
Abstract
Catalyzed reporter deposition (CARD) is a technique that allows amplification of routine immunolabeling in light microscopy. This procedure takes advantage of the horseradish peroxidase (HRP) from an HRP-avidin complex to catalyze the accumulation of reporter-conjugated tyramine (a phenolic compound) onto a surface displaying biotinylated antigen-antibody complexes. The large amount of labeled tyramine deposited allows the detection of an antigenic site with multiple reporter molecules. In this study we modified this amplification protocol to combine it with the immunogold technique for the ultrastructural localization of antigens in electron microscopy. We constructed various tyramide conjugates that permit the combination of this amplification method with a particulate colloidal gold marker. The new probes yield results of high specificity and enhanced intensity. Assessment of the level of resolution of the labeling has demonstrated that, in spite of the amplification, the resolution remains very good. Therefore, once associated, the immunogold and the CARD techniques lead to specific, high-resolution, sensitive and amplified signals that exhibit the advantages of both approaches.
Keywords
P
In 1989, Bobrow et al. introduced the catalyzed reporter deposition (CARD) for solid-phase immunoassays. This novel approach can enhance the signal up to 30-fold compared to standard membrane immunoassays (Bobrow et al. 1991). The method was later adapted to immunocytochemistry at the light (Adams 1992; Berghorn et al. 1994; Merz et al. 1995; Shindler and Roth 1996) and electron (Mayer and Bendayan 1997; Schöfer et al. 1997; Humbel et al. 1998) microscopic levels and to in situ hybridization (Kerstens et al. 1995; Raap et al. 1995; van Gijlswijk et al. 1996; Sano et al. 1998). The CARD amplification system relies on horseradish peroxidase (HRP) molecules, immunologically introduced at the specific antigen-antibody complex site, to catalyze the covalent attachment of a large number of derivatized tyramine molecules onto tissue sections. On oxidation by HRP in the presence of hydrogen peroxide, the phenol moiety of the tyramine forms a radical with a short half-life. This activated tyramine rapidly binds covalently to electron-rich amino acids of proteins immediately surrounding the site of the immunoreaction. Tyramine can be crosslinked to a variety of reporter molecules, such as biotin (Bobrow et al. 1989), digoxigenin (Plenat et al. 1997), trinitrophenyl (Hopman et al. 1998), FITC, rhodamine, and Cy-3 (van Gijlswijk et al. 1997). These deposited reporter molecules are then detected either by streptavidin, by specific antibodies, or directly by their fluorescent signal. Except for the directly detected fluorochromes, the streptavidin or the anti-hapten antibodies can be conjugated to particular markers for visualization of the antigenic sites.
The application of the biotinyl-tyramide amplification procedure developed by Bobrow, followed by streptavidin-gold as the detection step, should, in theory provide gold particle labeling of high intensity. In addition to the enhancement of the signal through the CARD technique, its combination with the gold marker should generate high-resolution labeling that can be quantitated.
In the present communication, new reporter-conjugated tyramine complexes, i.e., tyramide-IgG, tyramide-albumin-gold, and biotinyl-LC-LC-tyramide, were constructed and introduced into the immunogold labeling protocols to associate the advantages of the CARD technique with those of the colloidal gold particulate marker. The combination of this dual approach for the ultrastructural localization of antigenic sites led to specific amplified signals revealed by gold particles. The resolution of the enhanced signal was found to be high enough for localization of membrane-associated proteins. Moreover, the labeling intensity could be quantitated and demonstrated that the amplified signals were about 10-fold higher than those of the standard protein A-gold protocol.
Materials and Methods
Tissue Processing
Fragments of normal rat pancreatic and liver tissues were fixed by immersion with 1% (v/v) glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) for 2 hr at room temperature (RT). The pancreatic tissue was processed for low-temperature embedding in Unicryl as previously described (Bendayan 1995), and the liver tissue was postfixed with 1% osmium tetroxide and embedded in Epon 812 according to standard techniques. Thin sections were cut and mounted on Parlodion-carbon-coated nickel grids and processed for immunocytochemistry using different labeling protocols.
Antisera and Reagents
A rabbit anti-amylase antibody (Sigma-Aldrich Canada; Oakville, Ontario, Canada) was used to assess the different amplification protocols on pancreatic tissue. A rabbit anticarbamoyl phosphate synthetase (CPS) antibody (Bendayan and Shore 1982) was applied on liver tissue to assess the resolution of the labeling. The secondary antibody was a biotinconjugated goat anti-rabbit IgG (GAR-biotin) (Sigma-Aldrich). Streptavidin-gold (10 nm) was from Sigma. Streptavidin-HRP (SA-HRP), biotinyl-tyramide (BT), and the amplification diluent were provided by NEN Life Science Products (Boston, MA). Protein A- and protein A/G-gold (10 nm) were prepared according to previously described protocols (Bendayan 1995).
Preparation of the Probes
All coupling procedures were carried out at RT.
Biotinyl-LC-LC-Tyramide. The biotinyl-long chain-long chain-tyramide probe (B-LC-LC-T) was constructed through sulfosuccinimidyl-6′-(biotinamido)-6-hexanamido hexanoate (sulfo-NHS-LC-LC-biotin) (Pierce; Rockford, IL) and tyramine-HCl (Sigma-Aldrich). A total of 500 μl of 10 mM Sulfo-NHS-LC-LC-biotin in distilled water was added to 500 μl of 10 mM tyramine-HCl in 0.01 M PBS, pH 7.2. The solution was allowed to react for 30 min and was stored at 4C.
Tyramide-IgG. Synthesis of the tyramide-IgG probe was carried out by coupling tyramine-HCl with normal rabbit immunoglobulins (Dako Diagnostics Canada; Mississauga, Ontario, Canada) through the glutaraldehyde method (Harlow and Lane 1988). A total of 250 μl of 60 mM tyramine-HCl in PBS was mixed with the same volume of 20 mg/ml rabbit immunoglobulins in 100 mM NaCl. The crosslinking was performed with 250 μl of 0.2% glutaraldehyde in PBS slowly added dropwise over 30 min with constant stirring. Then the solution was allowed to react for 1 hr with stirring. The excess of glutaraldehyde was neutralized by adding 200 μl of 1 M glycine in PBS (200 mM final concentration) to the mixture which was incubated, under stirring, for 1 hr. The resulting solution was purified and separated through a Sephadex G-25 column (1 × 60 cm; elution with distilled water) (Pharmacia Biotech; Baie d'Urfé, Québec, Canada). The separation yielded two peaks and the fractions of the first peak, containing the tyramide-IgG complex, were pooled and evaporated to dryness with a Speed Vac Plus centrifugal vacuum concentrator (Savant Instruments; Holbrook, NY). The probe was resuspended in 500 μl PBS and stored at 4C.
Tyramide-BSA-Gold. The tyramide-BSA-gold probe was constructed by coupling tyramine-HCl to bovine serum albumin (BSA) (Sigma-Aldrich) and by tagging the tyramide-BSA complex with colloidal gold particles. The procedure involved the preactivation of 2 ml of 60 mM tyramine-HCl in PBS with 2 ml of 0.2% glutaraldehyde in PBS. Then 40 mg of BSA was added to the activated tyramine solution and the mixture was allowed to react for 1 hr with constant stirring. The reaction was stopped by adding 60 mg of glycine (200 mM final concentration) to the coupling solution for 1 hr. The reaction products were separated on a Sephadex G-25 column (1 × 60 cm; elution with distilled water) and yielded two peaks. The first peak was composed of three subfractions: a translucent fraction, a yellowish fraction, and a clear white one. The translucent subfraction, being the one containing the tyramide-albumin complex, was evaporated to dryness in the centrifugal vacuum concentrator and resuspended in 1 ml of distilled water. To couple this complex to gold particles, 9 ml of 10-nm colloidal gold particle suspension (pH 5.8) was added to 500 μl of the tyramide-albumin and centrifuged for 30 min at 25,000 × g. The precipitated tyramide-BSA-gold was recovered at the bottom of the tube, diluted with 500 μl of 0.02% polyethylene glycol in PBS, and stored at 4C.
Labeling Protocols
All labeling protocols were carried out by floating the grids with tissue sections face down on drops of the different reagents. All incubations were performed at RT unless otherwise specified.
Simple Protein A-Gold Protocol. Pancreatic tissue thin sections mounted on nickel grids were incubated for 30 min on a drop of 10 mM PBS containing 1% ovalbumin and then transferred to a drop of diluted anti-amylase (1:200 in PBS) for an overnight incubation at 4C. Tissue sections were then rinsed with PBS for 15 min, transferred to 1% ovalbumin for 15 min, and incubated for 30 min on a drop of the protein A-gold or the protein A/G-gold complex. The grids were thoroughly washed with PBS, rinsed with distilled water, dried, and stained with uranyl acetate before examination with a Philips 410 SL electron microscope.
Biotinyl-Tyramide/Streptavidin-Gold Protocol. Labeling was carried out by floating the grids on drops of 2% BSA in PBS for 15 min and then transferring them directly onto a drop of the diluted anti-amylase (1:200 in PBS) overnight at 4C. The grids were then rinsed with PBS for 15 min, transferred to the BSA solution for 15 min, and incubated on a drop of GAR-biotin diluted 1:100 in PBS for 60 min. Tissue sections were then washed with PBS for 15 min, transferred to the BSA solution for 15 min, and incubated on a drop of streptavidin-HRP diluted 1:500 with PBS for 30 min. The grids were rinsed for 15 min with PBS and incubated for 10 min with the biotinyl-tyramide diluted 1:50 in 1 x amplification diluent. After a 15-min wash with PBS, the tissue sections were incubated on a drop of 1% ovalbumin for 15 min and directly transferred to a drop of streptavidin-gold diluted 1:20 in PBS for 30 min. The grids were then washed with PBS, rinsed with distilled water, dried, and stained with uranyl acetate before examination in the electron microscope.
Biotinyl-LC-LC-Tyramide/Streptavidin-Gold Protocol. The same labeling procedure as described above for the biotinyl-tyramide/streptavidin-gold protocol was applied for the biotinyl-LC-LC-tyramide probe. The only difference was the use of the biotinyl-LC-LC-tyramide (dilution 1:50 in PBS) and the addition of hydrogen peroxide (H2O2) to the biotinyl-LC-LC-tyramide solution at a final concentration of 0.03%.
Tyramide-IgG Protocol. The tissue sections were incubated on a drop of PBS containing 1% ovalbumin for 30 min and transferred to a drop of the diluted anti-amylase (1:200 in PBS) for an overnight incubation at 4C. After rinsing in PBS for 15 min, the sections were incubated with 1% ovalbumin for 15 min and transferred onto a drop of GAR-biotin (1:100 in PBS) for 60 min. The grids were then washed with PBS for 15 min, transferred to the ovalbumin solution for 15 min, and incubated on a drop of streptavidin-HRP (1:500 in PBS) for 30 min. After rinsing for 15 min with PBS, the sections were transferred onto a drop of the tyramide-IgG solution containing 0.03% H2O2 and incubated for 10 min. The tissue sections were then washed in PBS for 15 min, transferred to a drop of 1% ovalbumin for 10 min, and then incubated with the protein A/G-gold complex for 30 min. At the end of this incubation, the grids were thoroughly washed with PBS, followed by distilled water, and allowed to dry. Before examination in the electron microscope, the tissue sections were stained with uranyl acetate.
Tyramide-BSA-Gold Protocol. The tissue sections were first incubated on a drop of 1% ovalbumin in PBS for 30 min and then transferred to the diluted anti-amylase (1:200 in PBS) for an overnight incubation at 4C. The sections were rinsed with PBS for 15 min, transferred to 1% ovalbumin for 15 min, and then incubated on a drop of GAR-biotin (1:100 in PBS) for 60 min. The grids were further rinsed with PBS (15 min), transferred to the ovalbumin (15 min), and incubated with streptavidin-HRP (1:500 in PBS) for 30 min. The sections were then washed with PBS (15 min) and incubated with the tyramide-BSA-gold (10 nm) probe diluted 1:10 in PBS containing 0.03% H2O2, for 10 min. The grids were thoroughly washed with PBS and distilled water, dried, and stained with uranyl acetate before examination.
Control Experiments
The specificity of the labelings was assessed through a number of control experiments. Two types of controls were carried out for (a) assessment of the specificity of the labeling, which included the use of the antigen-adsorbed antibody and the omission of either the primary or secondary antibody, and (b) the assessment of the specificity of the amplification, which included omission of one of the reagents such as the hydrogen peroxide, the streptavidin-HRP, or the conjugated tyramine.
Quantitation
Quantitative evaluations of the labeling over the zymogen granules as well as that over mitochondria, which reflects background staining, were carried out according to previously described protocols (Bendayan 1995). Electron micrographs were recorded and enlarged to a final magnification of x 42,000. Evaluation of the area of the zymogen granules and mitochondria was done by direct planimetry using the Videoplan 2 system (Carl Zeiss Canada; Montréal, Québec, Canada). The number of gold particles present in the granules and mitochondria was counted and the density of labeling was expressed as the number of gold particles/μm2. The labeling densities were evaluated for each of the five protocols and the amplification factor was calculated in reference to the labeling obtained with the simple protein A-gold protocol.
Assessment of Resolution
CPS is a mitochondrial enzyme associated with the inner mitochondrial membrane (Bendayan and Shore 1982). Because of its close association with the mitochondrial membrane, we selected it as a reference for a membrane-bound protein. The proximity of the labeling to the mitochondrial membrane was chosen as a reference for evaluation of the resolution. Sections of rat liver tissue were immunolabeled with anti-CPS using the simple protein A-gold and the tyramide-IgG protocols, as described above. To increase ultrastructural preservation, labelings were carried out on osmium-postfixed liver tissue embedded in Epon. The use of such material required an additional preliminary step in the labeling protocol, i.e., the use of metaperiodate (Bendayan 1995). Therefore, the tissue sections were incubated with a saturated sodium metaperiodate solution for 60 min and rinsed in water before the first antibody step. Evaluation of the resolution was assessed by measuring the distance between the gold particles and the inner mitochondrial membrane for both the simple and amplified protocols using the Videoplan 2 image analysis system.
Results
Four new tyramide protocols were assessed to evaluate the possibility of combining the CARD amplification approach with the immunogold technique to amplify immunocytochemical signals at the electron microscopic level. Working on a single tissue with the same antibody and maintaining the antibody's dilution as well as all other procedures constant allows the comparative study of the amplification signals obtained with the different probes. The immunolocalization of amylase in pancreatic tissue with the CARD amplification technique using either of the conjugated tyramine probes yielded labeling by colloidal gold particles over cellular compartments of pancreatic cells known to contain this antigen (Figures 1 and 2). The labeling was located over the rough endoplasmic reticulum, the Golgi apparatus, the condensing vacuoles, and the zymogen granules (Figure 2). Labeling was also found in the acinar lumen. Compared to the simple protein A-gold protocol, the use of any of the tyramide protocols yielded more intense labeling. However, differences were detected among the amplified signals obtained with the different CARD protocols. The amplified signal obtained with the biotinyl-tyramide was not as intense as those obtained with the other protocols, and background staining was present over mitochondria and nuclei. In contrast, the use of the biotinyl-LC-LC-tyramide probe led to much higher labeling, indicating that the incorporation of spacer arms between the tyramide and the biotin molecules overcomes the possible problems of steric hindrance presented by the biotinyl-tyramide probe (Table 1). Nevertheless, staining of amylase with the biotinyl-LC-LC-tyramide probe was also accompanied by relatively high background staining. Levels of background appeared to decrease from regions around zymogen granules to peripheral regions of the cell. The biotinylated probes therefore appear to display problems of diffusion leading to such background. These problems were solved by the addition of 10% dextran sulfate to the tyramide solution (van Gijlswijk et al. 1996), which indeed reduced the background staining. The other two protocols, the tyramide-IgG and the tyramide-BSA-gold, yielded labeling of very high intensity and specificity with adequate background stainings (Figures 1A and 1B).
Quantitative evaluation of the density of labeling for amylase over zymogen granules and mitochondria allowed us to perform a more thorough evaluation of the degree of signal amplification and of levels of background staining. Striking differences were observed among the protocols (Table 1). The immunogold labeling intensity obtained with the CARD system was four- to 13-fold higher than that of the conventional protein A-gold technique, depending on the type of tyramide probe used. The biotinyl-tyramide and the biotinyl-LC-LC-tyramide generated labeling with amplification factors of 4 and 12, respectively, compared to the standard protein A-gold. However, both amplification protocols gave high background over mitochondria. The shortest protocol, which uses the tyramide-BSA-gold, yielded excellent labeling intensity with an amplification factor 7 times that of the nonamplified signal. Moreover, the background over mitochondria remained very low. The method using the tyramide-IgG probe also showed a very high increase in the signal, with an amplification factor of 13 compared to the simple protein A-gold protocol. The background level over mitochondria remained excellent, demonstrating a very high specificity of the reaction.
Several control experiments were carried out to assess the specificity of the results obtained with the different amplification protocols (Table 2). Among them, antigen adsorption and omission of the first or second antibody served as controls for the specificity of the immunolabeling itself. Omission of one of the reagents needed for the amplification, such as the hydrogen peroxide, the streptavidin-HRP, or the tyramide, served as controls for the amplification protocols. The results obtained confirmed the high specificity of the labeling (Table 2). Among the controls of amplification, some resulted in absence of labeling and others displayed low signals. Omission of the biotinyl-tyramide or the biotinyl-LC-LC-tyramide gave low signals, which result from the normal binding of streptavidin-gold with the biotinylated secondary antibody. The tyramide-IgG protocol uses the protein A/G-gold complex to detect the Fc fragments of immunoglobulins. Therefore, even in the absence of the secondary antibody, the protein A/G-gold was able to recognize the first antibody giving a low signal, similar to that obtained with the standard protein A-gold protocol (Table 2).
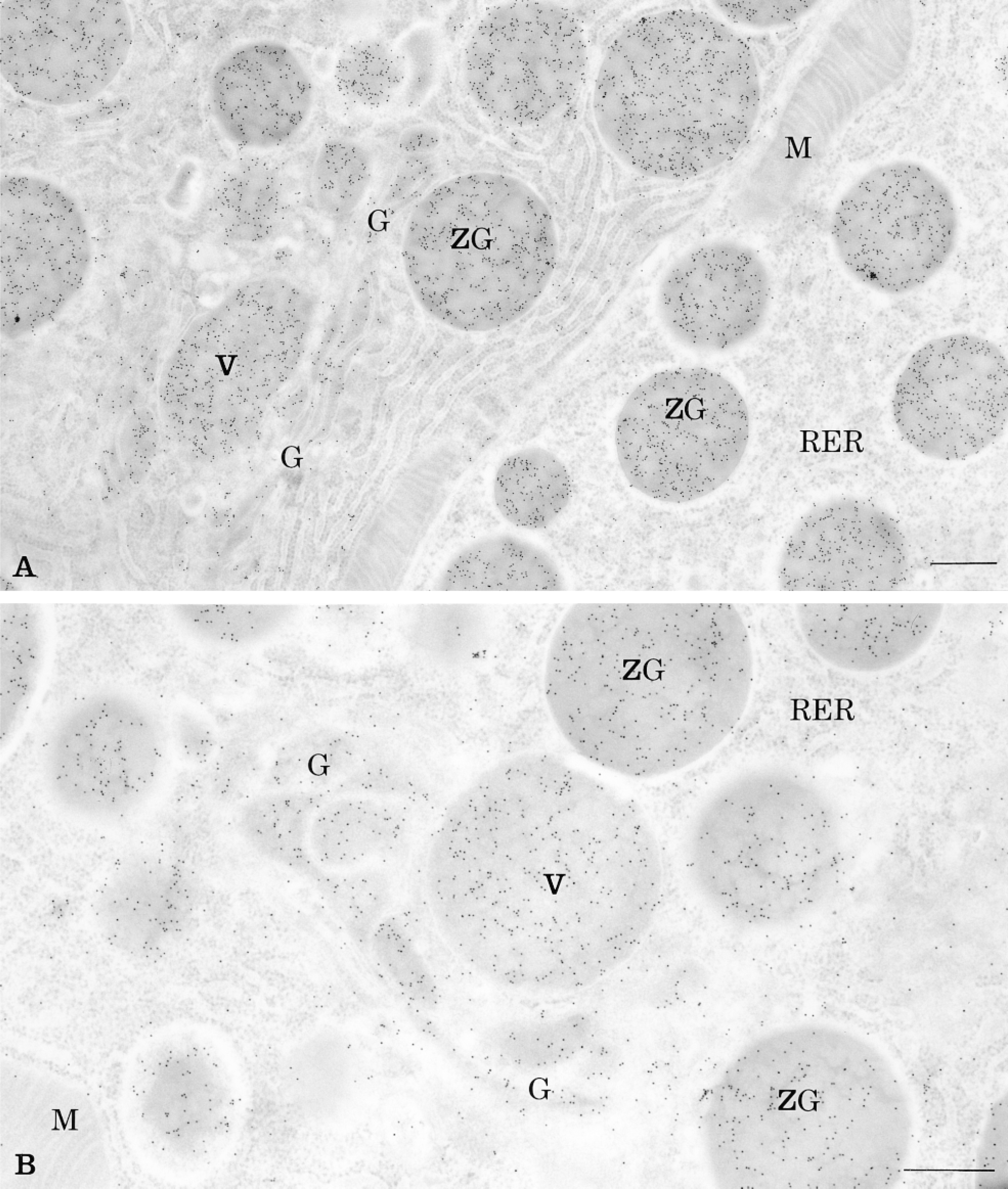
Thin section of rat pancreatic acinar cell labeled for amylase by the CARD technique using the tyramide-IgG (A) and the tyramide-BSA-gold (B) protocols. The amplified colloidal gold signal, revealing amylase antigenic sites, is present over the rough endoplasmic reticulum (RER), the Golgi apparatus (G), the condensing vacuoles (V), and the zymogen granules (ZG). Mitochondria (M) are almost devoid of labeling. Bars = 0.5 μm.
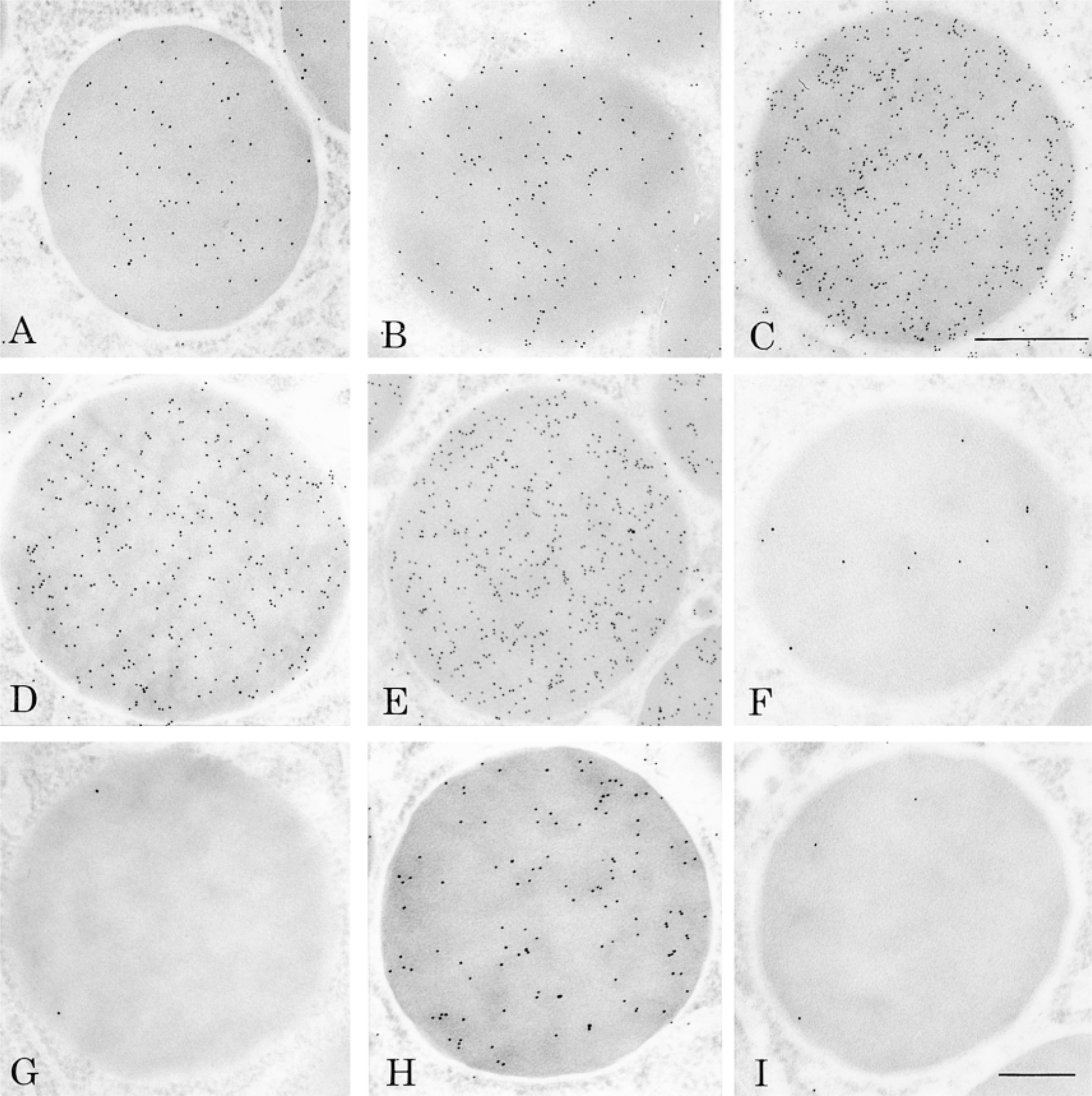
Zymogen granules labeled for amylase demonstrating the labeling obtained with simple (A) and amplified protocols (B-E). (A) The labeling obtained with the standard protein A-gold technique provides a positive control for comparison with the amplified protocols. Enhanced immunogold signals were obtained with the biotinyl-tyramide (B), the biotinyl-LC-LC-tyramide (C), the tyramide-BSA-gold (D), and the tyramide-IgG (E) protocols. (F-I) Results under control of specificity and of amplification. (F) Biotinyl-tyramide protocol; omission of the biotinyl-tyramide incubation. The signal is generated by the binding of streptavidin-gold to the biotinylated secondary antibody. (G) Tyramide-BSA-gold protocol; omission of H2O2 prevents deposition of the probe, leading to absence of labeling. (H) Tyramide-IgG protocol; omission of the anti-amylase primary antibody leads to absence of labeling. (I) Tyramide-IgG protocol; omission of the tyramide probe. The labeling is due to the binding of protein A/G-gold to the primary and secondary antibodies. A, C, D, E are at the same magnification. Bars: A,C-E = 0.5 μm; B,F-I = 0.25 μm.
The resolution of the labeling after amplification was assessed by the study of a mitochondrial membrane-associated protein, carbamoyl phosphate synthetase. This enzyme was previously demonstrated to be closely associated with the inner mitochondrial membrane (Bendayan and Shore 1982). Figure 3 shows the labeling obtained after incubation with concentrated (Figure 3A) and diluted (Figure 3B) anti-CPS antibody using the tyramide-IgG protocol. The distances between the cristae membrane and the gold particles were measured (Figure 4), and the distribution of these measurements allowed a good assessment of the level of resolution. Figure 4 shows that for both the simple (protein A-gold) and the amplified technique, the majority of the labeling is indeed located within 10 nm of the membrane. Figure 4 also shows that the average distance between the gold particles and the inner mitochondrial membrane is slightly higher for the amplified technique. The average distance between the gold particles and the membrane was found to be 8.7 nm for the simple protocol and 11.2 nm for the amplified protocol. This demonstrates that, in spite of the amplification, the resolution remains quite good.
Density of labeling for amylase in pancreatic cells: amplification protocols a
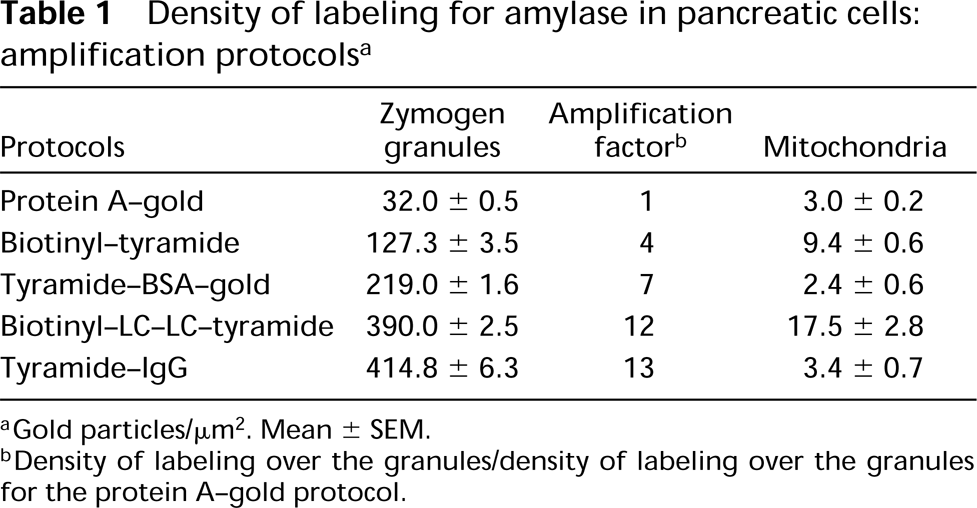

aGold particles/μm2. Mean ± SEM.
bDensity of labeling over the granules/density of labeling over the granules for the protein A-gold protocol.
Discussion
The CARD technique, first described by Bobrow et al. in 1989, was used to enhance the sensitivity of immunoassays and Western blots and to increase signal intensity (Bobrow et al. 1991). The CARD approach was then extended to immunocytochemistry at the light microscopic level (Adams 1992; Berghorn et al. 1994) and to in situ hybridization, in which it compared favorably to in situ PCR (Zehbe et al. 1997). Recently, the technique was broadened to immunoelectron microscopy (Mayer and Bendayan 1997; Schöfer et al. 1997). The technique uses HRP molecules introduced at specific antigenic sites to catalyze the accumulation of reporter-conjugated tyramine by a radicalar mechanism.
A number of different reporter-conjugated tyramines, useful in immunocytochemistry and in situ hybridization, have been described and are currently used (Plenat et al. 1997; van Gijlswijk et al. 1997; Hopman et al. 1998). In the present study, new tyramide probes were constructed and tested to combine the CARD with the immunogold approach and to exploit the advantages displayed by the gold marker in an amplification protocol. The studies and the assessment of the new protocols were carried out on wellestablished systems, the ultrastructural localization of amylase in pancreatic acinar cells and its distribution along the secretory pathway. Using this system under conditions in which tissue preparation and primary antibody dilution were maintained constant allowed a good assessment of the efficiency of each of the new protocols. Successful enhancement of the labeling intensity was registered with all the tyramide probes tested, although differences were detected among them in terms of quality of the end results. The biotinyl-tyramide generated a signal higher than the standard protein A-gold but, probably because of steric hindrance between the biotinyl-tyramide and the streptavidin-gold, the level of amplification remained low. The biotinyl-LC-LC-tyramide probe was therefore constructed to overcome the problem of steric hindrance among reagents. Spacer arms, which are 30-Å hydrocarbon chains, were incorporated between the tyramine and the biotin molecules. The use of this biotinyl-LC-LC-tyramide in combination with the streptavidin-gold complex resulted in a density of labeling threefold higher than that generated by the biotinyl-tyramide probe and 12-fold higher than the standard protein A-gold. However, both biotinylated probes, the biotinyl-tyramide and the biotinyl-LC-LC-tyramide, gave relatively high background levels that appeared to be due to diffusion problems. Indeed, the background was higher over those mitochondria located close to cell compartments containing amylase, indicating that either during incubation or during the washing steps the biotinylated reagents tend to diffuse away from their sites of deposition. To decrease this background, the addition of dextran sulfate was required (van Gijlswijk et al. 1996). The effect was concentrationdependent. Increasing the concentration of dextran to 20% abolished the diffusion artifact and the background but also reduced the amplification. A concentration of 10% dextran sulfate was found to yield the best results, compromising between amplification and background. The overall intensity of the labeling was lower with the use of dextran but still yielded results with very good signal-to-noise ratio.
Controls of the specificity of the immunolabeling and of the amplification protocols
aControl of specificity.
bControl of amplification.
cPositive result (without amplification) due to binding of the protein A-gold to the primary antibody.
To overcome the problems displayed by the biotinylated tyramine probes, other tyramide conjugates were developed to enhance the sensitivity of the labeling and to reduce background staining. The tyramide-BSA-gold probe shortens the CARD amplification protocol, the colloidal gold marker being directly coupled to the enzyme's substrate. It is directly catalytically deposited at the specific antigenic site without the need for further steps to detect the reporter molecules. The use of such a probe generated intense labeling with very low background staining. On the other hand, the highest immunocytochemical signal amplification was obtained with the tyramide-IgG complex. When activated by the HRP, this probe led to the accumulation of immunoglobulin molecules at specific antigenic sites. The accumulated tyramide-IgG molecules are then detected by the protein A/G-gold complex, which reacts specifically with the Fc fragment of IgGs, leading to very intense and very convincing labeling. Moreover, mitochondria and nuclei were almost devoid of labeling, reflecting the high specificity of the results. In fact, the specificity of the results generated by all the protocols tested was assessed by a number of control experiments and confirmed the validity of both the antibody steps and the amplification protocols.
Concerning the resolution of the amplified signals, one could wonder if, because of the various steps in the protocols, the distance between the gold label and the real antigenic site would increase significantly. To assess this, distances between gold particles and membrane antigenic sites were measured under labeling by the simple standard and the CARD-amplified protocols. Detection of a mitochondrial membrane-associated protein with the tyramide-IgG protocol has demonstrated that the amplified signal was still quite close to the membrane and that the resolution obtained with the tyramide-IgG probe, compared to the simple protein A-gold technique, remained very good.
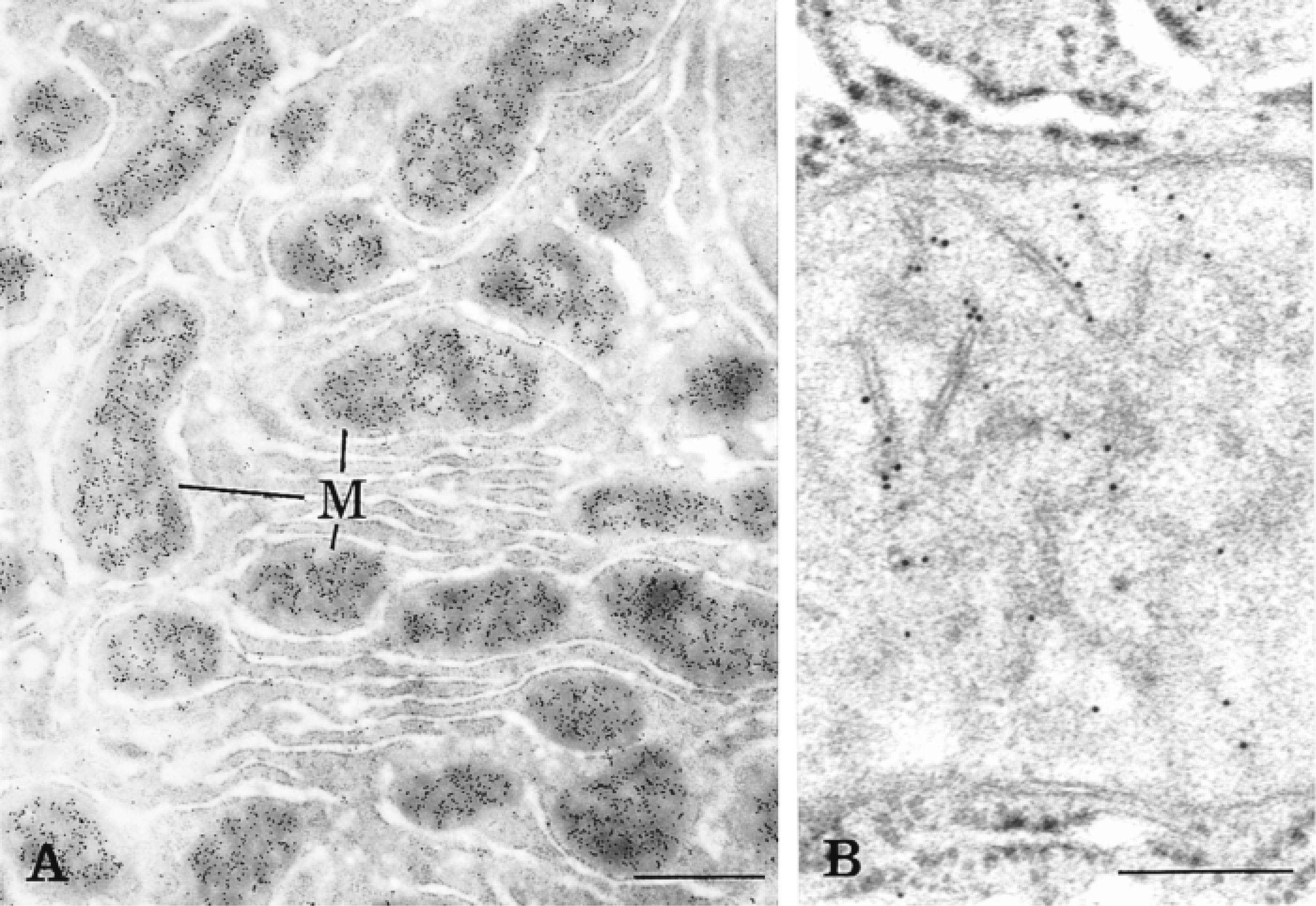
Thin section of rat liver tissue labeled for CPS by the tyramide-IgG protocol using a rather concentrated (A) and diluted (B) anti-CPS. The labeling by gold particles is present over mitochondria (M), particularly associated with the inner mitochondrial membrane. Bars: A = 1 μm; B = 0.25 μm.
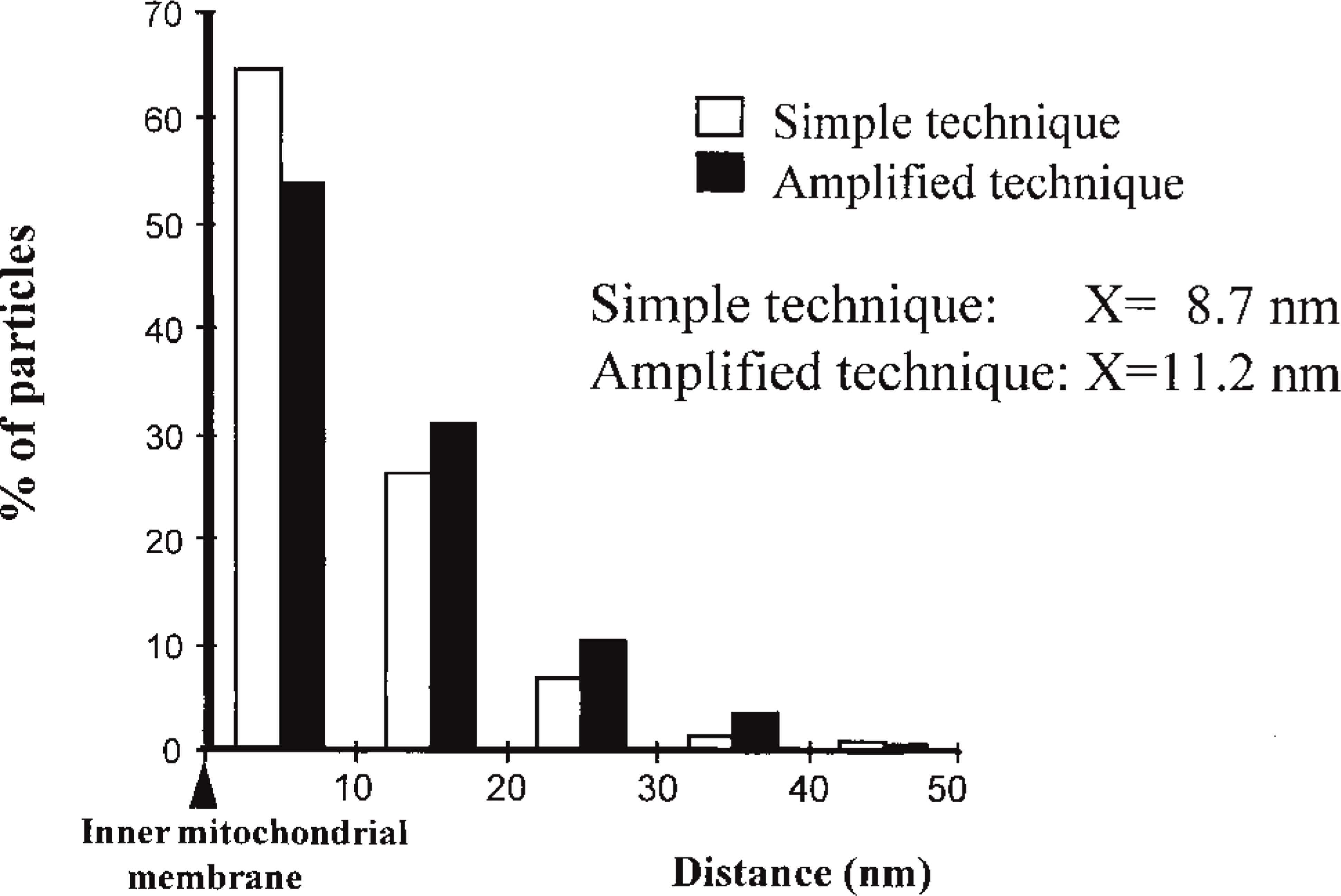
The evaluation of the resolution was assessed by measuring the distance between the gold particles and the closest membrane. Two labeling techniques were compared: the simple protein A-gold technique and the amplified tyramide-IgG technique. In general, the distance between the gold particles and the inner mitochondrial membrane appears slightly larger for the amplified technique.
The introduction of the gold marker by construction of various new tyramide conjugates and modification of the CARD technique have enabled us to take advantage of the properties of both protocols: the amplification of the signal on the one hand, useful for low labeling, and the resolution and possibilities of quantitation due to the particulate marker on the other hand. The end results have proved that this combination led to significant improvements in immunogold techniques.
Footnotes
Acknowledgements
Supported by grants from the Medical Research Council of Canada.
We thank Patricia Mayer from NEN Life Science Products (Boston, MA) for donating the Renaissance TSA indirect kit. We also thank Johanne Chaêney for secretarial assistance in the preparation of the manuscript and Jean Léveillé for the photographic work.