
Abstract
One limitation to housing rodents in individually ventilated cages (IVCs) is the ineffectiveness of traditional health monitoring programs that test soiled bedding sentinels every quarter. Aerogen transmission does not occur with this method. Moreover, the transmission of numerous pathogens in bedding is uncertain, and sentinel susceptibility to various pathogens varies. A novel method using particle collection from samples of exhaust air was developed in this study which was also systematically compared with routine health monitoring using soiled bedding sentinels. We used our method to screen these samples for the presence of murine norovirus (MNV), a mouse pathogen highly prevalent in laboratory animal facilities. Exhaust air particles from prefilters of IVC racks with known MNV prevalence were tested by quantitative reverse transcription polymerase chain reaction (RT–qPCR). MNV was detected in exhaust air as early as one week with one MNV-positive cage per rack, while sentinels discharged MNV RNA without seroconverting. MNV was reliably and repeatedly detected in particles collected from samples of exhaust air in all seven of the three-month sampling rounds, with increasing MNV prevalence, while sentinels only seroconverted in one round. Under field conditions, routine soiled bedding sentinel health monitoring in our animal facility failed to identify 67% (n = 85) of positive samples by RT–qPCR of exhaust air particles. Thus, this method proved to be highly sensitive and superior to soiled bedding sentinels in the reliable detection of MNV. These results represent a major breakthrough in hygiene monitoring of rodent IVC systems and contribute to the 3R principles by reducing the number of animals used and by improving experimental conditions.
Standardization of experimental designs is essential for high-quality biomedical research. Laboratory animal breeding or experimental facilities have to cope with different infectious agents known to impact animal welfare and experimental outcomes.1,2 Consequently, a standardized microbiological status is essential for ensuring the reproducibility of animal experiments and animal welfare. Currently, laboratory rodents are commonly housed in individually ventilated cages (IVCs). Because these cages provide biocontainment and bioexclusion, the spread of infectious agents between cages, and thus from infected mice to sentinels, is prevented when cages are handled properly.3–5 Therefore, health monitoring in IVC-housed rodent colonies by sentinel monitoring has become a challenging task.
Recently, the Federation of European Laboratory Animal Science Associations (FELASA) updated its recommendations for health monitoring programs in laboratory animal facilities. 6 Accordingly, if using contact sentinels is not feasible and colony animals are not available, it is recommended to expose a sufficient number of sentinel animals to soiled bedding and monitor the animals quarterly. This method may be reliable for some infectious agents; however, those not easily transmitted by the fecal–oral route or with a low survival rate in bedding remain undetected.7–10 Infections with viruses and some bacteria are normally detected by serological methods. However, the production of antibodies in the sentinel animal may take days or weeks, which means that early infections can remain undetected. 11 Moreover, sentinel animals only show seroconversion if they are exposed to an infectious dose of the unwanted agent. 12 Therefore, infections with low prevalence in the colony may remain unrecognized.
The FELASA recommendations mention testing exhaust air, from either a single cage or the complete rack via polymerase chain reaction (PCR). This is particularly important for pathogens known to be non transmissible via soiled bedding, and has been successful for fur mites, murine hepatitis virus (MHV), Helicobacter spp, Pasteurella pneumotropica and Sendai virus.13–16
In the present study, exhaust air prefilter quantitative reverse transcription PCR (RT–qPCR) was compared with serological monitoring of soiled bedding sentinels for the detection of murine norovirus (MNV) in a proof of principle field study with unknown prevalence, and also in an IVC rack with known MNV prevalence in order to determine detection limits. MNV was chosen because of its high prevalence worldwide17–21 and as the most commonly detected viral agent in laboratory mice. 6 Since MNV infections do not cause clinical signs in immunocompetent or even most immunodeficient mice, new infections may remain undetected even when highly prevalent, and soiled bedding sentinels cannot be reliably used to detect infections. 22
Animals
The MNV-negative colony consisted of genetically-modified mice and wild-type littermates with a mixed or C57BL/6 genetic background without pathological phenotypes. Mice were of different ages (2–18 months) and sexes, and represented a typical mouse colony in our facility. Sentinels and MNV-positive mice were female AVM:ICR bred in-house at different ages (2–18 months).
Analysis of particles from the exhaust air prefilter was more sensitive than soiled bedding sentinel serology for the detection of MNV infections at low prevalences.
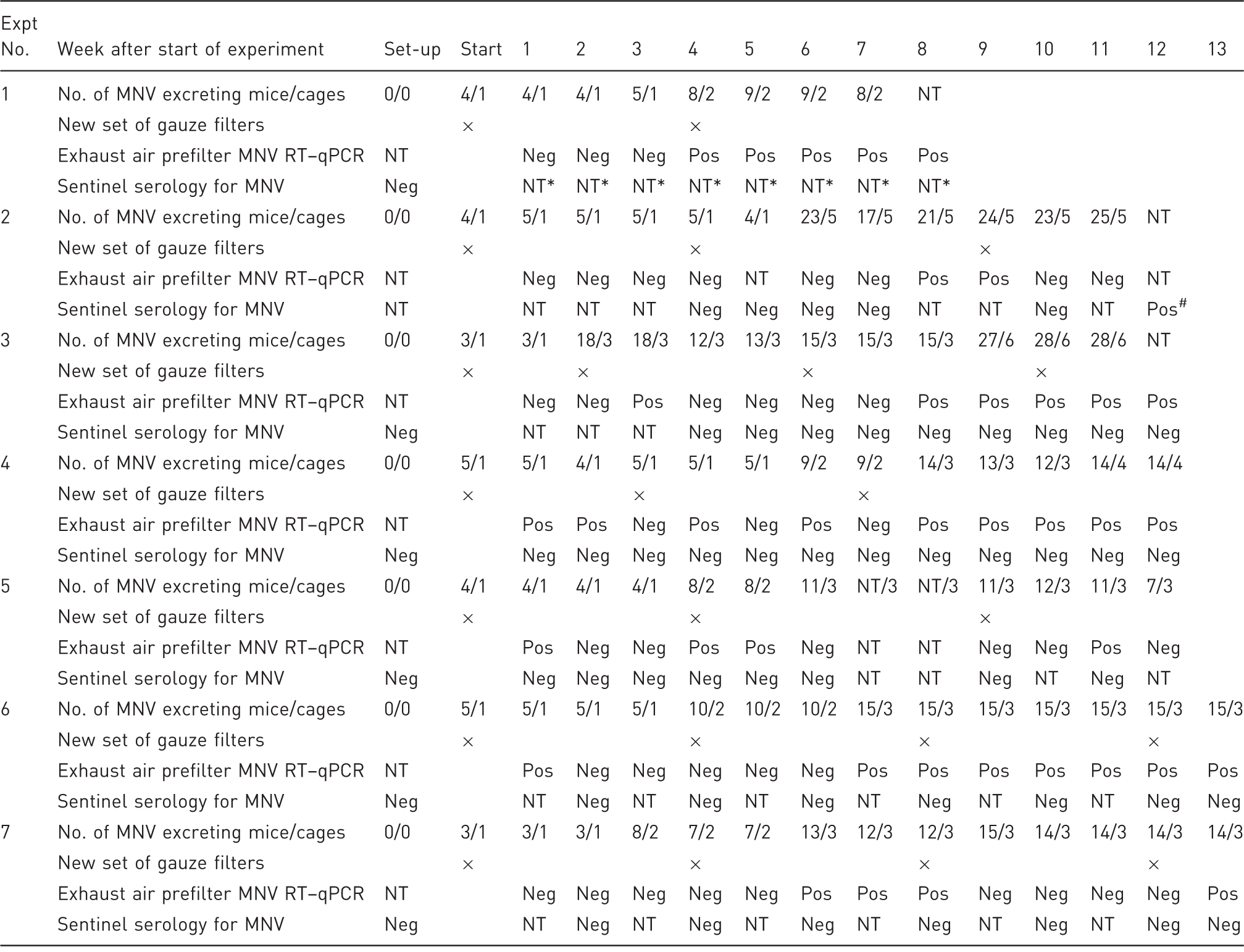
Expt 1 evaluated the general feasibility of MNV testing by particle sampling. MNV could be detected in the exhaust air prefilter from weeks 4–8. Expts 2–7 compared the detectability of MNV in the exhaust air prefilter with that by sentinel serology in five 12-weekly (Expts 2–5) and two 13-weekly (Expts 6–7) testing rounds. Seroconversion of soiled bedding sentinels was only detected in Expt 2 at the very end of the study in two soiled bedding sentinels. In the remaining experiments none of the soiled bedding sentinels showed seroconversion. However, RT–qPCR analysis of exhaust air prefilters detected MNV RNA in the exhaust air at every 12- and 13-weekly testing round at almost every four-weekly prefilter change interval. MNV was not detected only in the first prefilter change interval in Expts 2 and 7 and in the third interval in Expt 7. Expt: Experiment, MNV: murine norovirus, RT–qPCR: quantitative reverse transcription–polymerase chain reaction, Neg: tested negative for MNV, Pos: tested positive for MNV, NT: not tested. *Sentinel serology was not part of the objective of Expt 1. #Two out of four soiled bedding sentinels tested positive.
Materials and methods
Routine health monitoring
For routine health monitoring, one cage containing two sentinels plus one back-up per room were given soiled bedding once a week from cages in the same room. Soiled bedding was collected from each cage in the room (5 racks with 63 cages each; 315 cages in total) during routine cage changes. The sentinel cage was filled with equal amounts of soiled and fresh bedding. Soiled bedding sentinels were examined quarterly for all FELASA-listed organisms, 6 which included MNV serology performed by an external diagnostic laboratory.
Field study
Exhaust air particle RT–qPCR and routine soiled bedding sentinel monitoring were both undertaken and compared over a two-year period in mouse colonies housed in 13 rooms in our barrier facility, with an assumed high MNV prevalence. Sentinel serology was obtained quarterly by routine health monitoring, as described above, and exhaust air particle RT–qPCR was performed at the same time points. 6 At each stage when the sentinels were taken for monitoring, prefilters had been exposed to exhaust air for 1–4 weeks. All known MNV-positive animal rooms in the barrier facility (experimental and breeding rooms) were included in this study. During a two-week period, all colony mice made available for bleeding by scientists were tested for MNV antibodies to assess MNV seroprevalence.
Detection limit study
The relative sensitivities of MNV detection by exhaust air prefilter RT–qPCR and of sentinel serology were compared in six of the 12–13-week testing periods with a known MNV prevalence in the test rack. The study set-up consisted of one clean and autoclaved IVC rack with 63 cages connected to a cleaned and disinfected air handling unit containing a new exhaust air prefilter. The rack was occupied by three types of mice: (i) the MNV-negative colony in 54 cages as tested by feces RT–qPCR; (ii) two singly-housed soiled bedding sentinels; and (iii) increasing numbers of MNV-positive mice (Table 1) tested by individual feces RT–qPCR. No breeding occurred, and sick mice were replaced to keep the colony size constant. Additionally, this colony was reused in the six rounds of testing. At the start of each round of testing, one cage containing five mice naturally infected with MNV was added to the rack. The MNV-positive mouse colony providing the test mice was maintained by cohousing uninfected mice with mice positive for MNV by feces RT–qPCR. Each of the MNV-positive mice in the test rack was tested weekly for continued MNV excretion by feces RT–qPCR. The two sentinels housed individually in two cages received a 1:1 mixture of soiled bedding from the other 61 cages in the rack and fresh bedding during weekly cage changing.
Sample collection
Three fresh individual fecal pellets from each MNV-positive mouse were collected weekly by transiently placing the mice into an empty cage. In addition, one pooled cage sample of 20 fecal pellets was collected from each cage with MNV-positive mice. Fecal samples were tested for MNV via RT–qPCR. MNV-negativity of the colony mice was confirmed every three weeks by testing pooled fecal samples from each of the seven columns of the rack (10 pellets for cages with 1–2 mice, and 20 pellets for cages with 3–5 mice). Individual fecal samples (three pellets) as well as pooled soiled bedding samples (20 fecal pellets) were collected weekly from the sentinels and the two sentinel cages for RT–qPCR analysis. Sentinel blood samples were collected at least every four weeks via facial vein puncture (100 µL) and tested for MNV by serology in a commercial diagnostic laboratory.
In accordance with the manufacturer's instructions, the prefilter was exchanged every four weeks. To collect particle samples from the exhaust air prefilters, four gauze pieces (2 cm × 2 cm) were pinned onto the prefilter surface within the air-handling unit directly above the opening of the exhaust air hose (Figure 1). Every week, one gauze piece was removed and RNA was extracted. After four weeks, the last gauze piece was removed together with the prefilter, and the prefilter was tested by cutting out a 2 cm × 2 cm piece from the dustiest area for RNA extraction. Consequently the exposure time of the gauze to the exhaust air varied from one week (first gauze) to four weeks (last gauze).
Gauze pieces pinned onto the exhaust air prefilter. With every new prefilter inserted into the air handling unit, four gauze pieces were pinned onto the prefilter to enable weekly collection of particle samples without having to change the exhaust air prefilter on a weekly basis.
RNA extraction from feces
For RNA extraction, 500 µL of milli-Q-water (Milli-Y PLUS UF; Millipore, Billerica, MA, USA) were added to a single-mouse fecal sample consisting of three pellets in a 1.5 mL tube and shaken at maximum speed on a thermomixer (Thermomixer Comfort; Eppendorf, Hamburg, Germany) at room temperature for at least 20 min, until fully suspended before centrifuging at 6000
For processing of multicage samples, 30 mL of milli-Q-water were added to samples in 50 mL tubes and shaken at maximum speed on a thermomixer at room temperature for at least 30 min until the feces was completely suspended. Samples were then centrifuged and processed as above.
RNA extraction from filter material
RNA from the filter material was extracted using a QIAamp Viral RNA Mini Kit. The manufacturer’s instructions were adapted for the filter material: 1120 µL of AVL buffer + carrier DNA were added to the filter material and shaken at maximum speed on a thermomixer for 20 min. The filter material was then pressed to the bottom of the tube using a sterile pipette tip and 700 µL of the supernatant were processed according to the manufacturer’s instructions.
Quantitative reverse transcription PCR
The RT–qPCR assay was performed using an Agpath-ID™ One-Step RT–PCR Kit (Ambion, Kassel, Germany) in a 25 µL reaction volume including a 5 µL template. Primers and probe were used as described in Muller et al.: 23 forward primer: 5′-AGAGGAATCTATGCGCCTGG-3′; reverse primer: 5′-GAAGGCGGCCAGAGACCAC-3′; probe: 5′-Fam-CGCCACTCCGCACAAACAGCCC-Dabcyl-3′ with 800 nmol primers and 200 nmol probe.
PCR amplification was carried out using a 7500 Real Time PCR System (Applied Biosystems, Darmstadt, Germany). Thermal conditions were: reverse transcription at 50℃/30 min, initial step at 95℃/15 min, followed by 40 cycles of 95℃/15 s and 55℃/60 s. Fecal samples were tested in duplicate, and prefilter samples in triplicate. Data were collected and analyzed using Sequence Detection Software Version 1.2.2 (Applied Biosystems).
For quantitative analysis, an RNA standard flanking the RT–qPCR amplicon was constructed by linearizing the DNA plasmid. A ten-fold dilution series (3 × 108 to 3 × 103 copies in 5 µL) was included in each run to generate a standard curve. The 92 bp PCR product of the RT–qPCR was within the sequence of the PCR product used for the RNA standard.
Results
Analysis of exhaust air prefilter particles was more sensitive than routine soiled bedding sentinel serology for detecting MNV in a field study
Results from more than three years of routine MNV health monitoring with sentinel serology and RT–qPCR analysis of exhaust air prefilters for MNV in 13 rooms in the animal facility.
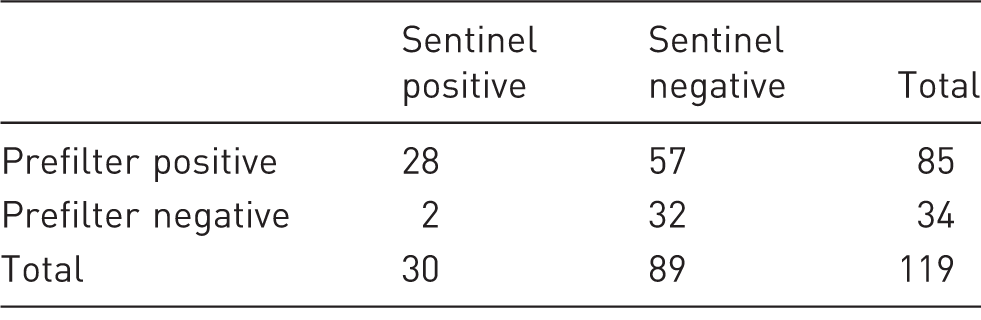
Prefilter = quantitative reverse transcription polymerase chain reaction (RT–qPCR) results of exhaust air prefilter; Sentinel = sentinel serology; MNV = murine norovirus.
MNV RNA was detected in the exhaust air prefilter of an IVC rack even at a low prevalence
Next, we evaluated the sensitivity of prefilter particle RT–qPCR. Our analysis described above determined colony MNV seroprevalence only at one time point in a small sample size, and no information on the prevalence of virus-excreting animals was available. In order to test a realistic scenario (i.e. an infection that started with one mouse infecting its cage mates and then infecting mice in other cages) the number of mice shedding MNV was increased gradually from four to nine. MNV shedding was confirmed weekly by feces RT–qPCR, and not all infected mice excreted MNV in all instances (Table 1, Expt 1). The MNV shedding mice housed in one or two cages were placed in a rack with 63 IVC cages of an otherwise MNV-negative mouse colony. Four gauze pieces pinned on the exhaust air prefilter (Figure 1) allowed testing of filter material without having to change the prefilter completely. Every week one gauze piece was collected and tested by RT–qPCR. The fourth gauze piece was collected together with a sample from the actual prefilter at the end of the regular four-week prefilter exchange period. At this time new prefilter gauze pieces were installed. Positive RT–qPCR results were obtained with the gauze pieces collected after 4–8 weeks, with 8–9 MNV excreting mice in the IVC rack, respectively (Table 1, Expt 1). The MNV shedding mice housed in one or two of 63 cages represented cage MNV prevalences of 1.6% and 3.2%, respectively. The experiment was stopped after eight weeks.
Five MNV-positive cages in an IVC rack induced seroconversion in sentinels
Next, the number of MNV shedding mice was increased to determine how many cages of MNV-positive mice were needed in a rack for a positive sentinel serology result. During a 12-week testing period, one (weeks 1–5) and five (weeks 6–12) cages with MNV shedding mice were placed in the test rack of the otherwise MNV-negative colony. Blood samples were collected from two (weeks 1–12) plus two additional (weeks 6–12) soiled bedding sentinels in weeks 4–7, 10 and 12 or weeks 7, 10 and 12, respectively, and were tested for MNV antibodies. Seroconversion was not detected at weeks 4–10, and only two of the four sentinels had seroconverted by week 12 (Table 1, Expt 2)
Exhaust air particle RT–qPCR detected MNV infection at much lower prevalence than sentinel serology
Based on the results of the previous two experiments, five additional experimental rounds were performed, monitoring soiled bedding sentinels and exhaust air particles for a period of 12–13 weeks for each round. The number of MNV-positive cages was increased gradually from one to six cages with three to 28 MNV shedding mice in the test rack (Table 1, Expts 3–7). Exhaust air prefilters were changed and replaced by new ones equipped with four gauze pieces at least every five weeks. The time schedule for sample collection and the test results are depicted in Table 1. Despite the high numbers of MNV shedding mice (up to 7.9% MNV-positive cages) in the test rack at the end of each of the five monitoring periods, serological testing of the sentinels was negative for MNV in all cases. By contrast, the exhaust air prefilters tested positive in all five experimental rounds at multiple time points. In addition, exhaust air particle RT–qPCR detected five or fewer MNV shedding mice within only one week in three of the five experiments (Table 1, Expts 4–6). Exhaust air particle RT–qPCR detected MNV at least once in all but three filter exchange intervals of four weeks (Table 1, Expt 3, interval 1 and Expt 7, intervals 1 and 3).
MNV RNA was found in the soiled bedding by PCR
Murine norovirus (MNV) RNA in sentinel feces and soiled bedding within the sentinel cage.
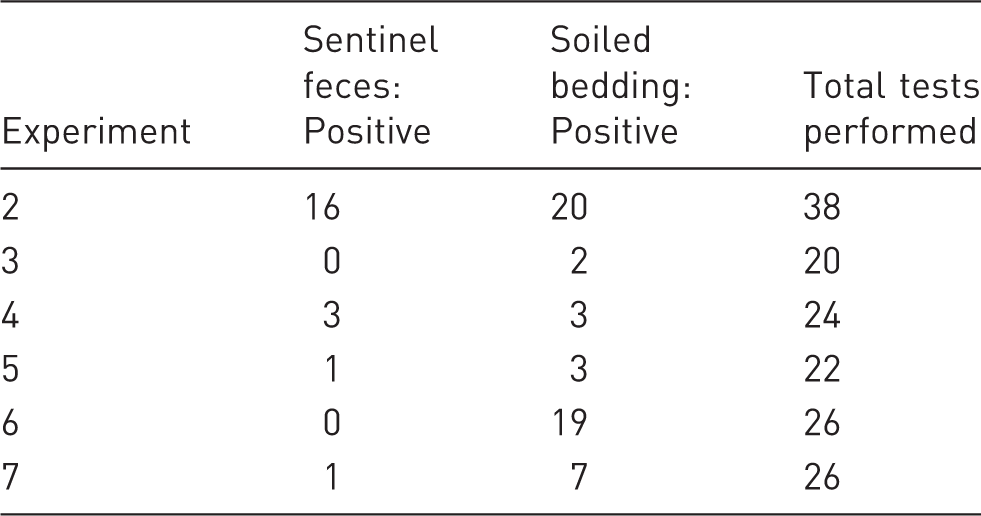
Sentinels discharged MNV RNA without being infected
To determine whether MNV RNA in the soiled bedding entered the sentinels through the fecal–oral route by coprophagy, we performed weekly tests of feces from the sentinels after the exposure to soiled bedding (156 tests in total). Although only two of the sentinels seroconverted to MNV, we found discharges of MNV RNA at 21 time points in our experiments (Table 3).
Biocontainment over a period of three years
The MNV-negative colony used for the seven experimental rounds was tested every three weeks in pooled fecal samples. All tests over a three-year period remained negative. Old mice were replaced with young animals whenever necessary. On average MNV-negative mice stayed in the test rack for over one year.
MNV detection by exhaust air particle RT–qPCR turned negative two weeks after removing MNV-positive mice from the IVC rack
Finally, we investigated how long MNV was detectable in the exhaust air prefilter after removing MNV-positive mice from the rack. We tested this in four rounds when removing MNV shedding mice from the test racks after the monitoring period. Three cages with MNV-positive mice had been in the rack for five weeks or more. The exhaust air prefilter was changed the same day the MNV-positive mice were removed and was equipped with four gauze pieces, which were then collected once a week for RT–qPCR. In two of the experimental rounds, MNV RNA was not detected at all; in the other two rounds, MNV RNA was detected only at weeks 1 and 2.
Discussion
Health monitoring of laboratory rodent colonies housed in IVCs remains a challenging task, and an effective method that is applicable to all relevant infectious agents has yet to be established. Here we examined whether MNV could be reliably detected in exhaust air prefilters and whether this method was superior to routine health monitoring regimes consisting of serological analysis of soiled bedding sentinels. By screening a mouse colony with a known prevalence of MNV, we demonstrated that MNV detection by sentinel serology was insufficient at low prevalence rates (in less than five MNV-positive cages out of 63 in total). Sentinel animals did not seroconvert, despite the presence of detectable MNV RNA in soiled bedding and sentinel feces. A reliable health monitoring program, however, depends on detecting infections already at low prevalence rates in order to implement appropriate measures.
The first part of this study aimed to test the efficacy of MNV detection in exhaust air prefilters under field conditions. Animal rooms known to be MNV-positive, but with unknown prevalence rates, were equipped with gauze pieces placed on exhaust air prefilters and analyzed after routine prefilter changes, i.e. after four weeks. A direct comparison showed that analysis of exhaust air particle RT–qPCR was a superior method to analysis of soiled bedding sentinels, which was consistent with the results of the second part of our study. Soiled bedding sentinels tested positive in rooms with high MNV prevalence, where the viral load in the soiled bedding was high enough to trigger infection and seroconversion. Other rooms with lower MNV prevalence only tested positive with exhaust air particle RT–qPCR. Since the exhaust air prefilter turned negative no more than two weeks after removal of the MNV-positive mice, the positive test results were not artefacts resulting from previous infections. In some cases, both methods failed to detect MNV. Since some of the rooms tested were experimental rooms in which live MNV-negative mice were frequently imported from commercial breeders, we might reasonably assume that in those cases no MNV infection was present in the tested interval.
The second part of our study was carried out in an IVC rack with a known MNV prevalence. This was crucial not only to assess the detection limits of both methods (sentinel serology versus exhaust air particle RT–qPCR), but also to evaluate the feasibility of using filter material from within the exhaust air flow. Testing of gauze pieces placed on the exhaust air prefilter reliably and repeatedly detected MNV infections at cage prevalence rates as low as 1.6% (1 MNV-positive cage out of 63). In our study we analyzed gauze pieces on a weekly basis. All six testing periods showed that monthly prefilter analysis is sufficient to reliably detect infection in the three-month time window traditionally used for monitoring of soiled bedding sentinels. Since the exhaust air prefilter in most IVC systems needs to be changed monthly, the collection of prefilter material for health monitoring is not associated with any additional work. In experimental round 7, in two of four testing periods (weeks 1–4 and weeks 9–12) the exhaust air prefilter did not detect the MNV infection in the rack; however, at weeks 5–8 and week 13 the infection was again evident. In experimental round 3 the exhaust air prefilter was changed after two weeks instead of the usual four weeks, making it more likely that the filter analysis was MNV-negative because of the shorter exposure. Subsequent tests were positive, and with the traditional monthly change this filter might have tested positive as well. Therefore, caution should be used in the analysis of prefilters that are changed at shorter intervals during particle monitoring.
At low prevalence rates the transmission of infectious agents may depend on uniformity of the airflow and optimal placement of gauze pieces. 13 To this end, we placed all gauze pieces directly above the opening of the exhaust air hose. This guaranteed that most particles in the exhaust air came into contact with the gauze pieces. Uniformity of airflow should be adjusted using the control panel provided by the manufacturer, and regular maintenance is crucial for keeping within the desired airflow parameters. Depending on the IVC system used, it may be necessary to choose different sampling sites, since determining the optimal place for sampling is crucial for detection of unwanted agents. 14 However, even after choosing the appropriate sampling site, the sampling technique can also affect the outcome. Nucleic acid extraction protocols and the choice of a suitable PCR protocol are additional key factors that impact the sensitivity of agent detection. While Ouellet et al. failed to detect Pasteurella pneumotropica in prefilter samples using a conventional PCR assay, 24 we reliably detected this bacterium in the prefilter with just one infected cage in a rack, with 63 cages in total using specific real-time PCR. 16 Manufacturers of IVC rack systems have recently developed sampling devices for exhaust air ducts, potentially simplifying and standardizing sampling. Moreover, by using appropriate PCR protocols, this method may easily be adopted for detecting other pathogens as well.
The analysis of soiled bedding given to sentinels addressed whether this method was effective for the transfer of MNV. Although MNV RNA was repeatedly detected in soiled bedding samples, the viral load was probably not high enough to induce seroconversion. Obviously, sentinels discharge MNV RNA without being infected if the amount of infectious agent is too low. Additional points include the environmental stability of MNV particles in bedding or intermittent viral shedding. Only intact viral particles will infect an animal and induce seroconversion, whereas degraded RNA will not. Notably, however, analysis by PCR can give positive results even where viral particles are no longer infectious.
MNV is highly stable in fecal samples if stored at ambient temperature and if long-term persistence of infectious MNV has been detected. 25 Therefore, the lack of seroconversion of sentinels does not seem to correspond with viral particle degradation, but rather with the infectious dose in the sentinel cage being too low to cause seroconversion. This seems highly probable since MNV-infected mice are known to consistently shed viruses in their feces, 22 and MNV RNA was detected in the feces of sentinels.
We were able to maintain our MNV-negative colony for over three years, despite housing MNV-positive mice in the same rack. Importantly, proper handling of IVC cages, i.e. opening cages under a laminar flow and limiting the direct contact of personnel with animals by using disinfected forceps, eliminates cross contamination. Because the animal caretakers in our study knew the locations of all the MNV-positive cages, we assume that the risk of cross-contamination might possibly be higher under field conditions.
In summary, using MNV as a representative unwanted viral organism that is present in many laboratory animal units, we were able to demonstrate that routine health monitoring performed by sentinel serology does not reliably detect infections at low prevalence rates. Exhaust air particle RT–qPCR is demonstrably more sensitive for detecting MNV at low prevalence rates. For routine use, it is sufficient to test an IVC rack at every prefilter change, normally carried out at one-month intervals, to reliably detect MNV infections. This method requires minimal additional work, while guaranteeing dependable and reproducible results that have already been shown to work with other viruses and bacteria.13,15,16
Footnotes
Acknowledgements
The author(s) thank the animal caretakers for excellent support and the AG Kiermayer for providing MNV-negative mice.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.