
Abstract
New plastic resins are gradually replacing traditional paraffin-embedding in situ hybridization (ISH) strategies. One unique resin that has not been fully investigated or exploited with respect to light microscopic ISH is a methacrylate mixture. The advantage of this resin is its ability to be removed from tissues postsectioning, dramatically increasing hybridization signal compared to that obtained in other plastics. The goal of this study was to evaluate the general applicability of the methacrylate embedding acetone de-embedding (MEADE) technique for ISH investigations. Several high-resolution, high-sensitivity ISH protocols are described, using both end-labeled oligonucleotides and randomly primed DNA probes (200–400
Keywords
I
Methacrylates have been used in the past as embedding media for immunolabeling and ISH studies. Although sensitivity of RNA detection was reduced (Jamrich et al. 1984), morphological detail of methacrylate-embedded tissues was superior to that of frozen or paraffin-embedded sections. Sensitivity was substantially improved in later studies by removing the methacrylate resin with acetone before probing (Harris and Rubin 1987; Gubler 1989; Baskin et al. 1992). This MEADE technique was initially applied to localize tubulin distribution in sea urchin eggs by immunofluorescence. Further refinement allowed similar detection of actin and tubulin in both plant and fungal species (Baskin et al. 1992,1994; Czymmek et al. 1996).
Kronenberger et al. (1993) extended the application of MEADE technology with the use of digoxigenin (DIG)-labeled rRNA probes and phosphatase-labeled secondary antibodies for colorimetric detection in Arabidopsis thaliana. They reported superior preservation of tissues and ultrastructure in fragile seedling tissues as well as high signal development. Despite the success of this study, methacrylate de-embedment has not been applied in other published ISH studies.
Our specific goals in this study were to extensively examine the utility of the MEADE technique in association with light microscopic ISH. Our research group required a plastic embedding protocol that would provide high morphological integrity and allow localization of low copy transcripts in marine invertebrate larvae. MEADE ISH was first applied to detect bacterial endosymbionts in the gills of the protobranch bivalve Solemya reidi. Although a previous ISH study localized dense concentrations of these symbionts in paraffin sections of adult gill filaments, the resolution and sensitivity of ISH were not sufficient to localize low-copy symbionts in poorly preserved paraffin-embedded larval tissues. MEADE ISH was applied in a second system to localize byssal thread collagen transcripts in the common blue mussel, Mytilus edulis. Future detection of transcripts for the collagen precursor preCol-P in M. edulis larvae also requires a sensitive ISH protocol with high resolution.
Both DIG- and biotin-labeled oligonucleotide probes and DIG-labeled double-stranded, randomly primed DNA probes were tested with MEADE ISH. Indirect chromogenic and fluorescent methods, as well as signal amplification by catalyzed reporter deposition (CARD) with the Renaissance Tyramide Signal Amplification (TSA) Kit, were successfully applied. The results have demonstrated the broad applicability of the MEADE technique and will enhance future ISH studies with methacrylate resins.
Materials and Methods
Tissue Preparation
S. reidi gill filaments and M. edulis foot tissue were fixed overnight in 4% paraformaldehyde (pH 7.6), 0.5 M NaCl, 1 × MOPS (pH 7.0) at 8C. Tissues were washed in 1 × MOPS/0.5 M NaCl, aldehyde-quenched in 0.15 mM NH4Cl in wash buffer, and dehydrated through a graded ethanol series (50–100%). From 100% EtOH, samples were transferred through an ethanol-methacrylate series (2:1, 1:1, 1:2 at −20C) before two or three changes in 100% methacrylate [80% butyl-methacrylate, 20% methyl-methacrylate, 0.5% (w/v) benzoin ethyl ether, and 10 mM DTT] and overnight storage (Baskin et al. 1992,1994; Kronenburger et al. 1993).
Polymerization occurred in gelatin capsules 15 cm from the UV light source (two bulbs, 6 W each) at −8C for 12 hr. Block brittleness was reduced by placing six sheets of paraffin between a UV light and the samples. Wet sections (1.5–2 μm thick) were transferred onto 10-well hydrophobically coated glass slides with bioadhesive surface treatment (Erie Scientific; Portsmouth, NH), flattened with chloroform vapors, and dried.
Probes
S. reidi Probes. Oligonucleotide probes were labeled with either DIG-ddUTP or biotin. The bacterial specific probe 338R (GCT GCC TCC CGT AGG AGT; Amann et al. 1990) was used as a positive control to assess the presence and accessibility of bacterial target. The symbiont-specific oligonucleotide SR-1254 (CAA CGG TTG GAC GCT TCC; Cary 1994) was derived from 16S rRNA sequence. The DIG-labeled non-homologous probe 13B-1242R (G GAA GGA CAA AGA GAA GCA; Cary et al. 1997) was specific to a member of the episymbiotic bacterial community associated with the hydrothermal vent polychaete Alvinella pompejana and served as a negative control. The nonhomologous biotinylated probe Mefp-3.5T [CCA CCT CTG TAA CGT CTT GGA GG(AG) CCA TA; provided by R. Floriolli] was specific to Mytilus foot protein 3. DIG-labeled probes were prepared by 3′ end-labeling reactions (Boehringer Mannheim; Indianapolis, IN). Oligonucleotide probes were synthesized with a 5′ biotin moiety (GenSet; La Jolla, CA).
M. edulis Probes
Template for preCol-P probes was prepared by PCR amplification of cDNA inserts from preCol-P clones. A nonhomologous probe was prepared by similar amplification of the plasmid vector pBluescript (Stratagene; Palo Alto, CA). Double-stranded, DIG-dUTP-labeled DNA probes were synthesized by random priming using a Genius System Nonradioactive DNA Labeling and Detection Kit (Boehringer Mannheim). Efficiency of labeling was determined by dot-blot hybridizations. Average length of random primed probes was 200–400
MEADE In Situ Hybridizations
Although both S. reidi and M. edulis tissues were prepared using MEADE techniques, two hybridization protocols are described below because of different optimization requirements. All hybridizations were performed on 20–50 sections per probe treatment, with sections taken from three to five different embedded block faces. Results were examined with a Nikon Microphot FXA epifluorescent microscope fitted with a dual bandpass filter set specific for visualization of fluorescein and propidium iodide.
S. reidi
A 10-min incubation in fresh 100% acetone removed the methacrylate resin from embedded tissue sections. Slides were washed in NaPBTw (20 mM Na2HPO4, 20 mM NaH2PO4, 0.9% NaCl, 0.1% Tween 20) (5 min), acetylated in 0.1 M triethanolamine (pH 8.0) with 0.5% acetic anhydride (3 min), washed, and prehybridized in hybridization buffer (HB1) (2 × SSC, 5 mM EDTA, 1 × Denhardt's solution, 0.5 mg/ml sheared herring sperm DNA, 0.1% Tween 20) (20 min). Hybridization occurred after addition of 1 ng/μl labeled probe in HB for 7–12 hours at 46C. Sections were washed (15 min each) in 2 × SSC, 0.1% Tween 20 at 46C and 0.2 × SSC, 0.1% Tween 20 cooling to room temperature (RT). Hybrid detection was performed according to one of the following strategies.
Strategy 1: NBT/BCIP Chromogenic Detection of DIG
Sections were blocked (15 min at RT) with 2 mg/ml BSA in NaPBTw, incubated with alkaline phosphatase (AP)-conjugated anti-DIG antibody (Boehringer Mannheim; 1:2000 in blocking buffer with 10% sheep serum at 37C; 3 hr), and washed in NaPBTw (5 min) and AP buffer (100 mM Tris-HCl, pH 9.6, 100 mM NaCl, 50 mM MgCl2; 2 min). The AP-conjugated antibody was detected with 4.5 μ/ml nitro-blue tetrazolium (NBT)/3.5 μl/ml 5-bromo-4-chloro-3-indolylphosphate (BCIP) (Boehringer Mannheim) and 1 × levamisol (Vector Laboratories; Burlingame, CA) in AP buffer for 1–2 hr. Sections were rinsed, counter-stained with pyronin Y–Methyl green, and examined.
Strategy 2: CARD Amplification/ Fluorescent Detection of Biotin
Endogenous biotin was blocked with the Avidin/Biotin Blocking Kit (Vector Laboratories) after tissue acetylation and before hybridization. For hybrid detection, protocols for the TSA-Indirect Kit (NEN Life Science Products; Boston, MA) were followed according to the manufacturer's instructions. After application of streptavidin–horseradish peroxidase (HRP), sections were incubated with biotinyl tyramide (8 min) and then streptavidin–fluorescein (FITC). Propidium iodide served as counterstain (Molecular Probes; Eugene, OR).
M. edulis Optimization
Optimal length of acetone treatment was investigated by incubating tissues in 100% acetone for 0–18 minutes and comparing hybridization signal using detection Strategy 3 for chromogenic development (described below). Sections were also subjected to proteolytic digestion with proteinase K (Sigma Chemical; St Louis, MO) (0.2, 2, 10, and 20 μg/ml at 37C), pepsin (Sigma) (0.2 and 2 μg/ml), or dilute hydrochloric acid (0.2 M) solutions before hybridization for ultrastructural/signal comparison.
M. edulis
Slides were de-embedded in acetone (12–15 min), washed in NaPBTw, and incubated in 10 μg/ml proteinase K (4 min, 37C). The reaction was stopped by incubation in 2% glycine in NaPBTw, and then sections were acetylated, washed, and prehybridized in HB2 (50% formamide, 4 × SSC, 1 × Denhardt's solution, 0.2 mg/ml sheared herring sperm DNA, 5% dextran sulfate). Dilutions of 1:500 DIG-labeled preCol-P probe or nonhomologous probe were used for overnight hybridizations (42C). Tissues were washed in 2 × SSC and 0.1 × SSC at 42C. When possible, all solutions were prepared with 0.1% diethyl pyrocarbonate-treated water (DEPC; Sigma). Blocking and detection of the DIG hybrid complex are described below.
Strategy 3: Vector Blue Chromogenic Detection of DIG
Sections were preblocked with 2% blocking buffer (Boehringer Mannheim) and incubated with AP conjugated anti-DIG Fab fragments (Boehringer Mannheim; 1:200 for 1.5–3 hr at 37C). After washes in 100 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.05% Tween 20, and development buffer [10 mM Tris-HCl (pH 8.2)], hybrids were detected with Vector Blue Substrate Kit reagents (Vector Laboratories; 20 min). Sections were counterstained with 1% Neutral Fast Red (Sigma) and viewed.
Strategy 4: CARD Amplification/Sandwiched Vector Blue Chromogenic Detection of DIG
Tissues were preblocked with the Avidin/Biotin Blocking Kit and then 2% blocking buffer. After detection of probe-target hybrids with biotin-SP-conjugated anti-digoxin (Jackson ImmunoResearch Laboratories; West Grove, PA) (1:200 for 1.5–3 hr at 37C), TSA-Indirect Kit protocols were followed for application of streptavidin–HRP and biotinyl–tyramide (6 min). Tissues were washed, incubated with an AP-conjugated biotin-avidin complex (Vectastain ABC-AP Kit; Vector Laboratories), and developed chromogenically with Vector Blue Substrate.
Strategy 5: CARD Amplification/ Fluorescent Detection of DIG
Methods were followed as above in Strategy 4. However, after the application of biotinyl–tyramide and TSA washes, hybrids were detected with streptavidin–FITC and counterstained with propidium iodide (Molecular Probes).
Results
To test the general applicability of MEADE in conjunction with ISH, a variety of probing and detection strategies were performed. To facilitate comparisons among these five detection strategies, the major steps of each are outlined in Figure 1.
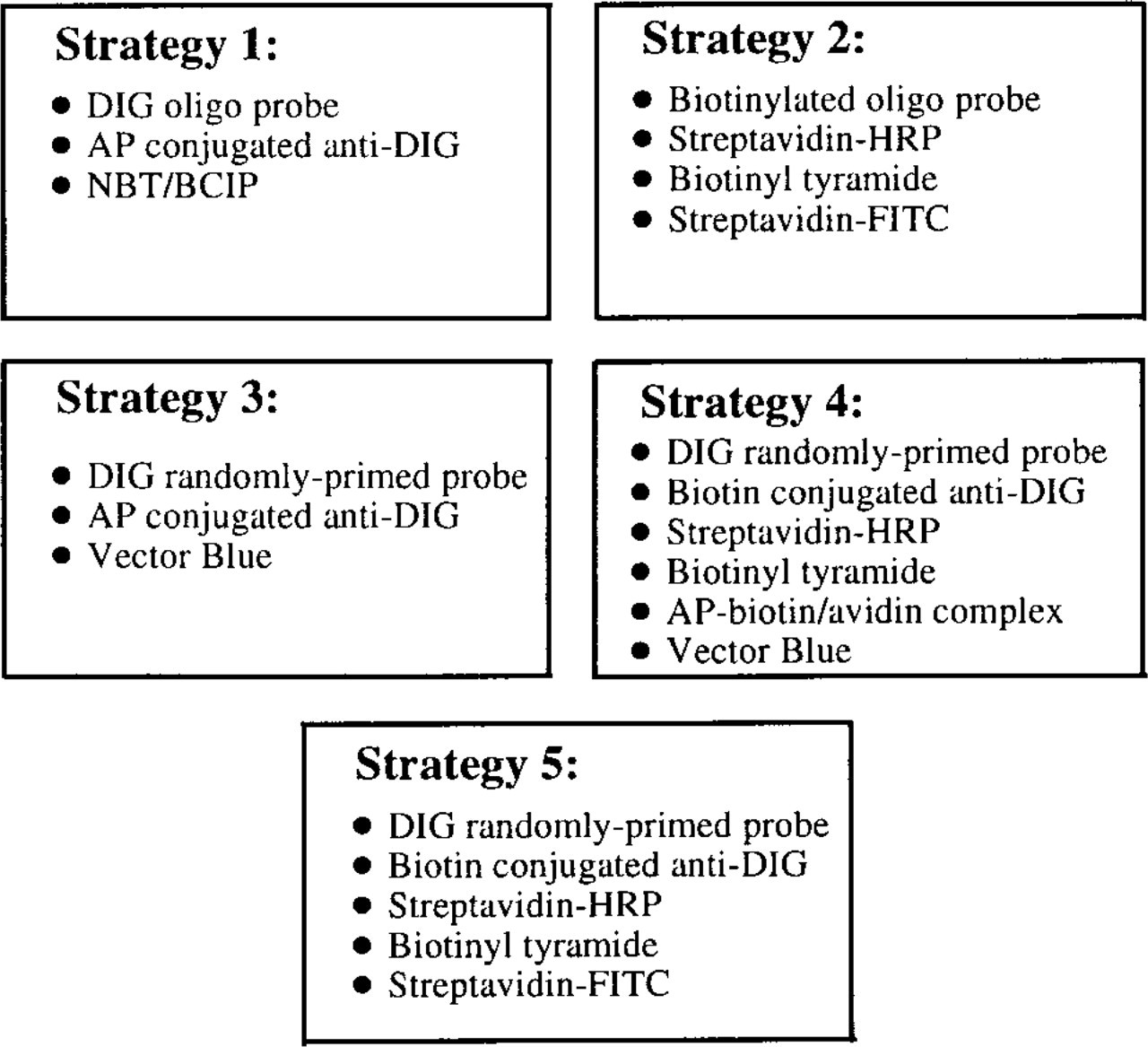
Summary of detection strategies. Strategies 1 and 2 were performed with gill tissues of S. reidi. Strategies 3, 4, and 5 were performed with foot tissue sampled from M. edulis. DIG, digoxigenin or digoxin; AP, alkaline phosphatase; HRP, horseradish peroxidase; and FITC, fluorescein. The AP–biotin/avidin complex is the functional component of the Vectastain ABC-AP Kit; streptavidin–HRP, biotinyl–tyramide, and streptavidin–FITC were components of the TSA-Indirect Kit.
The MEADE technique provided excellent resolution and integrity of cellular morphology in semithin sections of fragile S. reidi gill filaments. For structural orientation, tightly packed ribbon bands represent the gill lamellae of a single gill filament. The lamella is approximately two cells in width and is composed only of vascular tissue and an outer layer composed of bacteriocytes (containing symbionts) and thin intercalary cells that lack symbionts. Higher magnifications of the gill lamellae show large host cell nuclei surrounded on both sides by smaller, granule-like symbiotic bacteria (Figure 2).
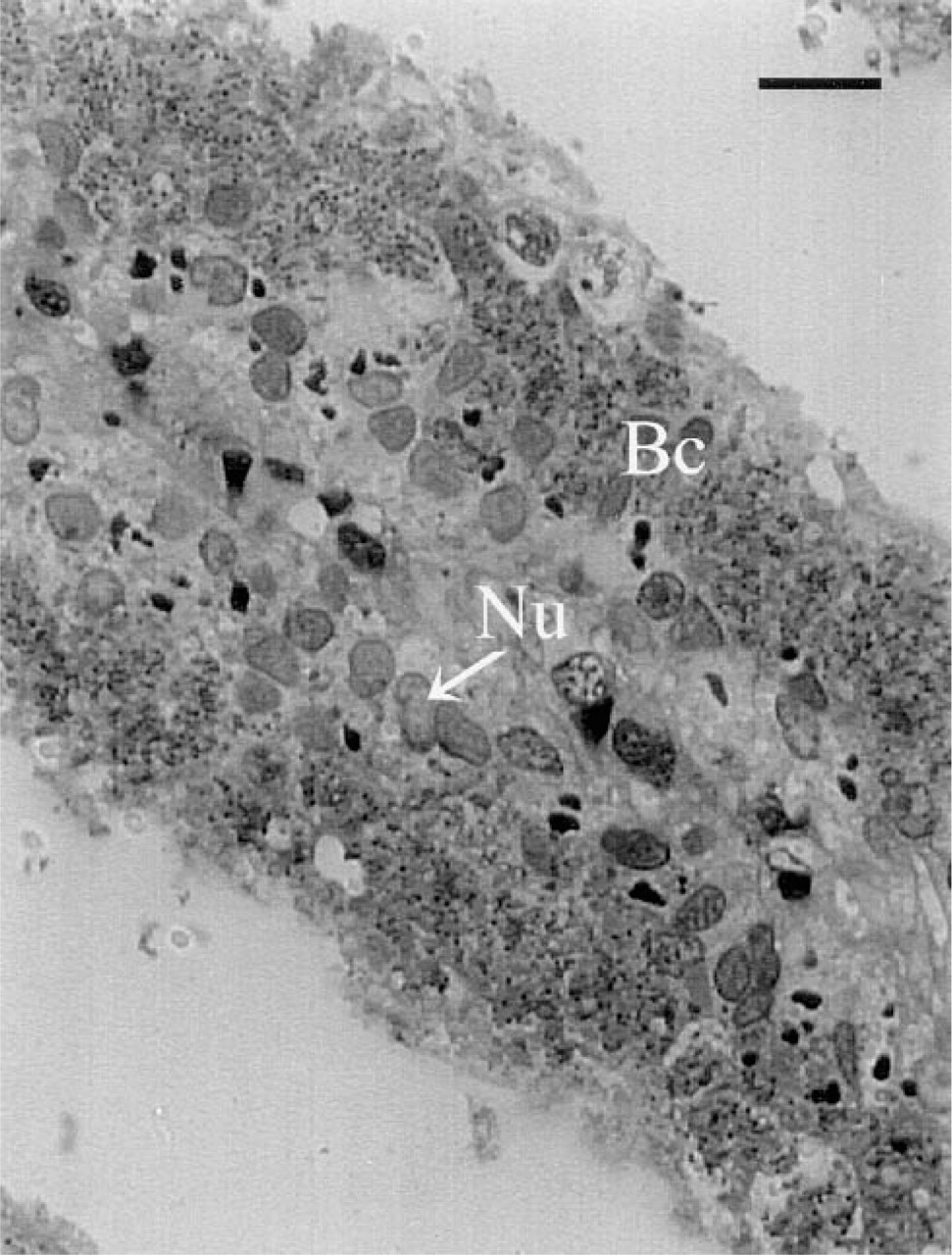
Morphology of methacrylate de-embedded gill filaments. One- to 2-μm thick sections were cut with glass knives, de-embedded via acetone treatment, and counterstained with 1% Neutral Fast Red. Excellent structural preservation was observed in the fragile lamellar tissues. A single lamella is shown. Large host cell nuclei (Nu) within the vascular tissue are clearly differentiated from surrounding smaller granular bacteria (Bc) in undifferentiated bacteriocytes. Bar =13.8 μm.
Hybridization results using each detection strategy are shown in Figures 3 and 4. The 16S rRNA of bacterial endosymbionts of S. reidi was successfully localized within host bacteriocytes along the proximal portions of the gill lamellae (Figure 3). Figures 3A and 3B were developed using Strategy 1 for chromogenic detection of the hapten DIG. The NBT/BCIP reacts with the conjugated AP to produce a purple precipitate wherever the hybrid complex is localized within the tissue. The nonhomologous probe produced minimal background nonspecific labeling (Figure 3A), whereas strong positive labeling of bacterial symbionts was noted in tissues hybridized with the symbiont-specific probe SR-1254R (Figure 3B). Figures 3C and 3D were each developed using CARD amplification and fluorescent detection (Strategy 2). Figure 3C shows hybridization results with the nonhomologous probe Mefp-3.5T. The propidium iodide counterstain (specific for nucleic acids) allows large host cell nuclei to be easily distinguished from surrounding granular bacterial symbionts. The green fluorescent precipitate in Figure 3D corresponds with positive labeling of the endosymbionts, after tissue hybridization with the symbiont-specific probe SR-1254R.
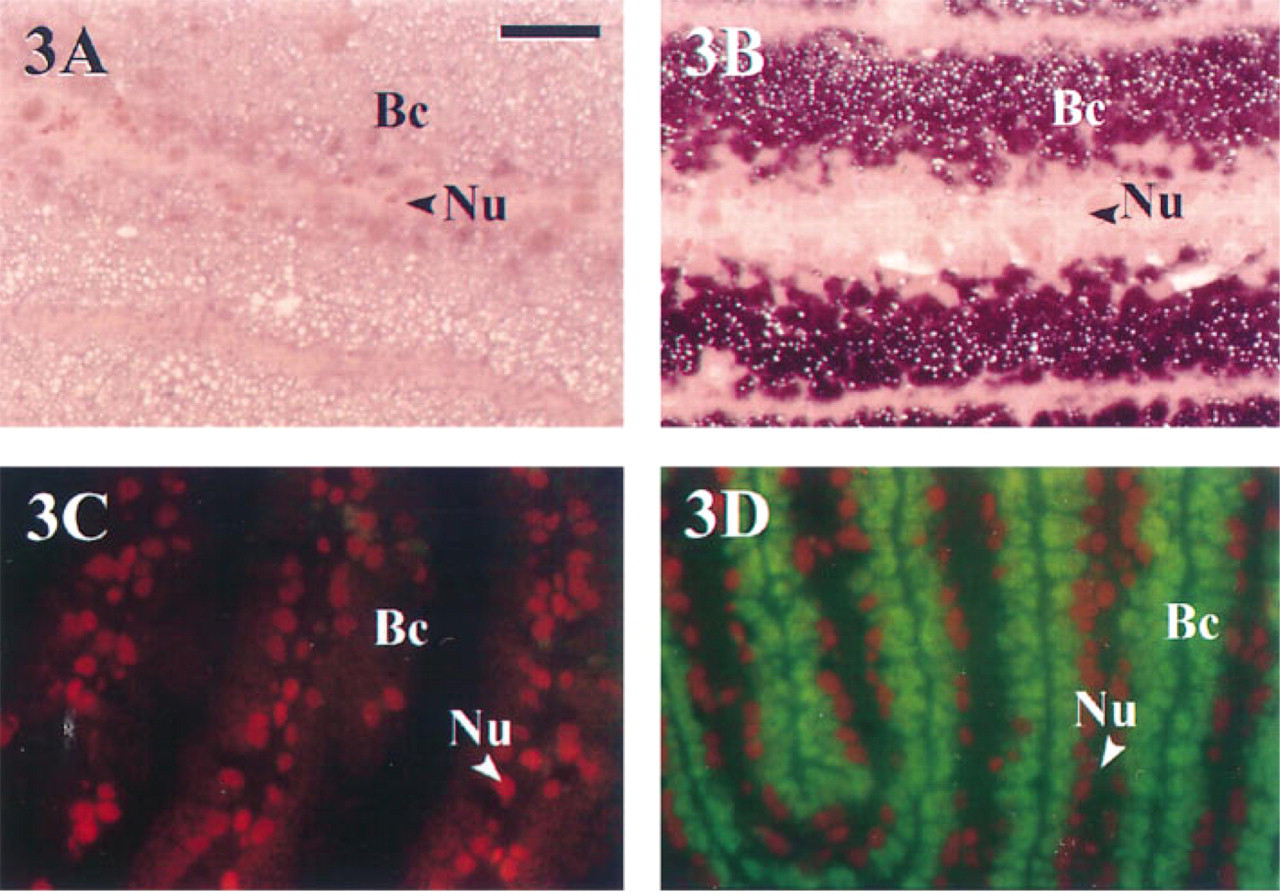
Detection of bacterial endosymbionts in gill filaments of S. reidi. All tissue sections were 1–2 μm thick and de-embedded via acetone treatment. Magnification is the same in all photographs; notation is equivalent to that in Figure 2. A and B were developed using Strategy 1 for the chromogenic detection of DIG using NBT/BCIP and were counter-stained with pyronin Y–methyl green. (
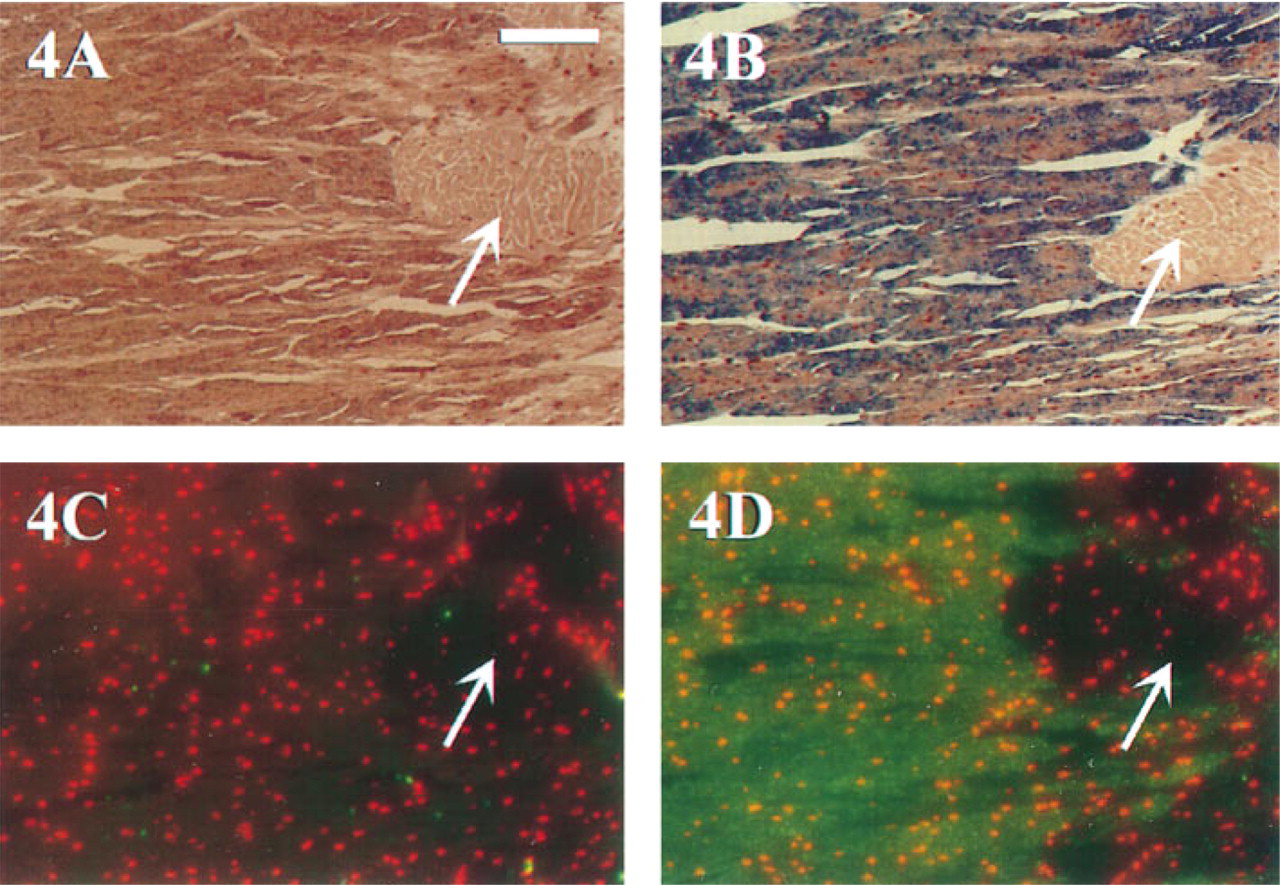
Detection of transcripts for the byssal precursor preCol-P mRNA in M. edulis. All tissue sections were 1–2 μm thick and de-embedded via acetone treatment. Magnification is the same in all photographs. Arrows indicate non-labeled muscle tissue. A and B were both probed and developed chromogenically using the Vector Blue substrate. Blue precipitate corresponds to regions of preCol-P expression within the glandular foot tissue. (
Several modifications were required to adapt the original MEADE ISH procedure for M. edulis tissues and double-stranded, randomly-primed DNA probes (200–400
MEADE ISH reactions were conducted to localize transcripts of the byssal collagen precursor, preCol-P, in M. edulis foot tissue. Foot tissue is mostly glandular, with some striated muscle running throughout (Figure 4). Probe hybrids were localized in patches throughout most of the glandular tissue; the globe-shaped muscle tissue located in the right portion of each section did not contain any preCol-P transcripts and therefore served as an internal negative Control (Figures 4A–4D). Figures 4A and 4B were both developed chromogenically. Figure 4A was detected using Strategy 3; faint labeling of preCol-P transcripts was observed. In contrast, Figure 4B was detected using Strategy 4 with CARD amplification. Although not shown, nonhomologous probe controls produced negligible background staining in each hybridization. Hybridizations were conducted on M. edulis foot tissue and were developed using Strategy 5 for CARD amplification/fluorescent detection of DIG (Figures 4C and 4D). Labeling in Figure 4D is similar to that shown in Figure 4B. Localization of transcripts for preCol-P using CARD amplification and chromogenic or fluorescent detection was substantially increased compared to chromogenic detection alone (Figure 4A).
Discussion
This study was undertaken to evaluate the utility of the methacrylate embedding, acetone de-embedding (MEADE) technique for light microscopic ISH studies. This technique was examined in conjunction with various marine systems, hapten-labeled probes, and detection strategies. Despite documentation of superior cellular detail and high hybridization signal (Kronenberger et al. 1993), no other investigations have attempted to use MEADE ISH with nucleic acid probes, and signal amplification methods remain unexplored with respect to this resin. Our interest in addressing these questions stemmed from our inability to examine transcript level expression in marine larvae without high cellular detail and a high-sensitivity ISH technique for light microscopy.
In addition to the methacrylate resin, two alternative plastic resins (LR White and Lowicryl K4M; Ted Pella, Tustin, CA) were initially examined to determine which provides maximal structural preservation and integrity of tissues. Neither LR White nor Lowicryl K4M allows tissue de-embedding before hybridization. The methacrylate resin permitted optimal retention of cell morphology and visualization of fine structural detail in semithin tissue sections of adult S. reidi gill filaments. It also provided maximal ISH labeling efficiency (results not shown). The reduced labeling of rRNA sequences found in LR White- and K4M-embedded filaments compared to the de-embedded methacrylate tissue was expected because of differences in probe accessibility through the embedding resins. Specifically, probe hybridization in LR White and Lowicryl K4M sections is probably restricted to those sites at the surface of the tissue section (“surface-only” labeling).
We developed the MEADE hybridization protocol using short digoxigenin (DIG) end-labeled oligonucleotide probes specific for the 16S rRNA of bacterial endosymbionts in the gill tissue of S. reidi (a single reporter molecule theoretically maximized probe penetrability, compared to the potential steric hinderance resulting from incorporation of multiple hapten molecules conjugated onto larger probes). A chromogenic method (Strategy 1) enabled successful hybrid detection, with strong hybridization signal and low levels of background (Figures 3A and 3B). Once the initial ability to perform ISH with the methacrylate de-embedded tissue was confirmed, it became the goal of this study to examine the possible limitations of the technique by testing a variety of probes, tissues, and detection strategies.
The versatility of the MEADE ISH protocol was first examined with respect to fluorescence and the application of biotinylated oligonucleotide probes. The TSA-Indirect Kit relies on catalyzed reporter deposition (CARD) amplification of biotinyl tyramide. The newly immobilized biotin source is then detected by standard indirect methods using fluorescent steptavidin conjugates. The technique has proved extremely sensitive for in situ hybridizations and signal amplification of low target signal (Kerstens et al. 1995; NEN Life Science Products).
In our study, hybridization signals were strong and clearly distinguishable from low background staining and the propidium iodide counterstain (Figures 3C and 3D). Although labeled regions corresponded to those identified with DIG detection and NBT/BCIP development, direct comparison between chromogenic and fluorescent detection systems was not possible because of the different qualities of each reaction.
MEADE in situ reactions were tested using a second marine system and longer DIG-labeled randomly primed, double-stranded DNA probes (average length 200–400
Strategy 1 (NBT/BCIP, Figure 3B) and Strategy 3 (Vector Blue, Figure 4A) development are directly comparable because both rely on an anti-DIG Fab fragment to bind to the DIG hapten. The antibody's conjugated AP molecule reacts chromogenically with the Vector Blue substrate and NBT/BCIP. However, in M. edulis, dissimilar counterstaining made the blue hybridization signal preferable to the purple NBT/BCIP. In addition, greater signal resolution and localized development were noted in this tissue with the Vector Blue substrate.
Both Strategy 4 and Strategy 5 utilized CARD signal amplification. Strategy 4 integrated catalyzed reporter deposition of biotinyl tyramide with the Vectastain ABC-AP Kit. This kit employs a preformed AP–biotin/avidin macromolecular complex (the ABC-AP complex), which can then be chromogenically developed as in Strategy 3. Detection in Strategy 5 was directly comparable to Strategy 2 using the TSA Kit. Chromogenic and fluorescent detection produced similar results, each yielding substantial signal amplification (Figures 4B and 4D). All detection strategies (Strategies 3–5) reproducibly localized the mRNA of the byssal collagen precursor in glandular foot tissue.
To conclude, the results of this study have shown the versatility of the MEADE technique in conjunction with light microscopic ISH. The methacrylate resin was ideal for fragile tissues that required high retention of cellular morphology, and the semithin (1–2 μm) sections reduced microscopic out-of-focus blur. Hybridizations were successfully completed in two different marine systems to localize mRNA and rRNA transcripts. Hybrids were detected with several different probe molecules (oligonucleotide and randomly primed), probe labels (biotin and DIG), signal amplification (CARD), and detection strategies (chromogenic and fluorescent). MEADE ISH will undoubtedly prove invaluable for future studies that aim to localize transcripts at the cellular and/or subcellular level throughout early embryological stages of development in other marine larvae. Moreover, we expect that advances in fluorescent sandwiching, cyanine fluorochromes (Cy5 and Cy7), and charge-coupled device (CCD) cameras will soon be applied to the MEADE ISH protocols for even more qualitative and quantitative studies.
Footnotes
Acknowledgements
Supported by grants from the National Science Foundation (OCE-9016373 and OCE-9217950 to SCC) and the National Institute of Health (3-2-21-3501-06 to JHW).
We would like to thank the captain and crew of the R/V Alta (Bamfield Marine Research Station, British Columbia, Canada) for their perseverance in collecting S. reidi specimens for this study. NEN Life Science Products generously donated materials for our use as well as technical assistance. Kirk Czymmek provided a critical review of the manuscript and Monroe Givens was helpful with reproduction of digital images.