0.9978) and a detection limit of less than 10 copies/µl, proving the assay suitable for nucleic acid detection in sample material with very low virus concentrations. Our assay showed an outstanding diagnostic sensitivity (100% of tested samples) and was further proven to be highly specific for the detection of murine parvoviruses (except MKPV) without showing any cross-reactivity to other common pathogens, with the exception of a highly related rat parvovirus (H-1). We consider this inter-species cross-reactivity acceptable, as rat parvoviruses should not be present in samples obtained from mouse colonies.
Systematic analysis of different sample material of mice oronasally infected with MVMi revealed positive qPCR results in all tested sample types of both inoculated as well as contact sentinel mice. Of all examined sample types, the two kinds of faecal samples (cage and fresh faeces) contained the highest viral concentrations and thus proved to be most suitable for reliable pathogen detection, while bedding material and EAD filter material also had high viral counts. All in all, environmental samples had the highest viral counts in the acute infection period, whereas animal-derived samples had the highest viral counts in the chronic phases of infection, without mouse strain differences.
In general, all eight mice tested positive for parvoviral DNA by qPCR, whereas only one mouse seroconverted (ICR), as indicated by a positive reaction in the IFA. These results are in line with those of others, who could also show that seroconversion rates in infected animals are often poor28,29 and that viral shedding significantly decreases several weeks after infection.31,37 Notably, Besselsen et al. observed that parvovirus can persist in infected animals despite the onset of immunity. 14 The same observation was confirmed by Janus et al., who showed that viral shedding can be reactivated by immunosuppression in persistently infected mice. 31 Therefore, undiagnosed low shedding animals are an especially significant problem for husbandry units and thus the ability to detect very small traces of parvoviral DNA by routine screening programmes is of key importance. In our set-up, environmental sampling strategies proved to be a promising approach for a routine parvovirus screening panel.
In the case of enzootic parvovirus infections the successful hygienic rederivation of positive embryo material into surrogate dams (embryo transfer) was described,47,48 but there seems to be a remaining risk of unintentional contamination of the surrogate dams and infection of their progeny due to virus particles firmly adhering to the zona pellucida of the embryos.50,51 So, within the second part of this study (Part B), we used our novel qPCR assay for a strategic risk assessment procedure during a scheduled rederivation process of a historically parvovirus-positive mouse colony we imported into our animal facility. Here, we combined the qPCR based analysis of embryo transfer-related sample material with the stepwise examination of different animal-derived and environmental sample material of the surrogate dams as well as of the rederived progeny. The process was supplemented by regular serological analysis.
As a first precautionary measure during the embryo transfer, we analysed the discarded embryo material as well as the washing media for remaining parvovirus particles and demonstrated that the transferred embryo transfer material still contained parvovirus, despite extensive washing procedures. This is in line with others who could also detect parvovirus DNA in contaminated embryo transfer material. 51 However, results of another study suggested that the detected parvoviruses were not necessarily infectious any more, as the authors managed to obtain seronegative, virus-free progeny regardless of the positive PCR results. 48 Nevertheless, due to these positive findings we were not able to rule out possible infection of the surrogate dams, which is why they were strictly isolated after the embryo transfer instead of being directly transferred into the breeding barrier unit. Subsequent environmental sampling procedures also strongly indicated that the surrogate dams were indeed infected by parvovirus-positive embryos, which is why all animals were relocated to a quarantine unit for comprehensive follow-up analyses. As expected, the majority of samples were found to be positive for parvoviral DNA, with the highest copy numbers measured in cage faeces. Remarkably, despite positive results in lymphatic tissue, none of the animals tested positive for parvovirus-specific antibodies via IFA, suggesting that surrogate dams became parvovirus carriers without seroconverting. As a direct consequence of the positive qPCR results, the rederived progeny were further kept under strict quarantine husbandry and subjected to a regular follow-up screening protocol, consisting of environmental and animal-derived sampling procedures to monitor the development of infection in the potentially contaminated colony. Regular testing revealed parvoviral DNA in numerous sample materials, indicating long term shedding of low viral quantities. Again, environmental samples (particularly cage faeces) dominated in the acute phase of infection, whereas animal-derived samples (especially fresh faeces and lymphatic tissue) were more suitable to detect ongoing infection in the chronic phase. However, only two out of 110 tested sera were found to be positive for parvovirus-specific antibodies, indicating that the progeny became persistently infected with extremely low seroconversion rates of under 2%. With our innovative, highly sensitive qPCR assay we were able to detect parvovirus at all stages of the rederivation process, beginning with the embryo washing media up to the progeny, whereas traditional virological diagnostics failed to reveal early infection of the colony.
In future rederivation procedures, our established risk assessment procedure could be further extended by pre-testing embryo-donating animals to select for virus free embryo transfer material right from the start. In this context it has to be critically pointed out that molecular detection methods generally have the disadvantage of being prone for false positive results. 41 Moreover, positive PCR findings do not automatically indicate actual infections, as it is impossible to distinguish between infectious material and residual nucleic acids, possibly still present on surfaces after autoclaving procedures. In this context, molecular assays might even detect traces of pathogens in the animals’ faeces after ingestion of food containing residual (non-infectious) parvoviral DNA. Therefore, an alternative explanation for the low viral quantities identified in the faecal samples would be the introduction of parvovirus DNA via commercial mouse chow. To rule out such artificial findings, we analysed dissolved pellet samples out of unopened food batches and obtained negative results via qPCR throughout the health monitoring process (data not shown). Thus, it seems extremely unlikely that we consistently found food derived parvovirus DNA instead of persisting virus infection throughout the whole monitoring procedure. Furthermore, since the animals were kept under strict quarantine conditions, we consider a parvovirus introduction via other sources, such as potentially contaminated materials, also as highly improbable.
To further address the question of whether the virus material detected was actually infectious, we finally performed an in vivo infectivity test with homogenized material that had tested positive with our qPCR assay (low viral quantities of approximately 15 copies/µl). Parvoviral DNA could be detected in the majority of sample material and, remarkably, also in the enlarged lymphoid tissue of the naïve recipient mice at the end of the experiment, indicating infectiousness of the detected virus material. Once more, none of the animals produced parvovirus-specific antibodies in this experimental set-up, which was consistent with the rest of our findings where less than 2% of the mice seroconverted.
Altogether, our novel qPCR assay proved to be a powerful diagnostic tool that enables fast and highly sensitive screening for parvovirus contamination in mouse colonies. A risk-orientated sampling strategy allows for the reliable detection of parvovirus DNA in a broad variety of different animal-derived and environmental materials, requiring significantly lower sample sizes than traditional methods such as conventional PCR and serology. To the best of our knowledge, this is the first study describing the use of a generic parvovirus qPCR assay within a strategic risk assessment procedure using environmental sample material during an embryo transfer. Since the herein described assay cannot differentiate between distinct parvoviruses, strain specific analyses for MVM and MPV have to be additionally performed to complete parvovirus diagnostics. As a follow-up analysis, specific strain characterization will be extremely helpful to answer questions related to infection epidemiology. All in all, our qPCR assay facilitates a risk-orientated, systematic screening procedure of mouse colonies using environmental sample material. This highly sensitive technique substantially improves parvovirus diagnostics and therefore directly contributes to research validity in line with the 6Rs.
Supplemental Material
sj-pdf-1-lan-10.1177_00236772211062861 - Supplemental material for Why serology just is not enough: Strategic parvovirus risk assessment using a novel qPCR assay
Supplemental material, sj-pdf-1-lan-10.1177_00236772211062861 for Why serology just is not enough: Strategic parvovirus risk assessment using a novel qPCR assay by Ann-Kathrin Iwantschenko, Florian Roegener, Wiebke Garrels, Martina Dorsch, Wiebke Köhl, Christian Riehle, Norbert Ghyselinck, Betty Féret, Nils-Holger Zschemisch, André Bleich and Stephanie Buchheister in Laboratory Animals
Footnotes
Acknowledgements
We thank Ulrike Freischmidt, Regine Lube, Melvin Przesdzink-Wiechers and Isabell Wittur for their excellent technical assistance and Erin Colleen Boyle, PhD, for English editing and proofreading our manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.