
Abstract
During the inflammatory response to an implanted biomaterial, monocytes undergo a striking phenotypic progression of differentiation into macrophages, which may subsequently fuse to form foreign body giant cells (FBGCs). Taking advantage of an in vitro system of cytokine-induced FBGC formation together with the optical slicing capabilities of a confocal microscope, we investigated the cytoskeletal reorganization and adhesive structure development during this dramatic morphological progression. Human monocytes demonstrated diffuse cytoplasmic staining of adhesive structural proteins. Punctate filamentous (F)-actin structures appeared along the ventral cell membrane of macrophages and were identified as the core of podosome adhesive structures by the distinctive ring staining of vinculin, talin, and paxillin around the F-actin. Cytokine-induced FBGCs were characterized by a restriction of podosomes to the extreme periphery of the ventral cell surface. Although macrophages and FBGC contained equivalent amounts of F-actin, significantly more F-actin was located within 1 μm of the ventral plasma membrane in FBGCs compared to macrophages. Taken together, these results provide new information on the dynamic cytoskeletal reorganization and adhesive structure development that occur during phenotypic progression from human monocytes to macrophages to FBGC. Furthermore, they suggest the acquisition of functional specializations on FBGC formation, which may enhance our understanding of chronic inflammatory processes.
Monocyte-derived macrophages are extremely versatile cells which are believed to be critical mediators of the host reaction to biomedical material implants (Anderson 1993). In response to the persistent presence of the foreign material, adherent macrophages may fuse to form multinucleated foreign body giant cells (FBGCs; Fais et al. 1997; Chambers and Spector 1982), the functional capabilities of which are not yet well understood. Importantly, the relationship between the acquisition of functional specializations and the striking morphological alterations during monocyte differentiation into macrophages as well as during the formation of FBGCs is as yet unclear.
Cytoskeletal participation has been demonstrated for many monocyte/macrophage functions that are critical during the inflammatory response, including migration (Yin and Hartwig 1988), attachment and adhesion (Lehto et al. 1982; Amato et al. 1983; Yin and Hartwig 1988), spreading (Reaven and Axline 1973; Lehto et al. 1982), phagocytosis (Reaven and Axline 1973; Oliver and Berlin 1982), and secretion (Tapper 1996). Cytoskeletal requirements for macrophage fusion and cytoskeletal organization in FBGCs, however, are essentially unknown. Macrophages, which are morphologically distinct from monocytes (Dougherty and McBride 1989), acquire significant functional capabilities on their development (Henson 1980; Nathan 1987; Johnston 1988; Stachura 1989). It is reasonable to hypothesize that cytoskeletal polarizations may also occur during macrophage fusion to form FBGCs that mediate the acquisition of as yet undescribed functional specializations in FBGCs.
Interestingly, several different types of adhesive structures (Lehto et al. 1982; Zambonin-Zallone et al. 1983; Marchisio et al. 1987; Ono et al. 1993) have been described in macrophages during in vitro culture. Focal contacts (Zambonin-Zallone et al. 1983; Ono et al. 1993) are rectangular structures that contain colocalized vinculin, talin, and F-actin (Burridge et al. 1988). The latter exits focal contacts as stress fibers, which appear as thick cables of actin connecting two distant focal contacts. Close contacts (Lehto et al. 1982) are closely related structurally to focal contacts but apparently lack stress fibers (Burridge et al. 1988). Podosomes (Lehto et al. 1982; Marchisio et al. 1987) are distinct, circular structures characterized by a ring of co-localized vinculin and talin surrounding an F-actin core. However, with continuing advances in this area, including the identification of new adhesive structural proteins, our understanding of the precise nature of adhesive structures formed by human monocytes/macrophages, FBGCs, and other cell types is still evolving. It is not known which adhesive structures support the mobility necessary for macrophage apposition leading to fusion and/or the adhesive strength required for phagocytic functions directed at the underlying surface.
Recent advances in confocal fluorescence microscopy, principally its capability for qualitative and quantitative evaluation of three-dimensional organization, now facilitate examination of cytoskeletal reorganization and adhesive structure development during the remarkable phenotypic progression from monocytes to macrophages and particularly during macrophage-macrophage fusion to form FBGCs. Using an in vitro system of cytokine-induced human macrophage fusion and FBGC formation (DeFife et al. 1997), the goals of the present study were twofold: (a) to compare the organization of the cytoskeletal elements in monocytes, macrophages, and FBGCs, and (b) to identify the type and assess the organization of adhesive structures formed by adherent macrophages vs those formed by FBGCs.
Materials and Methods
Monocyte Isolation and Culture
Human blood monocytes were isolated from the venous blood of unmedicated donors by a nonadherent density centrifugation method as described (McNally and Anderson 1994). Isolated monocytes were judged >97% viable by Trypan Blue exclusion and >80% pure by staining for nonspecific esterase and peroxidase. Monocytes were suspended in a medium of RPMI-1640 (GIBCO; Grand Island, NY) containing 25% autologous serum and antibiotic/antimycotic mixture (GIBCO) and were cultured on dimethyldichlorosilane (Alfa Aesar, Ward Hill, MA; Healy et al. 1994; Woodhouse and Brash 1994)-treated or untreated coverslips (Thomas Scientific, Swedesboro, NJ; DeFife et al. 1997) that had been secured in the wells of 24-well tissue culture polystyrene plates (Falcon 3047; Becton Dickinson, Lincoln Park, NJ) with silicone rubber rings (Cole-Parmer Instruments; Niles, IL). Monocytes were added at a concentration of 5 × 105 per well in 0.5 ml medium to culture plates containing the coverslips and were allowed to adhere for 2 hr at 37C in a humidified atmosphere of 95% air and 5% CO2. Nonadherent cells were removed by aspirating the medium and rinsing the wells with warmed (37C) PBS (GIBCO) containing calcium and magnesium, and the remaining adherent monocytes were covered with 1 ml per well of fresh medium. Samples collected after the wells were rinsed are presented as Day 0.
On Days 3 and 7 of incubation, the medium was replaced with 25% heat-treated (56C water bath for 1 hr) autologous serum in RPMI and 10 ng/ml interleukin-13 (IL-13; R & D Systems, Minneapolis, MN) was added to induce macrophage fusion and FBGC formation as indicated. For microfilament disruption experiments, 5 μM cytochalasin D (Sigma Chemical; St Louis, MO) was added to Day 10 cultures for 2 hr before fixation. Samples were fixed by rinsing twice for 5 min each with warmed PBS and covering with 3.7% formaldehyde or 3.7% formaldehyde containing 0.5 μM taxol (Sigma) in PBS for 20 min. After three additional rinses with PBS, cells were permeabilized with 0.2% Triton X-100 (Sigma) in PBS for 5 min.
Cytoskeletal and Cytoplasmic Protein Fluorescence Staining
Confocal scanning laser microscopy (MRC-600; Bio-Rad, Hercules, CA) was used to image cells with double labels of rhodamine-phalloidin for F-actin and an indirect immunofluorescence tag for another cytoskeletal or cytoplasmic protein. Nonspecific sites were blocked with a 1:100 dilution of goat serum (DAKO; Carpinteria, CA) in PBS for 30 min at 37C. Blocking serum was removed and the primary antibody solutions were incubated with cells for 1 hr at 37C. Primary antibodies were diluted in PBS containing 3% BSA (Sigma) as follows: anti-tubulin 1:50 (Boehringer Mannheim; Indianapolis, IN); anti-vimentin 1:10 (Boehringer Mannheim); anti-talin 1:100 (Serotec; Oxford, UK); anti-vinculin 1:15 (Serotec); anti-paxillin 1:200 (Transduction Laboratories; Lexington, KY); anti-gelsolin, 1:500 (Sigma); and anti-focal adhesion kinase (FAK) 1:50 (Santa Cruz Biotechnology; Santa Cruz, CA). Anti-L-plastin antibodies were the generous gift of Dr. Samuel L. Jones (Washington University; St Louis, MO). Each monoclonal primary antibody had an isotype-matched control IgG (DAKO) and each polyclonal antibody had a species-matched nonspecific control IgG (DAKO) that was run in parallel. After the primary antibody incubations, the cultures were rinsed four times for 5 min each with PBS. Rhodamine-phalloidin (Molecular Probes; Eugene, OR) at a final dilution of 1:150 was added to BODIPY FL-conjugated goat anti-mouse IgG (Molecular Probes) that was diluted 1:100 in PBS. The secondary antibody solution was incubated with cells for 30 min at room temperature. After three additional 10-min washes in PBS, samples were removed from the culture plates and mounted on glass slides with Gel/Mount (Biomeda; Foster City, CA).
For confocal imaging, control stains were set to a black background. Positive samples were then viewed at the same laser intensity as well as aperture, gain, and blacklevel settings. Optical slices approximately 1 μm thick were taken for each sample, beginning at the coverslip surface and ending at the apical surface of the adherent cell. Presented images of cellular cytoskeletal organization are projections of all of the slices from a sample. Images depicting adhesive structure organization are single optical slices taken from the basal cell surface.
Quantitation of Cellular Fluorescence
Samples that were double labeled for F-actin and gelsolin were used for quantitative measurements. Confocal optical slices encompassing the entire volume of individual cells were isolated using three-dimensional reconstruction software (microVoxel; Indec Systems, Capitola, CA). The fluorescence intensity of F-actin from the entire cell was measured to compare the total F-actin content in macrophages and FBGCs. F-actin participation in adhesive structures was quantified by measuring the fluorescence intensity of F-actin that co-localized with gelsolin at the ventral cell surface of macrophages and FBGCs and expressing the results as a percentage of the total cellular F-actin. Sixty macrophages and 60 FBGCs from four different donors were measured, and the unpaired Student's t-test was used for statistical analysis (StatView; Abacus Concepts, Berkeley, CA).
Measurement of Cell Diameter
Cells were stained sequentially with May-Grünwald stain (Sigma) for 1 min, phosphate buffer (pH 7.4; Sigma) for 1 min, Giemsa stain (McNally and Anderson 1994) diluted 1:14 in deionized distilled water for 5 min, and two brief distilled water rinses. Cell areas were measured from a minimum of 200 total cells from four different donors using the morphometric software SigmaScan Pro (Jandel Scientific Software; San Rafael, CA). Results are presented as mean cell diameter ± SEM.
Results
Dynamic F-Actin Reorganization Accompanies Monocyte-to-Macrophage Morphological Progression
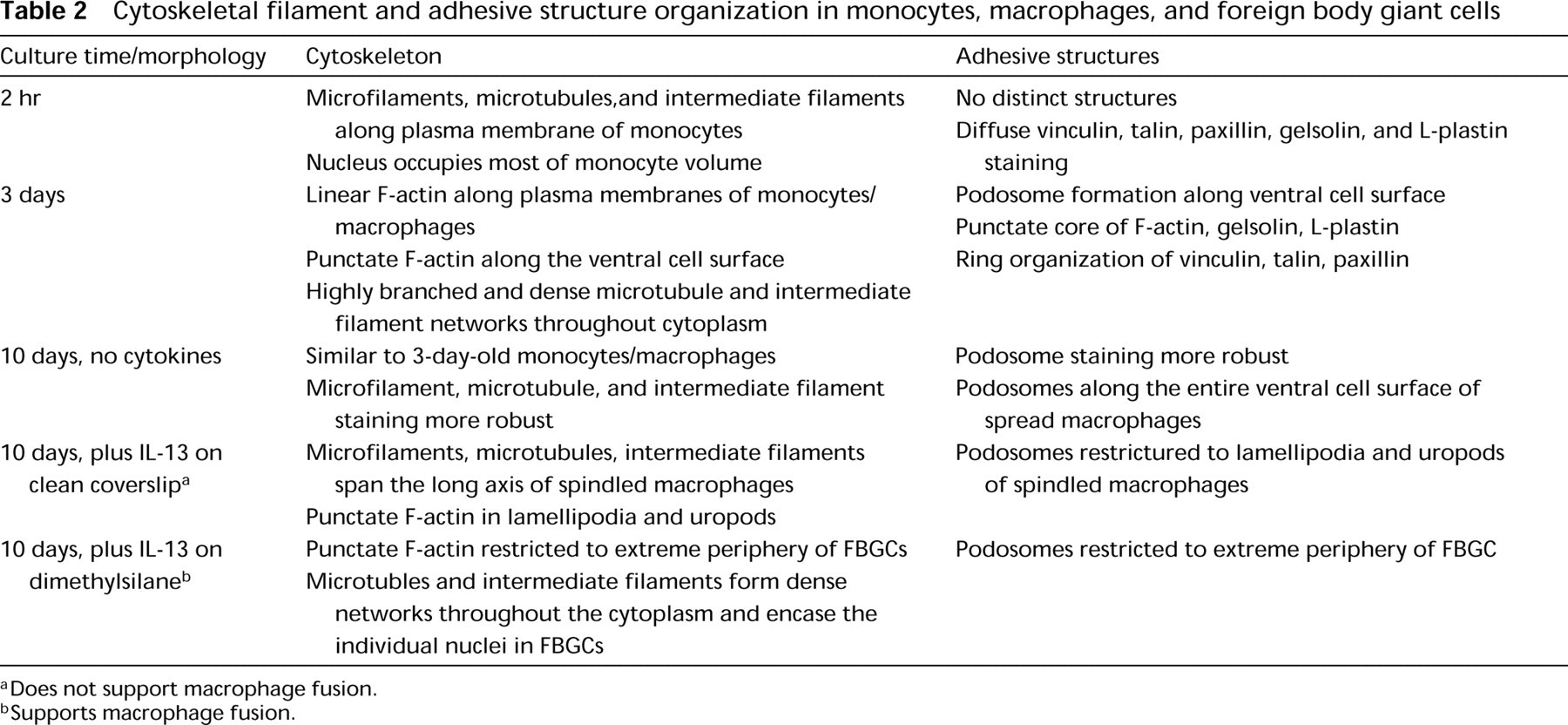
To provide a context in which to understand macrophage to FBGC cytoskeletal reorganization, we first sought to describe the monocyte-to-macrophage cytoskeletal changes occurring during a 10-day culture period.
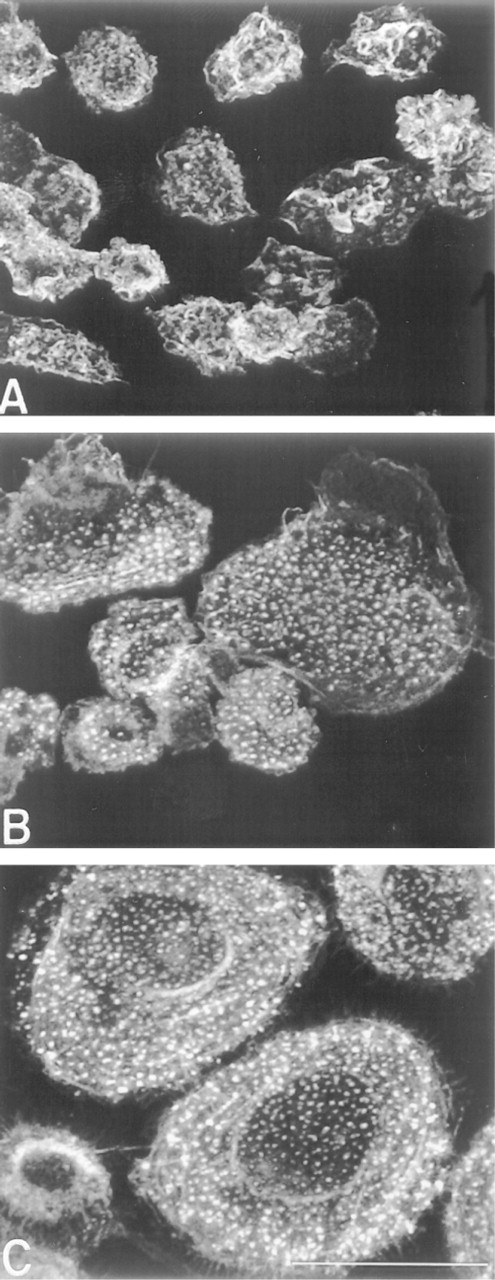
F-actin reorganization during monocyte-to-macrophage phenotypic progression. Freshly isolated monocytes were cultured on dimethylsilane-treated coverslips for 2 hr (
Two hours after freshly isolated monocytes were plated, they appeared fairly homogeneous as judged by phase-contrast microscopy. Most monocytes displayed a rounded morphology (mean diameter of 12.4 ± 0.1 μm) with characteristic kidney-shaped nuclei that occupied the majority of the cell volume (not shown). By confocal imaging, F-actin was observed to surround the nucleus in a diffuse pattern with concentrations in membrane ruffles, delineating the cell boundaries (Figure 1A). Some monocytes exhibited an elongated morphology suggestive of motility.
By 3 days of culture, monocyte/macrophage F-actin redistributed into intensely staining punctate foci that were restricted to the substrate-attached side of adherent cells (Figure 1B). Punctate F-actin was visible across the entire ventral cell surface in spread cells, whereas it was restricted to the lamellipodia and uropods of elongated cells. In monocytes/macrophages with either morphology, microfilaments were visible along the plasma membrane throughout the volume of the cell.
In the absence of added cytokine, most cells acquired a spread morphology by Day 10 of culture (Figure 1C). These macrophages resembled those at Day 3 but were larger in diameter (25.6 ± 0.57 μm).
Cytoplasmic Proteins Associate with Punctate Filamentous Actin to Form Podosomes
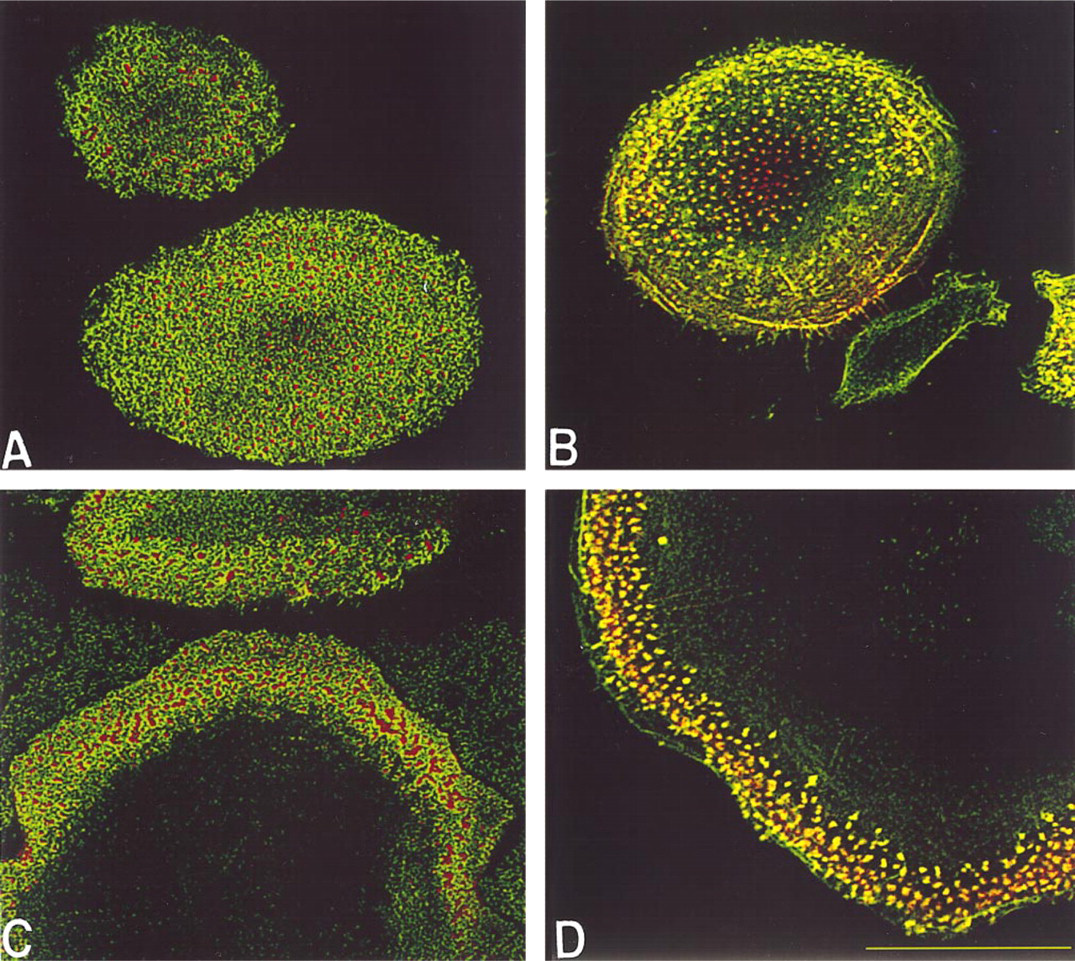
Development of podosome adhesive structures. Macrophages that had been cultured for 10 days on dimethylsilane-treated coverslips (
The punctate F-actin fluorescence along the ventral cell membrane is indicative of adhesive structure formation at these sites (Marchisio et al. 1987; Burridge et al. 1988); however, the type of adhesive contact cannot be identified by F-actin staining alone. To begin to characterize these areas and to more thoroughly describe the composition of adhesive structures that are acquired during macrophage development, we utilized antibodies to a variety of previously described adhesive structural proteins in combination with fluorescence microscopy.
Freshly isolated and cultured monocytes did not form prominent ventrally located punctate actin structures after 2 hr of culture. Likewise, cytoplasmic proteins known to participate in the formation of adhesive structures, i.e., vinculin, talin, and paxillin (Burridge et al. 1988; Turner et al. 1990), also did not organize into punctate structures at this early time point. Although these proteins were present, they were distributed diffusely throughout the cytoplasm, often co-localizing with microfilaments (not shown).
On development of punctate F-actin structures, vinculin, talin, and paxillin formed ring-like structures around the F-actin but did not co-localize with it, as demonstrated by paxillin in Figure 2A. Although vinculin and F-actin are considered hallmarks of focal contacts (Burridge et al. 1988), these adhesive structures were not observed, as judged by a lack of stress fiber formation and the lack of co-localization between vinculin and F-actin.
The lack of co-localization between F-actin and these structural proteins precluded their identification as focal contacts or close contacts. However, the arrangement of vinculin, talin, and paxillin in ring structures surrounding F-actin cores is consistent with podo-some adhesive structures (Lehto et al. 1982; Marchisio et al. 1987). To further confirm this, cultures were stained for gelsolin, which also has been described to be a component of podosomes (Marchisio et al. 1987) but not focal or close contacts. By Day 3, gelsolin reorganized into punctate structures that co-localized with F-actin along the ventral cell surface of mono-cytes/macrophages (Figure 2B). L-plastin, or fimbrin, which has been described in mouse macrophages (Messier et al. 1993), co-localized with punctate F-actin similarly to gelsolin (not shown).
The presence of paxillin prompted investigation for the presence of FAK, the activation of which has been described as an important step in the recruitment of paxillin to developing adhesive structures in rat embryo fibroblasts and mouse 3T3 cells (Burridge et al. 1992). However, FAK was not detected in monocytes or macrophages at any time during the 10-day culture period (not shown).
Induction of Macrophage Fusion to Form FBGCs Results in Further Cytoskeletal and Adhesive Structure Polarizations
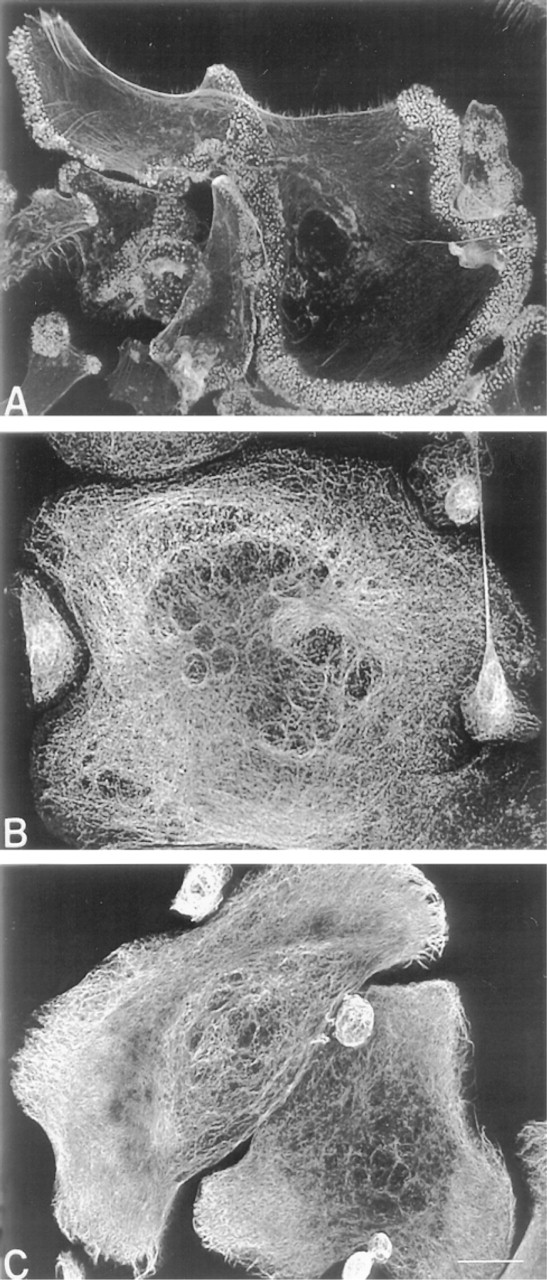
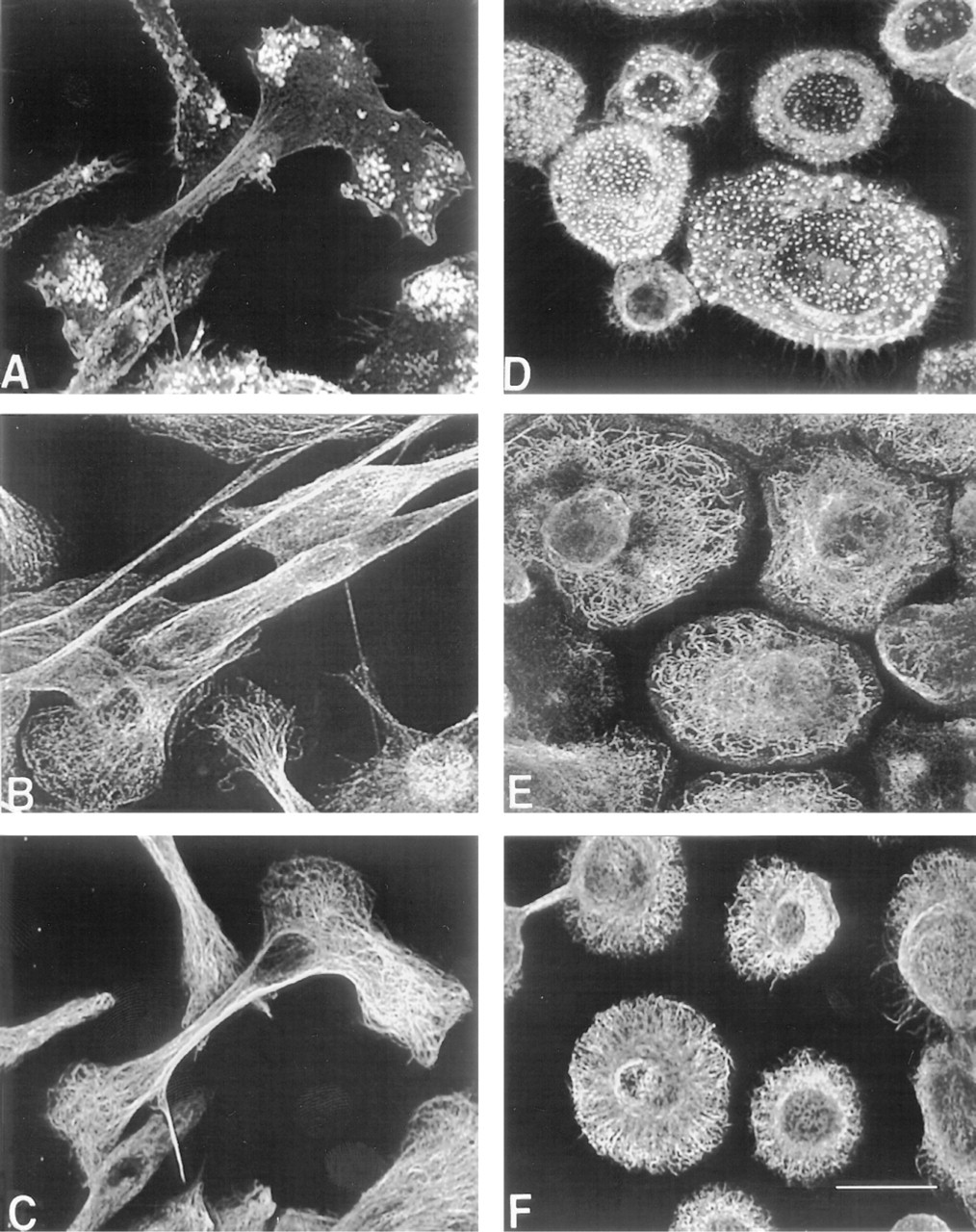
Effect of IL-13 on macrophage cytoskeletal organization. Monocytes/macrophages were cultured on dimethylsilane-treated coverslips for 10 days and were treated with IL-13 on Days 3 and 7 to induce FBGC formation. Fixed cultures were stained for F-actin with rhodamine-phalloidin (
Because IL-13 has been demonstrated to induce macrophage fusion and FBGC formation (DeFife et al. 1997), cultures were treated with IL-13 to examine the cytoskeletal and adhesive structures of these multi-nucleated macrophages.
In contrast to macrophages, punctate F-actin at the ventral cell surface was restricted to the extreme periphery of FBGCs and was very dense (Figure 3A). FB-GCs contained dense meshworks of microtubules that usually formed circles concentric to the plasma membrane (Figure 3B). Microtubules were present in the FBGC periphery but did not terminate in the podo-somes where punctate F-actin structures were concentrated. Furthermore, microtubules and intermediate filaments appeared to encase individual nuclei in FB-GCs. Very dense meshworks of intermediate filaments radiated throughout the cytoplasm and terminated in fibers along the plasma membrane (Figure 3C).
Vinculin, talin, and paxillin localized around the punctate F-actin, forming podosomes, although distinct podosomes were not always visible because of the dense and intense F-actin staining (not shown; and Figure 2C). Gelsolin remained co-localized with punctate F-actin in FBGC (Figure 2D).
To determine whether F-actin content and distribution were quantitatively different between macrophages and the morphologically distinct FBGCs, rhodamine-phalloidin-labeled F-actin fluorescence intensity was measured (Table 1). Although total F-actin, normalized for volume, was equivalent between macrophages and FBGCs, significantly more F-actin was located within 1 μm of the culture surface in FBGCs.
F-actin participation in podosome structures was quantified by measuring the fluorescence intensity of F-actin that co-localized with gelsolin at the ventral cell surface of macrophages and FBGCs (Table 1). Although the majority of the total F-actin in macrophages and FBGCs is localized to the ventral 1 μm of the cells, only up to one third of the F-actin is co-localized with gelsolin. Significantly more co-localization occurred in FBGCs.
To examine the tenacity of podosome-mediated adhesion, FBGC cultures were treated with 5 μM cytochalasin D, which disrupts F-actin. Microfilaments were disrupted throughout the cytoplasm. However, punctate F-actin at the ventral cell surface was resistant to cytochalasin D treatment (Figure 4), suggesting that podosome structural proteins protected the F-actin from depolymerization.
IL-13-Induced Alterations in Macrophage Morphology Are Culture Surface-dependent
Interestingly, we found that IL-13 induced a dramatic change in macrophage morphology on culture surfaces that did not support macrophage fusion and FBGC formation (Figure 5). When not treated with exogenous cytokines, cultures stained for microtubules and intermediate filaments showed no significant changes in organization throughout the 10-day culture period (not shown). Spread cells contained a centrally located concentration of tubulin from which highly branched and interconnected microtubules radiated towards and parallel to the cell periphery (Figure 5E). An extremely dense intermediate filament network was observed (Figure 5F).
On coverslips not treated with any silane, IL-13 did not induce FBGC formation, although macrophage adhesion was comparable to that on silane-treated coverslips. Instead, macrophages assumed an elongated, spindled morphology. Where spindles terminated on the coverslip surface, punctate F-actin structures formed (Figure 5A) and were identified as podosomes by vinculin, talin, paxillin, and gelsolin staining (not shown). Microfilaments, microtubules, and intermediate filaments extended along the long axis of these macrophages (Figures 5A-C). Microtubules and intermediate filaments were organized into meshworks where spindles terminated on the surface in an organization similar to that of spread macrophages (Figures 5B and 5C compared to Figures 5E and 5F, respectively).
Discussion
F-actin distribution in macrophages and FBGCs

p < 0.0001 compared to FBGCs.
Combined cells from four different donors.
The results presented here demonstrate that there is continued cytoskeletal rearrangement during the formation of FBGCs and that there are quantitative differences between the distribution of microfilaments in FBGCs and in macrophages. The increase in F-actin at the ventral cell surface of FBGCs may reflect the more spread morphology compared to macrophages, or it may indicate that FBGCs require more adhesive structures per area for adhesion than macrophages. In support of the latter, the fluorescence staining of punctate F-actin in FBGCs appeared much denser and more intense than that in macrophages. These quantitative differences in microfilament distribution between FBGCs and macrophages, but not overall polymerization as measured by equivalent amounts of total F-actin, suggest that additional functional specialization may be conferred by macrophage fusion to form FBGC. A summary of the results from this study is presented in Table 2.
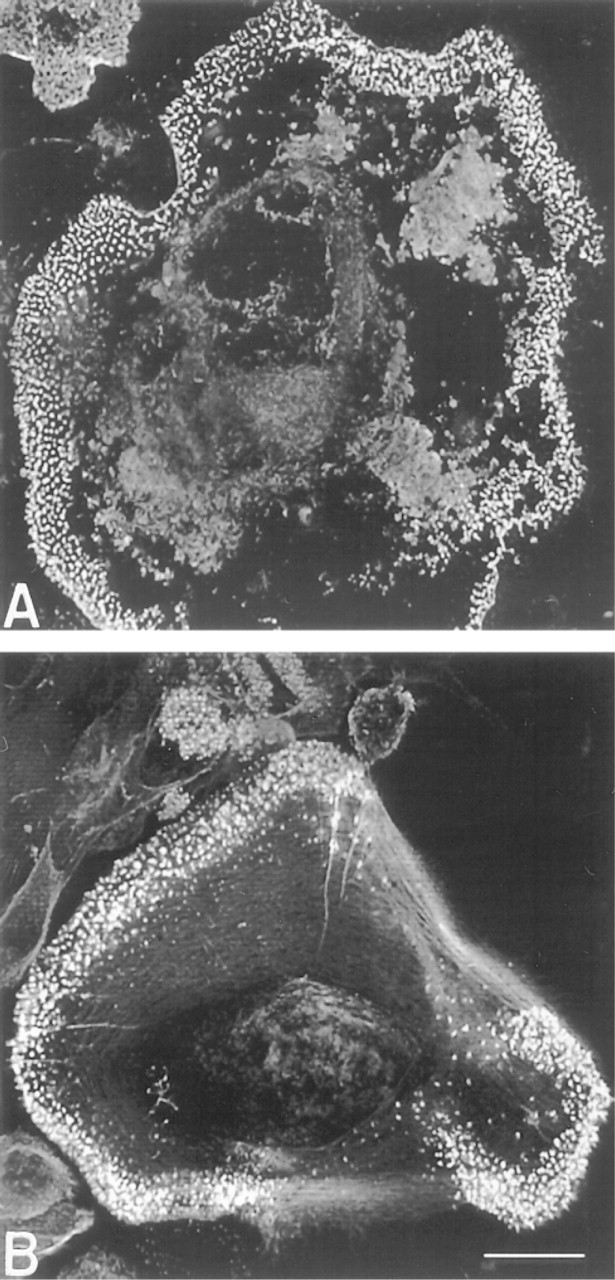
Effect of cytochalasin D on microfilament organization. Monocytes/macrophages were cultured as described for Figure 3 with the addition of 5 μM cytochalasin D for 2 hr before fixation. Rhodamine-phalloidin-stained F-actin, except for that contained in podosomes, was disrupted (
The cytoskeletal organization observed in monocytes and macrophages in this study confirms and extends to human monocytes/macrophages earlier reports on nonhuman monocytes/macrophages (Reaven and Axline 1973; Trotter 1981; Amato et al. 1983; Zambonin-Zallone et al. 1983; Marchisio et al. 1987; Yin and Hartwig 1988; Baba et al. 1991; Ono et al. 1993). Interestingly, our findings also indicate that macrophages develop podosomes but not focal contacts or close contacts. Vinculin and talin are previously described components of podosomes and were used to categorize these adhesive structures as podosomes. This is the first demonstration of the participation of paxillin in podosome formation by human macrophages. The observation that human monocytes do not express FAK at any point during the phenotypic progression to macrophages or FBGC formation is consistent with previous reports using human monocytes (Lin et al. 1994) and macrophage colony-stimulating factor-treated human macrophages (DeNichilo and Yamada 1996). In these previous studies, cells were maintained for 2 days or less. Therefore, the present results extend the previous findings that macrophages do not require FAK to establish podosomes during macrophage development over a 10-day culture period or after cytokine-induced macrophage fusion. Moreover, these studies confirm the localization of paxillin to podosome adhesive structures in the absence of FAK, implying that, unlike fibroblasts (Burridge et al. 1992), macrophages and FBGCs do not require FAK for paxillin localization to adhesive structures. Although integrin participation in podosome development has not yet been established, paxillin interacts with peptides that mimic the cytoplasmic domains of integrin β-subunits (Schaller et al. 1995) and localizes with focal contacts in an integrin-dependent manner (DeNichilo and Yamada 1996). Further experimentation is ongoing to identify specific integrin participation in macrophage podosome organization.
These adhesive structure results are in agreement with previous investigations (Lehto et al. 1982; Marchisio et al. 1987) but contradict others (Lehto et al. 1982; Zambonin-Zallone et al. 1983; Ono et al. 1993). This discrepancy may be attributed to differing culture conditions or sources of macrophages. Alternatively, it may point to premature assignment of the terms focal or close contacts in light of continually advancing technical capabilities. In these studies, interference reflection microscopy was used as a diagnostic tool in which black spots along the ventral cell surface were termed focal adhesions (Lehto et al. 1982; Zambonin-Zallone et al. 1983; Ono et al. 1993). However, podosome-substrate separation distance has not been reported, and therefore it cannot be ruled out that these areas may instead be podosomes. Furthermore, only vinculin and actin (Lehto et al. 1982) or microtubules and microfilaments (Zambonin-Zallone et al. 1983) were immunofluorescently stained in these studies, making interpretation or comparisons difficult. The most recent report of focal contacts in mouse macrophages exploited a combination of confocal fluorescence and interference reflection microscopy in which only F-actin was stained (Ono et al. 1993). Focal contacts were identified by the co-localization of F-actin and dark interference reflection marks. F-actin at the ventral cell surface appeared more punctate than elongated as in focal contacts of fibroblasts (Burridge et al. 1988), suggesting that these structures may have instead been podosomes.
Effect of IL-13 on macrophage cytoskeletal organization. Monocytes/macrophages were cultured on untreated coverslips for 10 days and IL-13 was added on Days 3 and 7 (
Cytoskeletal filament and adhesive structure organization in monocytes, macrophages, and foreign body giant cells
Does not support macrophage fusion.
Supports macrophage fusion.
Characterization of adhesive structures may be valuable for interpretation of macrophage function in chronic inflammatory responses. Focal contacts function in the stabilization of cell adhesion to an underlying substrate (Burridge et al. 1988) and, potentially, to enable directed locomotion (Abercrombie et al. 1971). Podosomes, or so-called “invadopodia,” may degrade extracellular matrix components through the activities of putative membrane proteases (Chen 1989). Therefore, the presence of a particular type of adhesive structure may reveal specific macrophage functional activity at chronic inflammatory sites. Podosomes may enable a macrophage to mobilize membrane-associated proteases (Chen 1989) and/or other components to the ventral cell surface. Furthermore, podosome spatial alterations during FBGC formation may imply additional functional polarizations. This may reflect frustrated phagocytosis (Henson 1980) via the formation of a closed compartment (Wright and Silverstein 1984; Heiple et al. 1990) between FBGCs and the culture material surface into which degradative enzymes and/or other products are secreted. This possibility is consistent with the enrichment of a lysosomal antigen at the ventral cell surface of FBGCs compared to mononuclear macrophages (Vignery et al. 1989) and with the occurrence of material surface cracking, an indicator of degradation, directly beneath adherent FBGCs but not beneath mononuclear macrophages (Zhao et al. 1991).
The failure of cytochalasin D to destabilize podosome structure provides insight into the strength of adhesion of spread macrophages and FBGCs to synthetic surfaces. Adhesion strength may result from the combined influences of the multiple protein components of podosomes, which could lend stability to these structures. Macrophage and FBGC trypsin resistance (personal observations) could be similarly explained because the action of trypsin depends on the disruption of F-actin and subsequent rounding of cells (Britch and Allen 1980).
Our findings also demonstrate that the nature of the culture surface with which monocytes interact substantially influences the subsequent phenotypic response to IL-13. This is similar to results with IL-4 (McNally and Anderson 1995). Because the monocytes were subjected to otherwise identical culture conditions, these surface-dependent morphological responses may be explained by quantitative differences in the adsorption of serum proteins and/or by adsorption-induced conformational alterations of potentially important adhesion proteins, such as fibronectin and vitronectin.
Taken together, these studies enhance our understanding of macrophage and FBGC cytoskeletal and adhesive structural support, which is likely critical for the acquisition of functional specializations by these cells during the chronic inflammatory response.
Footnotes
Acknowledgments
Supported by the National Heart, Lung, and Blood Institute, Devices and Technology Branch, Grant HL 55714, the Whitaker Foundation, and the Center for Cardiovascular Biomaterials at Case Western Reserve University.