
Abstract
Correct identification of bacteria is crucial for the management of rodent colonies. Some bacteria are difficult to identify phenotypically outside reference laboratories. In this study, we evaluated the utility of 16S ribosomal DNA (rDNA) sequencing as a means of identifying a collection of 30 isolates of rodent origin which are conventionally difficult to identify. Sequence analysis of the first approximate 720 to 880 bp of the 5′- end of 16S rDNA identified 25 isolates (83.33%) with ≥99% similarity to a sequence of a type strain, whereas three isolates (10%) displayed a sequence similarity ≥97% but <99% to the type strain sequences. These similarity scores were used to define identification to species and genus levels, respectively. Two of the 30 isolates (6.67%) displayed a sequence similarity of ≥95 but <97% to the reference strains and were thus allocated to a family. This technique allowed us to document the association of mice with bacteria relevant for the colonies management such as Pasteurellaceae, Bordetella hinzii or Streptococcus danieliae. In addition, human potential pathogens such as Acinetobacter spp., Ochrobactrum anthropi and Paracoccus yeei or others not yet reported in mouse bacterial species such as Leucobacter chironomi, Neisseria perflava and Pantoea dispersa were observed. In conclusion, the sequence analysis of 16S rDNA proved to be a useful diagnostic tool, with higher performance characteristics than the classical phenotypic methods, for identification of laboratory animal bacteria. For the first time this method allowed us to document the association of certain bacterial species with the laboratory mouse.
Accurate identification of bacterial isolates is a challenging task for clinical microbiology laboratories. Traditional phenotypic identification is the approach most used in clinical bacteriology. This can be difficult and time-consuming and the interpretation of the results can involve some amount of subjective judgment. 1 In addition, within some bacterial species there is also phenotypic variability among different strains, making them atypical for possible identification outside the reference laboratory. 2 The phenotypic identification of bacterial isolates originating from laboratory rodents is even more difficult, since many of them are not included in the commercially available databases. Moreover, these databases are often outdated and their taxonomy is not current. 3 In these conditions the capabilities of laboratories in the laboratory animal discipline to correctly identify bacterial species are well below what are considered to be acceptable limits for human diagnostic laboratories. 4
Identification of bacteria by partial 16S ribosomal RNA (rRNA) gene sequencing is considered to be more accurate than the conventional phenotypic methods.5,6 Bacterial 16S rRNA genes contain nine ‘hypervariable regions’ (V1–V9) that demonstrate considerable sequence diversity among different bacterial species and thus allow their differentiation. 7 The partial sequencing of V1–2–3 regions located at the 5′- end of the 16S rRNA gene proved to be sufficient and more sensitive than the other variable regions for identification of several groups of bacteria of veterinary origin to species level. 5 In comparison to the phenotypic identification, the 16S rRNA gene sequences are not restricted to a specific group of bacteria and the novel, not yet described, isolates can be assigned to a group of related bacteria. 8 Moreover, the results are not dependent on individual interpretation or variation within the strains. Unfortunately, identification of laboratory animal bacteria by genetic methods or by 16S rDNA sequencing is, to date, very seldom undertaken.
The main goal of the present study was to implement and to study the usefulness of partial 16S rDNA sequencing in the identification of bacterial isolates originating from laboratory rodents which proved to be difficult to identify by phenotypic methods or for which a more accurate identification was needed. A further objective of the study was to observe the diversity and the relevance of the identified bacteria for their host. The 16S rDNA sequencing proved to be an easy to implement and a very specific method for the identification of rodent bacteria. Using this strategy we have documented the association of laboratory mice with some bacterial species for the first time.
Materials and methods
Bacterial isolates and conventional identification methods
Identification of bacterial strains by phenotypic methods and 16S rRNA analyses.

*Rat isolate; **A taxonomic group that include related species which cannot be differentiated solely by 16S rRNA sequences; Bacillus aerophilus group** (B. altitudinis, B. stratosphericus, B. aerophilus); Escherichia coli group** (E. coli, E. albertii, Shigella flexneri, S. sonnei, S. boydii, S. dysenteriae); Ochrobactrum anthropi group** (O. anthropi, O. cytisi, O. lupini).
16S rDNA amplification and sequencing
DNA template for polymerase chain reaction (PCR) amplification of the 16S rDNA was extracted from pure colonies from the agar plates using a boiling procedure as described previously. 11 Briefly, two to three colonies of bacteria from the 48 h cultures were suspended in 300 µL deionized water and frozen at −20℃ for 10 min. Afterwards the suspension was heated at 98℃ in a thermo block for another 10 min and then centrifuged for 10 min at 13,000 × g. Subsequently 270 µL of the supernatant was transferred into a clean Eppendorf tube and stored at −20℃ until use.
The 16S rDNA was amplified using the universal primer pair BSF8/27 (5′-AGAGTTTGATCCTGGCTCAG-3′) and BSR1541/20 (5′-AAGGAGGTGATCCAGCCGCA-3′) described previously,5,12 located at the 5′- and the 3′- end respectively of the 16S rRNA gene (Figure 1). Additionally, the 16S rDNA universal reverse primer described previously
13
named UniR (5′-ACGTCRTCCMCACCTTCCTC-3′) in this study and located at positions 1175–1194 of the Escherichia coli 16S rRNA gene was used instead of the reverse primer BSR1541/20, in conjunction with the forward primer BSF8/27, when the first primer pair did not retrieve any 16S rRNA gene amplification or when secondary amplification products were present in the amplification tubes. All DNA amplification assays were done in 25 µL reaction volume. PCR mixtures contained 2.5 µL of template DNA, 0.5 µL of the four deoxynucleoside triphosphates (dNTPs) in a primary concentration of 10 mM (AnalytikJena, Jena, Germany), 10 pmol of each primer, 2.5 µL of 10 × InnuTaq PCR buffer; 1.5 µL of 25 mM MgCl2; 0.2 µL InnuTaq-DNA polymerase (1 U) (AnalytikJena). The assay was filled up to the final volume with water for molecular biology (Applichem, Darmstadt, Germany). PCR assays were carried out in an Eppendorf Mastercycler Gradient 5331 thermocycler (Eppendorf, Hamburg, Germany) programmed for the initial denaturation at 95℃ for 5 min and 30 cycles as follows: denaturation at 94℃ for 1 min, annealing at 55℃ for 1 min, extension at 72℃ for 1.5 min, and a final extension step at 72℃ for 7 min. Amplification products were analyzed by performing electrophoresis of 5 µL of the reaction mixture in a Midori Green (Nippon Genetics Europe, Düren, Germany) containing 1.5% agarose gel (Applichem) and photographed under ultraviolet (UV) exposure. A 100 bp DNA ladder (AnalytikJena) was used as molecular size standard.
Schematic localization of the primers on the 16S rRNA gene and of the resulting polymerase chain reaction (PCR) and sequencing products.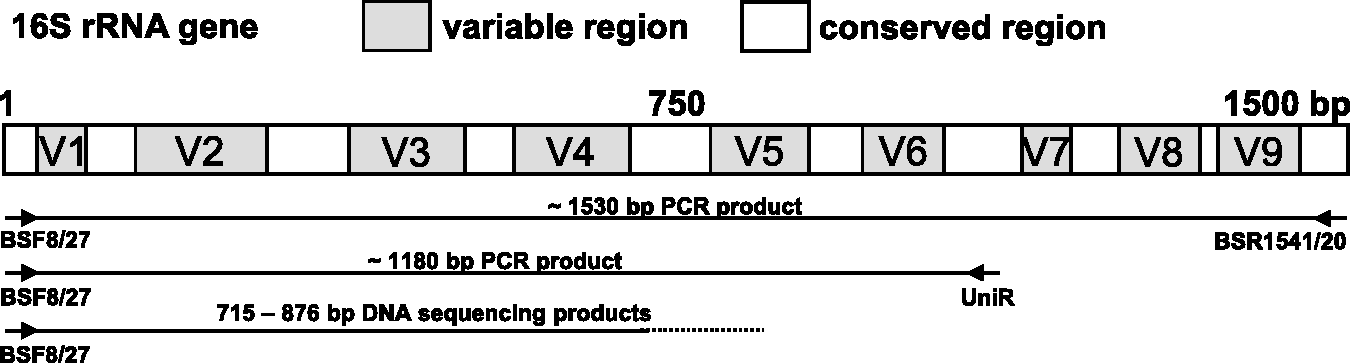
The PCR fragments representing part of the 16S rRNA gene region were purified using the QIAquick PCR purification kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Purified DNA samples were adjusted with water to a concentration of 50 ng/µL and then sequenced using the PCR primer BSF8/27. Sequencing was carried out with the BigDye® Terminator v3.1 Ready Reaction Cycle sequencing kit (LifeTechnologies, Darmstadt, Germany). The sequencing reaction mixture contained HiPerSolv water, 1 µL of BigDye mix, and 1.5 µL of 5 × sequencing buffer; 5 pmol of sequencing primer (BSF8/27); and 3 µL (150 ng) of cleaned PCR product in a total volume of 10 µL. Following an initial denaturing step (15 s at 96℃), the cycle sequencing reaction comprised 30 cycles of denaturation (10 s at 96℃/5 s at 50℃) and extension (2 min at 60℃) at a thermal ramping rate of 1℃/s. The sequencing products were purified by ethanol precipitation, dissolved in HiDi Formamid and analyzed on a 3130XL genetic analyzer (Life Technologies) in accordance with the manufacturer's instructions. The KB™ Basecaller software v1.4 (Applied Biosystems) ABI was used for base calling. Resulting trace-files underwent limited manual editing.
Analysis of the 16S rDNA
The 16S rDNA sequences were compared for identification with corresponding sequences of type strains using EzTaxon server at http://eztaxon-e.ezbiocloud.net/. 14 Identification to the species level was defined as a 16S rDNA sequence similarity of ≥99% with that of the type strain sequence in GenBank.2,5 A sequence similarity of ≥97% with that of the type strain retrieved an identification at the genus level, whereas a similarity score <97% was considered insufficient for genus identification. 5 A similarity value ≥95 but <97% was considered sufficient for family identification. If the unknown isolate was assigned to a species and the second classified species in the scoring list showed less than 0.5% additional sequence difference this was categorized as a ‘species with low demarcation to the next species’. 8
Results
Identification of the clinical isolates by morphological and biochemical profiles
The selected isolates underwent presumptive phenotypic identification based on colony morphology, ability to grow on special media, cell form after Gram staining, spore forming ability, respiratory type, and motility; and oxidase and catalase tests. For 22 out of the 30 isolates tested (73.33%) a presumptive identification at genus or family level was possible (Table 1). The presumptive phenotypic identification facilitated allocation of the isolates to specific API kits for further biochemical characterization. Thirteen out of 26 isolates tested (50%) retrieved at least a good identification at species level using the API strips (Table 1).
Identification of the clinical isolates by 16S rDNA sequencing
The primer pair BSF8/27 and BSR1541/20 able to amplify nearly the complete 16S rDNA (Figure 1) did not produce bands or provide secondary products from a few isolates. Using an alternative second primer pair BSF8/27 and UniR (Figure 1) it was possible to amplify sufficient 16S rDNA PCR products from those isolates (details not shown).
The 16S rDNA sequences obtained (Access no. HG917870–HG917899) using the primer BSF8/27 contained between 715 and 883 bp and covered in all cases at least the first four variable regions of the 16S rRNA gene. Most of the sequences covered the first part of the fifth variable region as well (Figure 1). The isolates were classified in three categories based on 16S rDNA sequence analysis. A total of 25 of 30 isolates (83.33%) possessed a 16S rDNA sequence similarity ≥99% to that of a type bacterial strain deposited in GenBank, and could thus be identified at species level or at least attributed to a smaller group of species which belonged to the same 16S rDNA taxonomic group. Three of the 30 isolates (10%) showed a sequence similarity ≥97% but <99% to that of a prototype strain from GenBank and were thus attributed to a bacterial genus. The remaining two of the 30 isolates (6.67%) displayed a sequence similarity ≥95 but <97% to the reference strains and were allocated to a family (Table 1).
Discussion
In the present investigation we analyzed the usefulness of 16S rDNA sequencing for the identification of selected rodent bacterial isolates.
Phenotypic identification represents the gold standard in laboratory animal bacteriology and it allowed us to identify presumptively 73.33% of the isolates tested to genus or at least to family level. However, the 16S rDNA sequence is regarded as a backbone in bacterial taxonomy. 15 Interestingly, the presumptive phenotypic identification was confirmed in all cases by the 16S rDNA sequencing but in only a few cases by the biochemical profile using the API strips (Table 1). Moreover, in some cases there was a discrepancy between the 16S rDNA sequencing and the API identification. For example, Bordetella hinzii, an opportunist pathogen in mice and humans,16,17 is not included in the API 20NE database, but it was identified as Bordetella avium using this test. Although Leucobacter chironomi 18 is not included in the API Coryne database, this bacterium was misidentified as Erysipelothrix rhusiopathiae by this assay. A particular situation is represented by the rodent Pasteurellaceae such as [Pasteurella] pneumotropica and [Actinobacillus] muris. Although described several decades ago, these species have not been formally classified under genera. 11 The brackets indicate that the species are not true members of Pasteurella and Actinobacillus respectively. They display variable phenotypes, and with the exception of [P.] pneumotropica are not included in the commercial phenotypic databases which do not seem to be reliable enough to allow their identification. 19 The API 20NE kit failed to identify or misidentified the three rodent Pasteurellaceae isolates included in this study. In contrast to the phenotypic methods, the 16S rDNA identification allowed us a more accurate identification of the Pasteurellaceae isolates (Table 1). The sequence analysis diagnostic obtained for the three rodent Pasteurellaceae strains studied here was confirmed in an [A.] muris 20 and a rodent Pasteurellaceae multiplex–PCR assay. 21
Overall, the 16S rDNA sequencing allowed us a more accurate identification of the selected laboratory rodent isolates than the phenotypic methods. The 5′- end sequencing procedure of the first four to five variable regions of the 16S rDNA used in this study was easy to implement and did not involve a significantly higher cost than the biochemical characterization. Therefore, we recommend supplementary testing using sequencing or another genetic method if a more accurate identification is indicated in laboratory animal bacteriology.
The 16S rDNA sequences allowed us to document association of mice with interesting bacterial species. Species relevant for the breeding and management of rodent colonies such as Pasteurellaceae 9 and Bordetella hinzii 16 could thus be diagnosed. We could hereby also document the presence of Streptococcus danieliae, a very recently described species, 22 as the dominant bacterium in the upper respiratory tract of the mice from our facility. Additionally, we document here for the first time the association of laboratory mice with human potential pathogens such as the Acinetobacter species, 23 Ochrobactrum anthropi, an emerging opportunistic pathogen in immunocompromised patients 24 and Paracoccus yeei. 25 From this perspective, the carrier role of the (laboratory) rodents for these infectious agents is plausible. The association of laboratory mouse with Leucobacter chironomi a chromate-resistant bacterium isolated from a chironomid egg mass is also notable. 18 To our knowledge this provides the first evidence of a mammal as an ecological niche for the Leucobacter species.
In conclusion, we showed in this study that the 5′- end sequence analysis of 16S rDNA is an easy to implement and valuable technique, with higher performance characteristics than the classical phenotypic methods in laboratory animal bacterial diagnostics. For the first time this method allowed us to document the association of certain bacterial species with the laboratory mouse, with possible implications in breeding management.
Footnotes
Acknowledgements
We gratefully acknowledge Sonja Green, Andrea Grunwald and Manuela Stockhausen for their excellent technical assistance.