
Abstract
Seventy paraffin-embedded cervical biopsy specimens and condylomata were tested for the presence of human papillomavirus (HPV) by conventional in situ hybridization (ISH) and ISH with subsequent signal amplification. Signal amplification was performed either by a commercial biotinyl-tyramide-based detection system [GenPoint (GP)] or by the novel two-layer dextran polymer visualization system EnVision+ (EV), in which both EV–horseradish peroxidase (EV–HRP) and EV–alkaline phosphatase (EV–AP) were applied. We could demonstrate for the first time, that EV in combination with preceding ISH results in a considerable increase in signal intensity and sensitivity without loss of specificity compared to conventional ISH. Compared to GP, EV revealed a somewhat lower sensitivity, as measured by determination of the integrated optical density (IOD) of the positively stained cells. However, EV is easier to perform, requires a shorter assay time, and does not raise the background problems that may be encountered with biotinyl–tyramide-based amplification systems. (
Keywords
I
Despite these improvements in sensitivity ISH usually detects only medium to high copy number nucleic acid targets. In contrast to ISH, in situ PCR (IS-PCR) allows the detection even of low copy number nucleic acids, but this method is much more cumbersome and is often hampered by poor reproducibility and morphology, especially when applied to paraffin-embedded tissue. Recently, a new system for signal enhancement in enzyme immunoassays (EIA) and immunocytochemistry (IHC) using the water-soluble dextran polymer conjugate EV has been described (Cartun 1995; Heras et al. 1995; Bisgaard and Pulzek, 1996; Sabattini et al. 1998; Vyberg and Nielsen, 1998). To evaluate this novel method for its signal enhancement capabilities in ISH, we tested 70 formalin-fixed, paraffin-embedded cervical biopsy specimens and condylomata for the presence of HPV infection, using either a commercial biotin-labeled DNA probe (PathoGene; Enzo, Farmingdale, NY) or a cocktail of 5′-digoxigenin-labeled oligonucleotide probes (Figures 1–3). EV was compared with both conventional enzyme-labeled anti-hapten detection and signal amplification using a commercial biotinyl tyramide-based detection system (GenPoint; Dako, Hamburg, Germany) (Speel et al. 1999). Thereby, we established an optimized protocol for the application of EV in ISH, the principle of which is displayed in Figure 4.
Materials and Methods
Sample processing from fixation up to permeabilization was performed as described by Wiedorn et al. (1999a).
Permeabilization was done either by incubation with 250 μg/ml Proteinase K (Roche Diagnostic; Mannheim, Germany) for 15 min at 37C or by incubation in Target Retrieval Solution (TRS) (S1700; Dako) at 95C for 30 min. Thereafter sections were incubated at room temperature (RT) twice for 5 min in 0.1 M Tris-NaCl, pH 7.5.
Quenching
If peroxidase/AEC was used as a detection system, inactivation of endogenous peroxidase was performed by incubation in peroxidase blocking reagent (S2001; Dako) for 30 min at 37C followed by a 1-min incubation each in 50%, 70%, and 90% ETOH at RT. Slides were dried for 5 min at 37C.
ISH
ISH was performed on a MISHA (Shandon; Frankfurt, Germany) in situ thermocycler using either a commercial DNA probe (PathoGene) according to the recommendations of the manufacturer or a cocktail of 5′-DIG-labeled oligonucleotides as described previously (Wiedorn et al. 1999a).
Evaporation control was performed as described elsewhere (Wiedorn et al. 1999a, b).
Posthybridization washes were performed according to the recommendations of the manufacturer for the commercial DNA probe or once in 2 × SSC, 0.1% SDS at RT for 10 min, followed by two washes in 1 × SSC, 0.1% SDS at 55C for 15 min.
Detection
Unless explicitly stated, all incubations were performed at ambient temperature. For probes handled with EV the procedure was as follows: three washes in 1 × TBST (S3306; Dako) for 1 min; 30-min incubation in protein block (X909; Dako) at 37C; 30-min incubation in primary anti-biotin antibody diluted in antibody diluent (S0809; Dako); either an unconjugated monoclonal mouse antibody (M0743; Dako) diluted 1:20 or an enzyme-conjugated polyclonal rabbit antibody diluted 1:30 (HRP conjugate P5106, AP conjugate D5105; both from Dako) was used; 3 incubations in 1 × TBST for 1 min; 30-min incubation with the EV conjugate at 37C. The primary antibody was detected using either EV–anti-mouse–HRP (K4001; Dako) or EV–anti-mouse–rabbit–AP (K4018; Dako), three incubations in 1 × TBST for 1 min. Staining was completed by incubation in NBT/BCIP, Fast Red, New Fuchsin for AP detection or AEC+ (K3469; Dako) for HRP detection, and counterstaining with eosin and hemalum, respectively, for 30 sec. Probe detection with GP was performed with one cycle of biotinyl–tyramide incubation as described previously (Wiedorn et al. 1999a, 2000).
Slides hybridized with the PathoGene probe were treated according to the recommendations of the manufacturer if not processed with EV. Slides hybridized with the DIG-labeled oligonucleotide cocktail were treated with anti-DIG–AP or anti-DIG–HRP (Roche Diagnostic) diluted 1:100 for 30 min at 37C after to the blocking reaction.
Controls
Several controls, including solution-phase PCR (Wiedorn et al. 1999a), were performed to ensure the specificity of the amplification reaction.
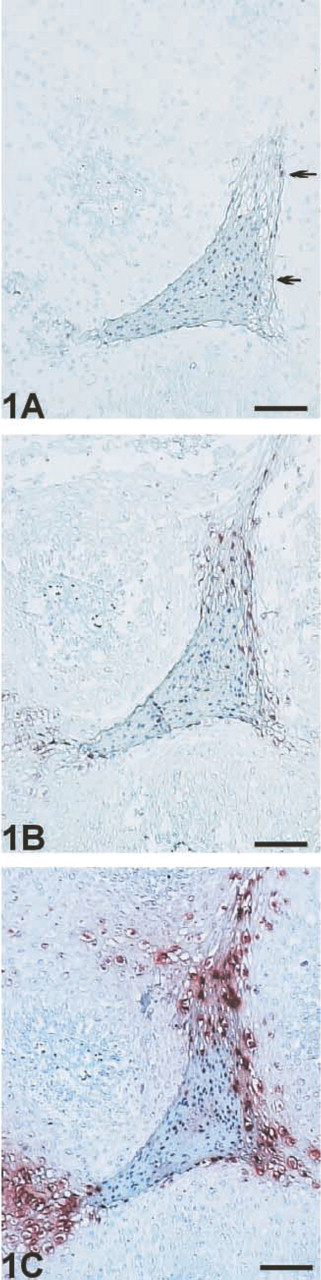
ISH with and without signal enhancement. (
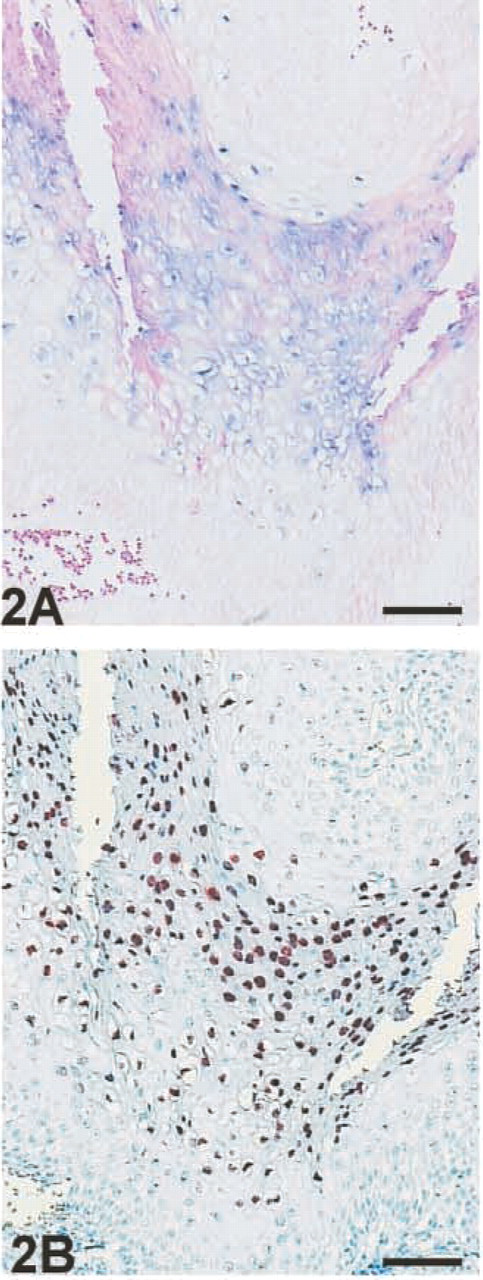
Comparison of signal intensity between EV–AP/NBT/BCIP and EV–HRP/AEC+; serial section of cone of cervix. (
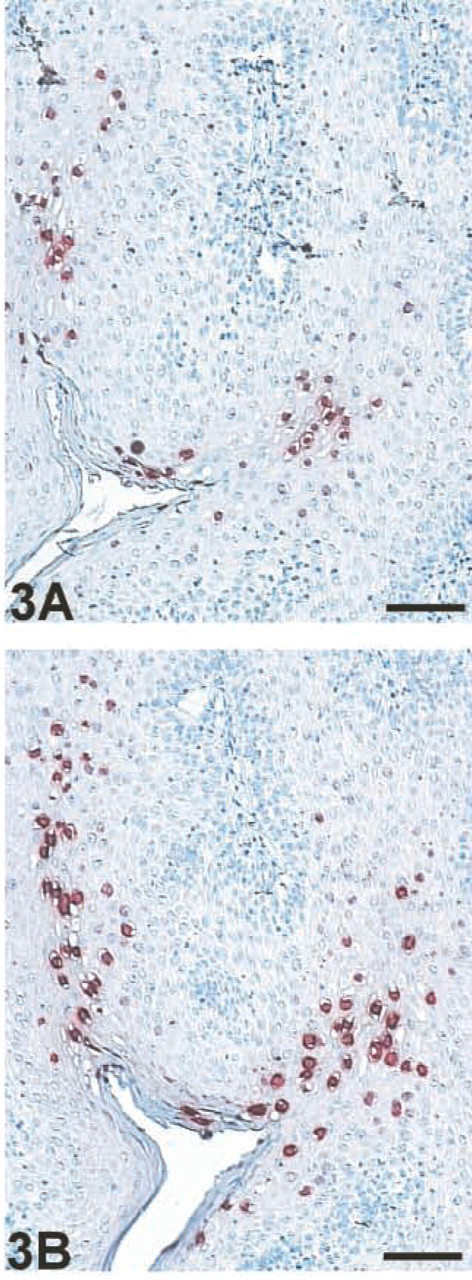
Comparison of signal intensity between EV–HRP (
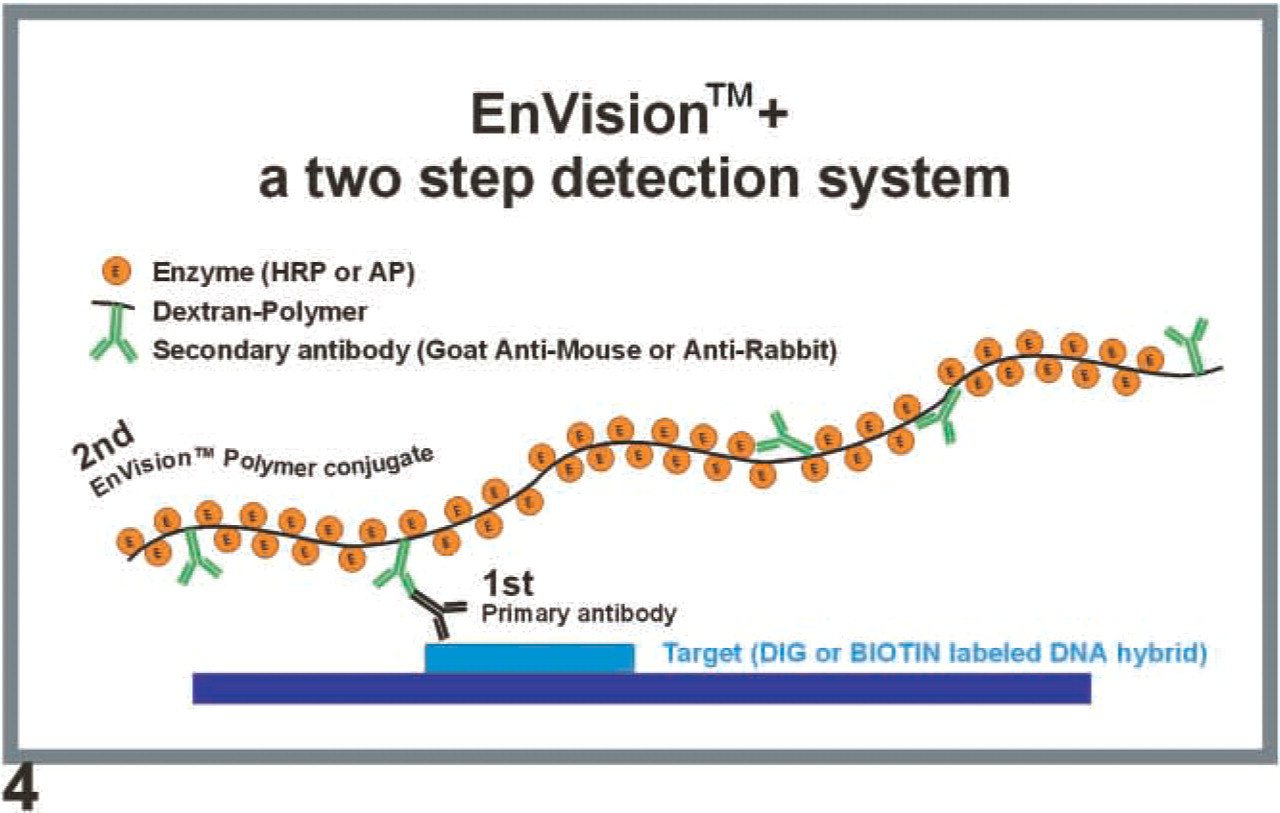
Principle of EnVision +. EV consists of a hydrophilic polymeric backbone (dextran) to which, in contrast to traditional conjugates, multiple (i.e., up to 100) enzyme molecules such as peroxidase or alkaline phosphatase as well as several secondary antibodies, have been coupled. During detection the tissue is first incubated with a primary mouse or rabbit anti-DIG or anti-biotin antibody, which adheres to the labeled DNA hybrid. In the subsequent incubation the goat anti-mouse or goat anti-rabbit antibody of the EV polymer conjugate binds to the primary antibody, delivering not only one or two enzyme molecules for the subsequent color reaction but up to 100 per polymer, thus dramatically increasing the signal intensity.
Determination of Staining Intensities
Staining intensities of the chromogen were determined by measuring the IOD using the image analysis software ImagePro Plus3.01 (Media Cybernetics; Silver Spring, MD), which was adopted for cell detection by applying the integrated macro programming and VisualBasic 5.0 (Microsoft Corporation; Munich, Germany) (Wiedorn et al. 1998).
Results
To ensure an optimal comparison between conventional ISH and ISH with subsequent signal enhancement by EV, in all experiments the same enzyme-conjugated primary antibody was used as the first layer. EV–HRP revealed a dramatic increase in signal intensity (Figure 1), exhibiting strong distinct signals without background staining. However, with EV–AP no specific staining was generated (data not shown). This phenomenon was also reproducible with HRP- or AP-conjugated antibodies from other manufacturers.
Only the application of unconjugated antibodies resulted in a significant increase in signal intensity using EV–AP, although the degree of amplification was less than that observed with EV–HRP (Figure 2) for all chromogens tested. In contrast to EV–HRP, EV–AP often generates a filamentous background staining even after short incubation periods with the chromogen. This background staining could be reduced by a 30-min pre-incubation with a protein block at 37C. This additional step was not required for EV–HRP.
Conditions initially optimized for ISH without signal enhancement could be maintained for EV–HRP detection. However, for EV–AP detection, either a prolonged incubation with proteinase K or a two- to threefold higher concentration of proteinase K was required for optimal results. For example, after doubling of the proteinase K concentration during permeabilization, EV–AP exhibited an increased signal, whereas signal intensity with EV–HRP decreased, as could be expected after overdigestion (data not shown). Nevertheless, even after optimization of the permeabilization step, EV–AP detection showed a lower sensitivity than EV–HRP (Figure 2) and often resulted in an inferior morphology due to the extended and more aggressive protease treatment.
When EV–HRP and EV–AP are compared, the former shows significantly shorter incubation periods for the chromogen to give equivalent signal intensities. However, prolonged incubation with the chromogen intensifies the diffusion artifact that is especially inherent to the slow NBT/BCIP reaction and therefore impairs spatial resolution (Figure 2). However, EV–AP in combination with NBT/BCIP exhibits stronger staining than Fast Red or New Fuchsin (data not shown).
The signal amplification power of EV was estimated to be approximately 50- to 100-fold. Forty percent of samples that were negative with conventional ISH showed a positive signal with an optimized procedure using EV–HRP. All positive results after signal enhancement could be verified by conventional solution-phase PCR as true positives (data not shown).
When the signal intensity achieved by EV–HRP was compared to that of the biotinyl–tyramide-based amplification system, GP exhibited an IOD of two- to 10-fold higher as determined by the Image Analysis Program IPP3.01 (Figure 3).
Comparing the commercial probe with the oligonucleotides, they showed the same results, with the exception that the latter usually exhibited a lower signal intensity (data not shown).
Discussion
Our study clearly demonstrates for the first time that the novel dextran polymer-based detection system EV, which up until now has only been reported to enhance sensitivity in immunoassays and immunohistochemistry, is also a potent tool for signal amplification in combination with ISH, thus dramatically increasing sensitivity and shortening assay times.
Whereas Vyberg and Nielsen (1998) stated that after adjustment of the primary antibody concentrations the staining results revealed by the tested multilayer techniques LSAB, VABC, and EV were almost identical with respect to staining intensity and number of cells stained, Sabattini et al. (1998) and Pileri and Sabattini (1997) showed that EV exhibited a superior sensitivity compared to the APAAP, ChemMate, LABC, and SABC methods. However, in comparison to the biotinyl–tyramide-based amplification system CSA, Sabattini found that for 53 of 54 tested antibodies EV appeared to be as effective as CSA. This is in contrast to our results, in which we could show that EV is clearly superior to conventional ISH but usually does not achieve the same sensitivity as the biotinyl–tyramide-based GP. In comparing sensitivity as measured by the determination of the IOD of the positively stained cells using the adopted image analysis software ImagePro Plus3.01 (Wiedorn et al. 1998), GP exhibited a two- to 10-fold higher IOD. Therefore, GP not only exhibits stronger signals but also shows more positively stained cells and often a greater stained area per cell, whereas with EV and GP we found a good spatial resolution with distinct signals.
However, in contrast to GenPoint, EV exhibited only minor unspecific background staining, especially when proteinase K was used for permeabilization, which did not interfere with correct interpretation as confirmed by solution-phase PCR. In this case, Gen-Point may yield high background levels, which can be reduced by applying TRS for permeabilization, although this will simultaneously decrease sensitivity. This result is in agreement with the findings of Vyberg and Nielsen (1998) who only in a few cases found faint non-specific staining when using EV.
Especially when used with HRP and AEC+ as chromogen, EV shows a distinct staining pattern of individual cells without loss of spatial resolution. However, this problem will likely arise in applying EV–AP, preferably when used in combination with NBT/BCIP, as a diffusion artifact that is inherent to the relatively slow NBT/BCIP reaction (DeBlock and Debrouwer 1993; Wiedorn et al. 1999a). However, diffusion artifact with subsequent loss of spatial resolution and background staining is not as strong as that observed with AP- and tyramide-based amplification systems (Wiedorn et al. 1999a).
In two other studies (Cartun 1995; Heras et al. 1995), EV exhibited a sensitivity comparable to that of the avidin–biotin techniques. However, Heras et al. (1995) also showed that, on the one hand EV was more sensitive than LSAB but, on the other hand, with some antibodies requiring proteolytic digestion or target unmasking EV revealed slightly weaker signals than LSAB, possibly due to problems of tissue penetration of the labeled polymer. These results correspond with the differences we have found between EV–HRP and EV–AP. We found that EV–AP usually requires two- to threefold the concentration of proteinase K during permeabilization than is necessary for EV–HRP to get optimal signal intensities. Because EV–AP, with a molecular weight of approximately 10,000 kD, is more than twice as large as EV–HRP, these results may be explained by an improved permeabilization of the nucleus, which may consequently improve the accessibilty of the DNA hybrids to the antibody and dextran polymer.
In conclusion, EV–HRP in combination with AEC + appears to be the method of choice with regard to handling, spatial resolution, and background staining. EV–HRP is also suitable for signal enhancement after IS–PCR using the same protocol. The use of EV will enable the application of ISH even for the detection of low-copy nucleic acids and therefore confine the requirement for the application of the much more cumbersome method of IS-PCR. It also enables shorter assay times compared to conventional ISH- or tyramide-based amplification systems and easier handling (with respect to GP), although not showing the same amplification power as GP, which will yield a two- to tenfold higher IOD.