
Abstract
With the ongoing progress in human genome projects, many genes are discovered whose function and/or expression pattern are not known. Most of these genes are expressed in relatively low abundance compared to housekeeping genes such as elongation factor-1α and β-actin. Gene expression is studied by Northern blot assays or by semiquantitative PCR methods. Another method is the visualization of transcripts in tissue or cell cultures by fluorescence in situ hybridization (FISH). However, for low-abundance RNA detection, this method is hampered by its limited detection sensitivity and by the interference of background signals with specific hybridization signals. Background signals are introduced by nonspecific hybridization of probe sequences or nonspecific binding of antibodies used for visualization. To eliminate background signals derived from both sources and to benefit from the peroxidase-driven tyramide signal amplification (TSA), we directly conjugated horseradish peroxidase (HRP) to oligodeoxynucleotides (ODNs) and used these probes to study in the bladder cancer cell line 5637 the expression of various cytokine genes which, according to Northern hybridization and reverse transcriptase-polymerase chain reaction (RT-PCR) assays, are expressed at levels up to 10,000-fold less than abundantly expressed housekeeping genes. The results show that reduction of probe complexity and the limited use of immunocytochemical detection layers strongly reduces noise signals derived from nonspecific binding of nucleic acid probe and antibodies. The use of the HRPODNs in combination with TSA allowed detection of low-abundance cytokine mRNAs by FISH.
Keywords
T
To determine the presence and abundance of an mRNA in a tissue or cell sample, a variety of techniques are available. Northern hybridization and the more sensitive reverse transcriptase polymerase chain reaction (RT-PCR) are most frequently used for this purpose. With appropriate calibration these techniques also allow relative and absolute quantitation of mRNA abundance, and spatial and temporal differences in mRNA expression can likewise be determined. However, they do have the basic limitation that only average mRNA abundance of a tissue or cell sample can be determined, which does not provide data on the frequency and the histo- and cytological characteristics of the mRNA-producing cells.
In situ hybridization (ISH) has the ability to visualize mRNA sequences at the (sub-)cellular level and has therefore become an important additional tool for gene expression studies (Singer et al. 1986; Lawrence et al. 1989; Dirks 1996). Abundantly present housekeeping gene mRNAs, such as those for actin, glyceraldehyde phosphate dehydrogenase, and elongation factors, can be detected at subcellular resolution with nonradioactively labeled cDNA or cRNA probes and immunocytochemical visualization techniques (Lawrence and Singer 1986; Dirks et al. 1993). Although the use of nonradioactive ISH, particularly its fluorescent variant (FISH), is well established, the radioactive variant is still often applied for mRNA detection because radioisotopic probes provide a higher detection sensitivity. However, near the limits of their detection sensitivity both radioactive and nonradioactive mRNA ISH procedures meet a fundamental specificity problem. Noise signals originating from nonspecifically bound probe and antibodies have features identical to specific signals originating from the targeted mRNA sequence (e.g., silver grains or fluorescent dots) and therefore cannot be discriminated from each other. Such a specificity problem is not encountered in Northern hybridizations, in which the molecular weight information of the hybridized mRNA is an important indicator of specificity even though a background hybridization smear may be present. Likewise, in RT-PCR assays, specificity is provided by the fragment size resulting from amplification with RNA species-specific primer pairs.
In ISH, the contribution of nonspecifically bound probe to the total amount of signals obtained is assessed indirectly by execution of control experiments with sense probes or probes specific for RNA targets known not to be expressed in the cells under study. In addition, the specific probe should be hybridized to a cell sample in which no expression of the sequence is expected and, finally, mock (no probe) hybridizations will show the level of noise introduced by the various immunological detection layers. All these negative control experiments give an indication of which fraction of the ISH signals in a cell reflects noise signals and which fraction represents genuine mRNA hybridization signals.
Noise signals derive from two sources: nonspecific probe binding and nonspecific binding of the various immunological detection layers used for visualization. Reduction of probe complexity can, in principle, reduce hybridization noise. However, a decrease in probe size inevitably leads to a decrease in signal generation capacity because a smaller number of labels can be attached to shorter probe sequences. Although restricting the number of antibody layers will keep the immunocytochemical noise to a minimum, this also reduces sensitivity.
Tyramide signal amplification (TSA) has been recently introduced and has proved to considerably increase the signal intensities in various immunocytochemical and FISH applications (Kerstens et al. 1995; Raap et al. 1995; Hunyady et al. 1996). TSA employs peroxidase-catalyzed deposition of fluorochromized or haptenized tyramides for localized signal enhancement thus combining the sensitivity features of fluorescence and enzyme-based assays (Bobrow et al. 1989; van Gijlswijk et al. 1997).
All these aspects concerning the sources of noise signals, the need for reducing them for low mRNA copy number detection, and the signal generation capacity of TSA have led us to develop a new FISH strategy in which oligodeoxynucleotides (ODNs) are directly conjugated to horseradish peroxidase (HRP), the enzyme driving the TSA reaction. In contrast to hapten-labeled ODNs, this direct HRP conjugation allows deposition of tyramides immediately after hybridization and thus avoids TSA amplification of nonspecifically bound HRPconjugated primary immunoreagents (van Gijlswijk et al. 1996; Schönhuber et al. 1997).
Previously, we have demonstrated that mRNA transcribed in abundance from the human cytomegalovirus immediate-early (HCMV-IE) gene cluster of rat 9G cells can readily be detected using a single ODN-HRP probe and TSA detection. In addition, the 50 tandem repeat copies of the integrated HCMV-IE gene cluster were detectable using a single ODN-HRP and TSA detection in metaphase spreads at high efficiency (van de Corput et al. in press). This and the ability to detect the 10-20 tandem copies of the 28S ribosomal DNA gene on human chromosome 22 (Shiels et al. 1997) with a single ODN-HRP (unpublished results) indicate that the TSA system can compensate for the reduction in number of label molecules that is inherently associated with the use of ODN probes.
The ODN-HRP/TSA approach may therefore have the potential to detect low-abundance mRNA sequences by FISH. The purpose of this study was to test this hypothesis and to assess the sensitivity limits of mRNA-FISH with ODN-HRP probes and TSA detection. We used the human bladder carcinoma 5637 cell line, which shows a wide range of expression levels of a variety of cytokine genes (Fogh 1978; Kaashoek et al. 1991). The results show that detection of low-abundance mRNAs by FISH is feasible.
Materials and Methods
Cell Culture
Exponentially growing human bladder carcinoma 5637 and human osteosarcoma U2OS cells were left untreated or were treated with 50μg/ml cycloheximide (CHX) (Sigma; St Louis, MO). After 4 hr of incubation, cells were washed with PBS and used for total RNA isolation. For FISH studies, cells were cultured on uncoated glass microscope slides (Dirks et al. 1993).
Semiquantitative Northern Hybridization and RT-PCR
Semiquantitative Northern hybridization was performed according to Kaashoek et al. (1991) using decreasing amounts of fractionated total mRNA (10, 1, 0.1, 0.01μg/lane) isolated from untreated and CHX-treated cells. Hybridization was done using cytokine and housekeeping gene cDNA probes labeled with [γ32P]dCTP by random priming (Boehringer Mannheim; Indianapolis, IN).
cDNA synthesis and PCR amplification of the specific cytokine cDNA sequences were performed according to Kaashoek et al. (1991). Briefly, for total cDNA synthesis 2μg total RNA was reverse-transcribed into cDNA using 400 U reverse transcriptase (BRL; Gaithersburg, MD) and 0.8μg oligo(dT) 15 (Boehringer Mannheim) for 1 hr at 37C. PCR amplification was done with 10-fold serial dilutions of cDNA with 5′sense and 3′anti-sense primers specific for various cytokine mRNAs. The PCR reaction was performed on a programmable Omni-Gene Thermal Cycler block (Hybaid Limited; Biozym, Landgraaf, The Netherlands).
Probes
Cytokine cDNA probes were labeled with digoxigenin-11-dUTP (Boehringer Mannheim) by standard nick-translation. 30-mer 5′-aminohexyl-oligodeoxynucleotides (amino-ODNs) were purchased from Eurogentec (Seraing, Belgium) (for nucleotide sequence, position, and genebank accession numbers see Table 1) and checked for absence of human sequence cross-homologies using the BLAST program. The amino-ODNs were conjugated to horseradish peroxidase (Pierce; Rockford, IL) using bifunctional crosslinkers and were purified by IE-HPLC as described by van Gijlswijk et al. (1996).
Just before hybridization, the ODN-HRP probes were diluted in hybridization mixture containing 40% deionized formamide, 2 × SSC (0.3 M sodium chloride, 0.03 M sodium citrate, pH 7.0), 10% dextran sulfate, 10 mM EDTA, and 2 × Denhardt solution to a final concentration of 100 ng/ml.
In Situ Hybridization and TSA Detection
RNA-FISH was performed according to Dirks et al. (1993) with a few modifications. The RNA targets were denatured before hybridization with 70% deionized formamide/2 × SSC at 80C for 2.5 min. Cells were washed with ice-cold 2 × SSC, dehydrated, and air-dried. Next, 10μl of hybridization mixture was applied to the cells and covered with an 18 × 18-mm coverslip. Hybridization was for 30 min at 37C in a moist chamber. After hybridization, coverslips were removed and cells washed briefly in 2 × SSC at 37C, followed by a stringent 5-min wash with 40% formamide, 2 × SSC, pH 7.0, at 37C. Finally, cells were washed three times for 2 min with 2 × SSC at room temperature and rinsed briefly in TBS (100 mM Tris-HCl, pH 7.5, 150 mM NaCl) containing 0.05% Tween- 20 (TNT). The in situ hybridized ODN-HRPs were detected using the biotin TSA plus reagent (Adler et al. 1997, a generous gift from NEN Life Science Products, Boston, MA). After an incubation of 30 min at 37C, slides were washed three times for 5 min in TNT followed by an incubation with mouse anti-biotin fluorescein (Sigma; 1:250) diluted in TBS containing 0.5% (w/v) Boehringer Blocking Reagent (Boehringer Mannheim) (TNB), followed by an incubation with rabbit anti-mouse fluorescein (Sigma; 1:500) diluted in TNB. After each antibody incubation, slides were washed three times for 5 min with TNT.
Nucleotide sequence of anti-sense and sense-specific HRP-labeled oligodeoxynucleotide probes used for FISH studies; nucleotide sequence numbering and gene bank accession numbers are according to the references a
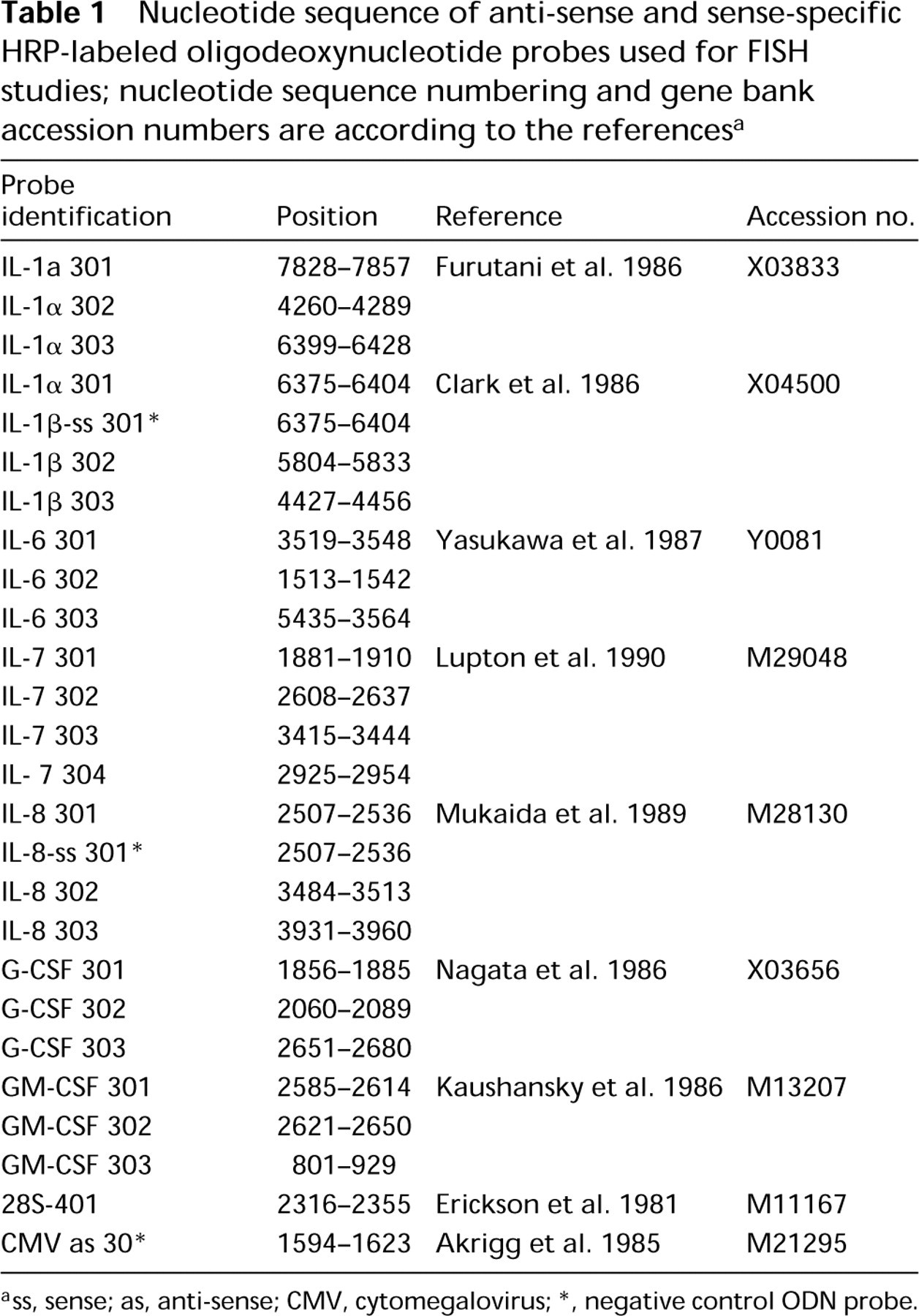
ss, sense; as, anti-sense; CMV, cytomegalovirus
negative control ODN probe.
For cytoplasmic and nucleolar staining, a 40-mer 28S ribosomal RNA-specific ODN probe was labeled with Texas Red (28S-TR). After the final immunological detection layer was applied, cells were hybridized with the 28S-TR probe for 20 min at 37C as described above. Cells were washed with 2 × SSC for 5 min at 37C, dehydrated, air-dried, and embedded in Vectashield (Vector Labs; Burlingame, CA) containing 40 ng/ml 4,6-diamidino-2-phenylindol.2HCl (DAPI) for nuclear counterstaining.
Slides were examined with a Leica DM microscope equipped with a single bandpass filter for fluorescein and Texas Red and × 40, × 63 objectives and a × 100 oil objective with 1.3 numerical aperture. Photographs were taken with Scotch 3M 640 ASA color slide film and slides were exposed for 30 sec.
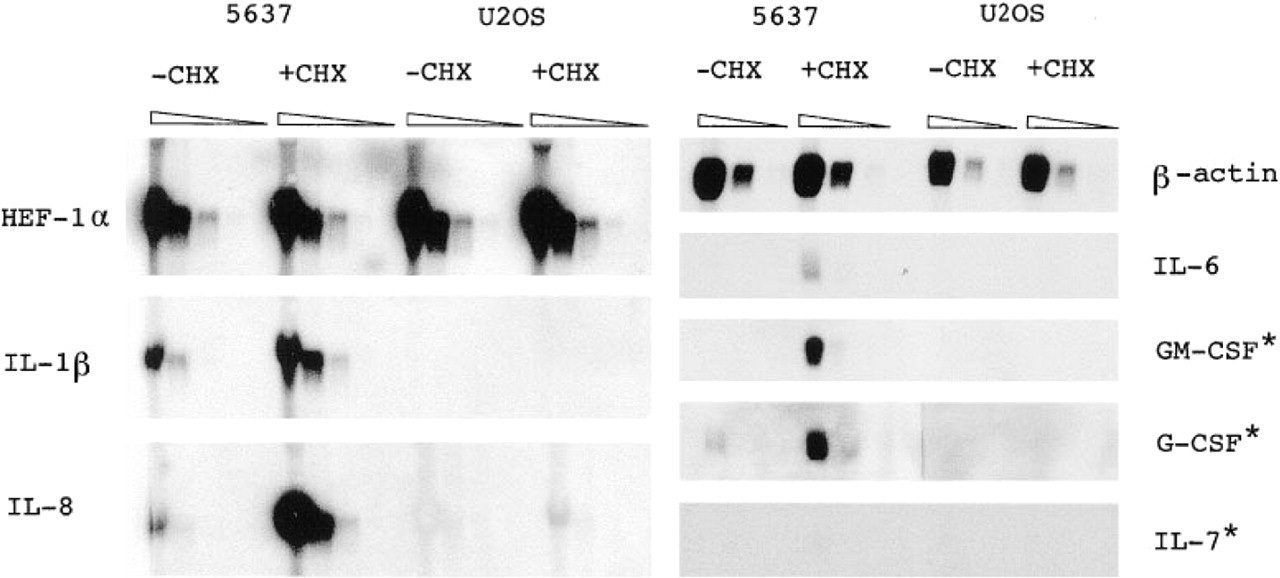
Semiquantitative Northern hybridizations. Northern blot analysis was performed on 10-fold decreasing amounts of fractionated total mRNA (10, 1, 0.1, 0.01 mg total RNA/lane) and probed with the various RT-PCR generated cytokine and housekeeping gene cDNAs. ∗, 4-day autoradiography; triangles, decreasing amounts of total RNA.
Results
At the beginning of this study we could not predict where the sensitivity limits of the RNA-FISH technique would lie. Therefore, we broadened the mRNA level range for methodological purposes by treating the cells with CHX, a protein synthesis inhibitor. The U2OS cell line, which does not express cytokines, was used as a negative cell control throughout this study.
Relative Quantification of mRNA Levels by Northern Hybridization and RT-PCR
Northern hybridizations were performed on 10-fold decreasing amounts of total RNA extracted from untreated and CHX-treated 5637 and U2OS cells. The results are shown in Figure 1.
Untreated 5637 cells showed distinct interleukin (IL)-1β and IL-8 mRNA bands after 24-hr autoradiographic exposure, whereas IL-6, granulocyte/macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), and IL-7 mRNA were undetectable. After 4 days of autoradiography a faint G-CSF mRNA band was detectable but the IL-6, GM-CSF, and IL-7 remained undetectable. CHX treatment resulted in increased IL-1β, IL-8, IL-6, and G-CSF mRNA expression. IL-7 mRNA remained undetectable, and GM-CSF mRNA transcripts were detected only after 4 days of autoradiography. U2OS cells showed no cytokine mRNA expression except for low IL-8 expression after CHX treatment. Expression levels of the two housekeeping genes did not change in 5637 and U2OS cells after CHX treatment. The quantitatively less reliable but more sensitive RT-PCR experiments confirmed the Northern hybridization results (see Figure 2). Because of its higher sensitivity, IL-6, G-CSF, GM-CSF and IL-7 mRNAs were detectable in untreated and CHX-treated 5637 cells. The RT-PCR for IL-8 clearly showed the CHX induction of IL-8 mRNA expression in U2OS cells. U2OS cells (CHX-treated or not) were weakly positive for a few cytokine mRNAs with undiluted total cDNA (IL-6, GM-CSF, and IL-7). The Northern hybridzation and RT-PCR studies showed that in untreated 5637 cells cytokine mRNAs can be detected at levels up to 10,000-fold less than an abundantly expressed housekeeping gene such as human elognation factor-1α (HEF-1α). (Quantitative interpretation of the Northern hybridzation and RT-PCR experiments is in Table 2).
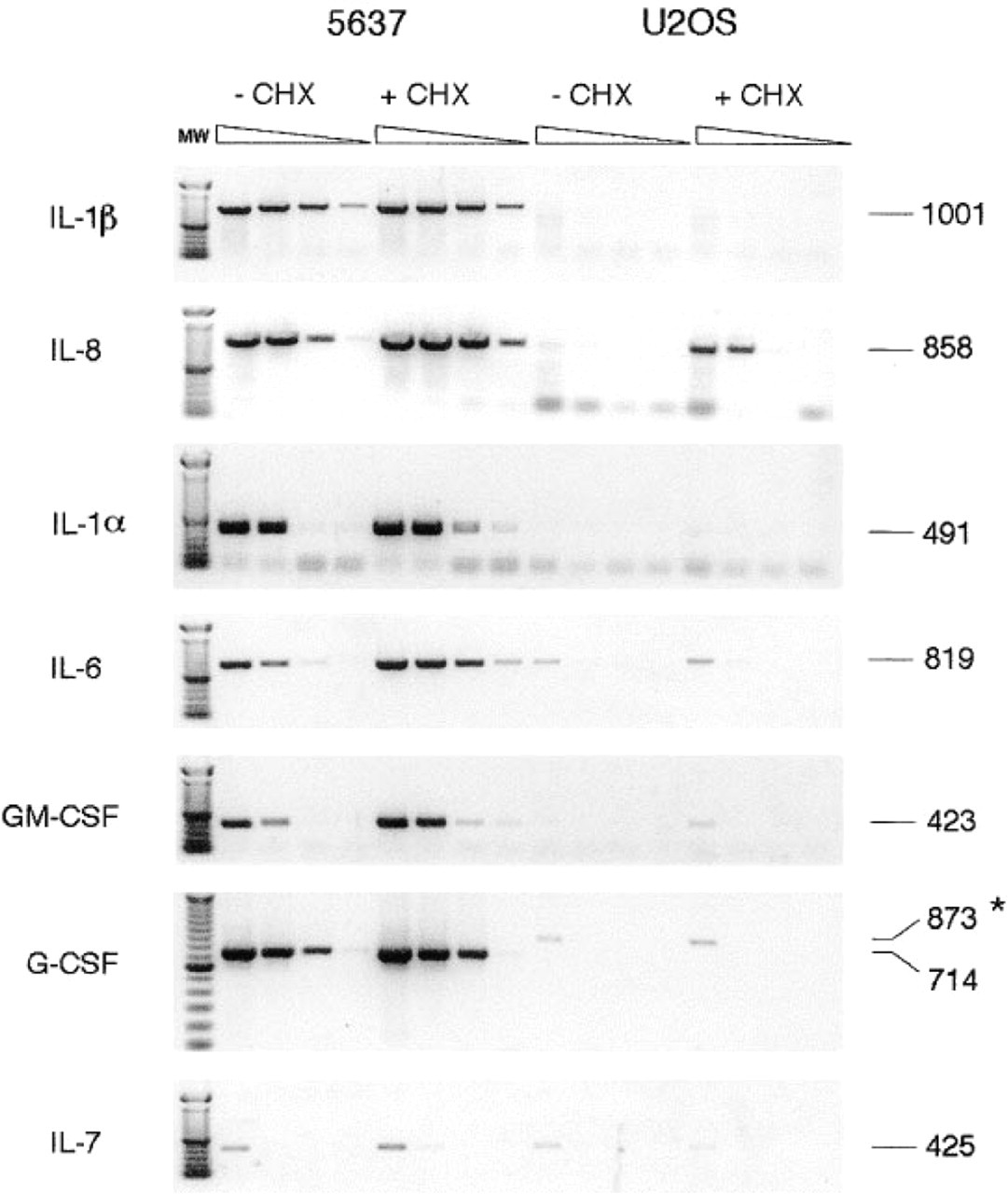
Semiquantitative RT-PCR. cDNA samples were obtained from untreated and CHX-treated 5637 and U2OS cells and were used in 10-fold dilution series for PCR amplification of HEF-1α and various cytokine mRNAs. The figure is a negative of ethidium bromide-stained gels. Numbers at right are fragment sizes in bp. Specific cytokine primer pairs were used that allow distinction of possibly contaminating genomic DNA. ∗, the 873-
Optimization of mRNA-FISH
Initially, we optimized the TSA detection procedure for RNA-FISH using either digoxigenin- or HRP-labeled ODN probes specific for IL-1β or IL-8. The following experimental parameters were tested: (a) the use of direct TSA reagents (fluorescein-, rhodamine-, and Cyanine3-tyramides) and indirect TSA reagents (biotin TSA and biotin TSA plus (Adler et al. 1997); (b) different TSA development temperatures (4C, 22C, and 37C); and (c) two different immunocytochemical biotin detection procedures (avidin-fluorescein and antibiotin-Ig-fluorescein).
The use of all direct TSA reagents led to an overall intense fluorescent staining of the cells and prevented detection of specific hybridization signals above this relatively high level of fluorescence. This staining could be caused by binding of the charged fluorescent moiety of the direct TSA reagents to the protein-rich cell matrix. Using the indirect biotin TSA reagents, such cytological staining was not observed. When the performances of biotin TSA and biotin TSA plus were compared, it appeared that the use of biotin TSA plus resulted in far more intense signals. Furthermore, TSA development at 37C was superior to development at 4C or 22C in terms of fluorescent intensity of the signals.
On hybridization of digoxigenin-labeled negative control ODN probe and deposition of biotin-TSA plus after anti-digoxigenin-HRP incubation, punctate fluorescent dots were observed when either avidin-fluorescein or mouse anti-biotin-fluorescein was used for visualization. Similar observations were made with cell control experiments. This clear background pattern could not be attributed solely to nonspecific hybridization of the digoxigenin ODN probe, because with mock (no probe control) hybridizations fewer but still clear background spots were observed. In contrast, hybridization of control ODN-HRP probes did not lead to the punctate background pattern, and we therefore concluded that the primary mouse antidigoxigenin-HRP detection layer contributes strongly to distinct background signals on cells and glass surface.
Semiquantitation of mRNA levels by Northern hybridization and reverse transcriptase-PCR of two housekeeping genes and various cytokines in untreated and CHX-treated 5637 and U2OS cells; HEF-1α RNA levels are arbitrarily set at 100 a
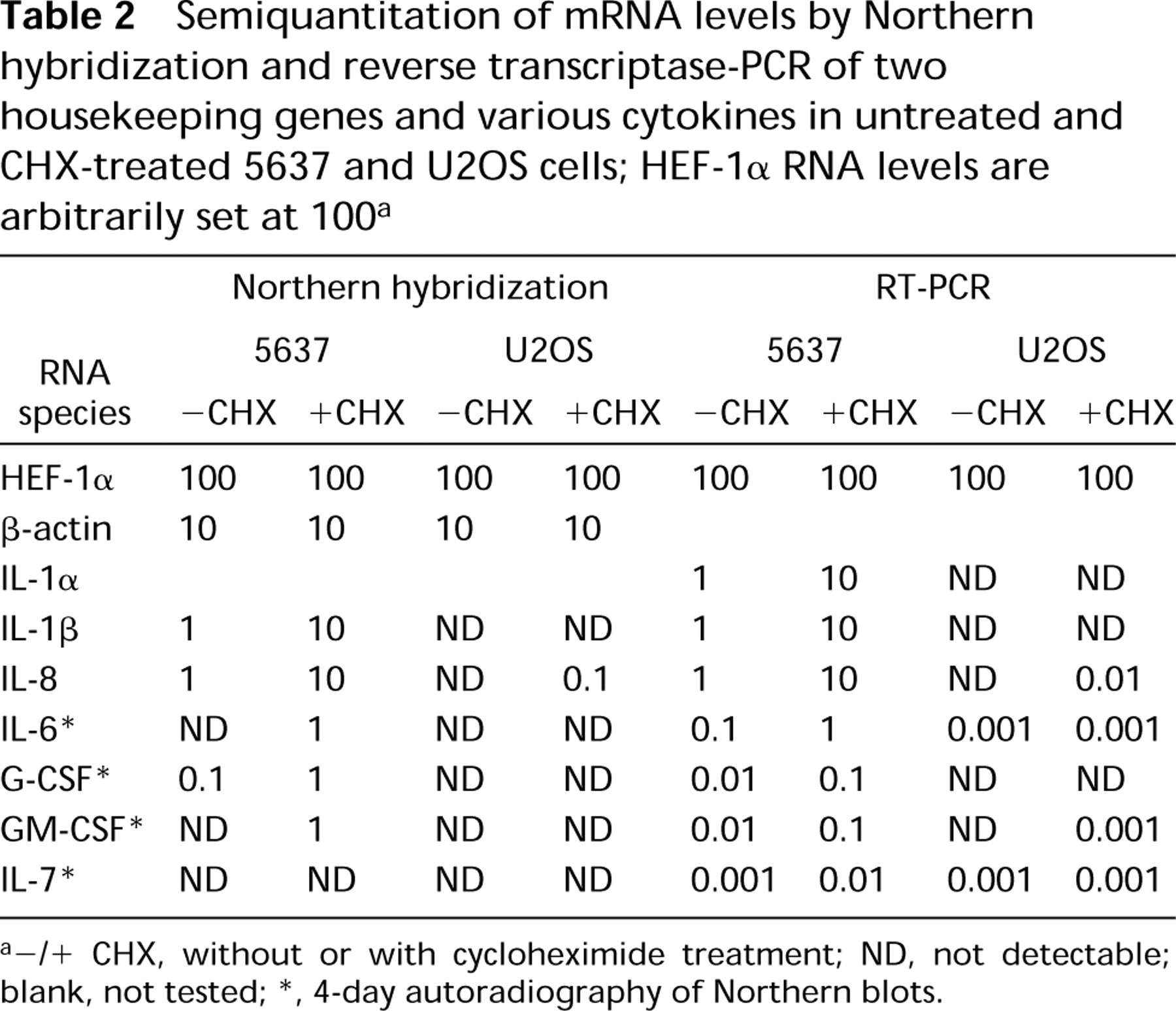
-/+CHX, without or with cycloheximide treatment; ND, not detectable; blank, not tested
4-day autoradiography of Northern blots.
We noted that avidin-fluorescein caused some nonspecific staining in 5637 cells, but this proved to be cell type-dependent.
Additional variables tested included ODN-HRP probe concentration, stringency and time of hybridization, and stringency and time of posthybridization washes. All ODNs were 30-mers with 43% GC content. When the formamide concentration in the hybridization or washing solution was raised above 45% and when hybridization times exceeded 30 min, loss of hybridization signals was observed. This loss is most likely caused by inactivation of the HRP moieties (van Gijlswijk et al. 1996).
These experiments led to the ODN-HRP/TSA RNA FISH protocol described in Materials and Methods (see also Figure 3 for a schematic representation).
Detection of Cytokine mRNA by FISH
5637 cells were probed with digoxigenin-labeled cDNA probes for IL-1α, IL-1β, IL-6, IL-8, and GM-CSF cytokine mRNAs, and hybrids were detected with a conventional two-layer immunological detection system. The most abundant of these cytokine mRNAs, IL-1β (Figure 4A) and IL-8, showed a heterogeneous expression pattern. The lower-abundance cytokine mRNAs showed no signals that exceeded the autofluorescence levels of the cells.
To enhance the fluorescence intensity of the IL-1β mRNA-specific hybridization signals, sheep anti-digoxigenin-HRP followed by biotin TSA plus detection was used instead of the conventional detection method (Figure 4B). Although strong signals were obtained, a negative probe control showed a considerable amount of noise signal (Figure 4C). The amount and intensity of the noise signal are such that they would interfere strongly with detection of specific signals of low-abundance mRNAs.
Next, we tested the use of a single ODN-HRP probe specific for IL-1β or IL-8 mRNA. When the hybridizations were followed by the biotin TSA plus detection, clear heterogeneous distribution patterns of these mRNAs were observed (yellow fluorescence in Figures 5A and 5B). The contrast between the specific hybridization signals and the background signal had improved considerably compared to the conventional RNA-FISH approach (compare Figures 4A and 5A). Hybridizations performed with negative control ODN-HRP (sense and nonspecific) probes revealed a small amount of background signal (Figure 5C).
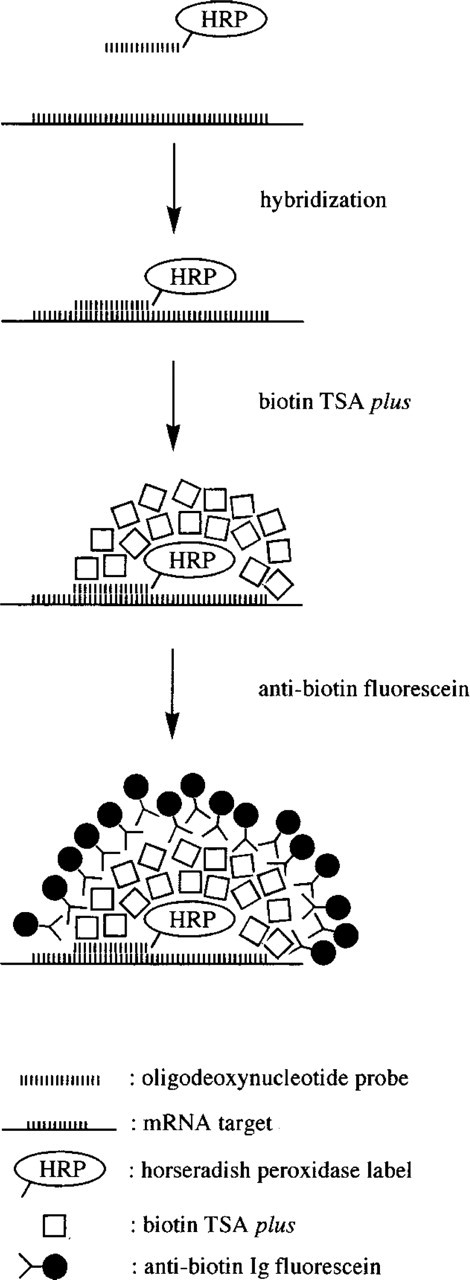
Schematic representation of the HRP-ODN/TSA mRNA-FISH procedure.
To assess the accessibility of cells to probes and to accomplish cytoplasmic and nucleolar counterstaining of the cells, in situ hybridization of a Texas Red-labeled ODN probe specific for 28S rRNA was performed after the ODN-HRP/TSA mRNA FISH. The cytoplasm and nucleoli of all cells revealed a positive 28S rRNA hybridization signal (red signal in Figures 5A–5E). To further ascertain that the heterogeneous expression patterns of the interleukin mRNAs were not the result of poor accessibility of a subpopulation of cells to nucleic acid probes and reagents, cells were also hybridized with a probe specific for elongation factor mRNA. After hybridization, all cells revealed hybridization signals of almost equal number and intensity, suggesting that all cells are well accessible to probes and antibodies (result not shown). Therefore, the apparent heterogenous cytokine RNA expression pattern cannot be attributed to methodological pitfalls.
FISH of the lower-abundance cytokine RNAs in untreated 5637 cells (i.e., IL-1α, IL-6, GM-CSF, G-CSF, and IL-7 in untreated 5637 cells) using a single ODN-HRP and TSA detection resulted in mRNA signals that had either low fluorescence intensity, as illustrated in Figure 6A for G-CSF mRNA, or no signal above background. Therefore, cocktails of ODN-HRP probes were used, which led to an increased fluorescence intensity of the hybridization signals of mRNAs already detectable with one ODN-HRP. Two of the three mRNAs not detectable with a single ODN-HRP could be visualized with cocktails. Figure 6B shows the intensity of G-CSF mRNA after hybridization with a cocktail of two ODN-HRP probes and Figure 6C after hybridization with a cocktail of three. IL-7 mRNA could be convincingly detected by FISH only with a probe cocktail of four ODN-HRPs (Figure 6D). GM-CSF mRNA was not detectable with the cocktail of three used. The use of probe cocktails led to an increased amount of background signal. However, this additional contribution to noise was outweighed by the gain in signal (compare Figures 6A and 6C).
Except for IL-8 hybridizations, no clearly cytokine mRNA-containing U2OS cells were observed, although a few cytokines showed faint RT-PCR bands. The increase in cytokine mRNA levels in 5637 cells induced by CHX treatment, as determined by Northern hybridization and RT-PCR, was confirmed by RNA-FISH experiments. In 5637 cells, CHX treatment generally resulted in an increase in fluorescence intensity of the specific hybridization signals and in the number of cytokine RNA-containing cells. GM-CSF mRNA, not detectable in untreated 5637 cell, could clearly be detected in a subpopulation of the CHX-treated 5637 cells. In line with the Northern and RT-PCR data, the IL-8 gene revealed strong induction of expression in a subset of the CHX-treated U2OS cells (Figure 5E). In untreated and CHX-treated 5637 cells, one or two nuclear RNA signals were observed next to clear cytoplasmic signals. These signals most likely represent cytokine transcription sites (indicated by arrows in Figures 5A and 5E).
The RNA-FISH results obtained with the ODN-HRP/TSA method are summarized in Table 3.
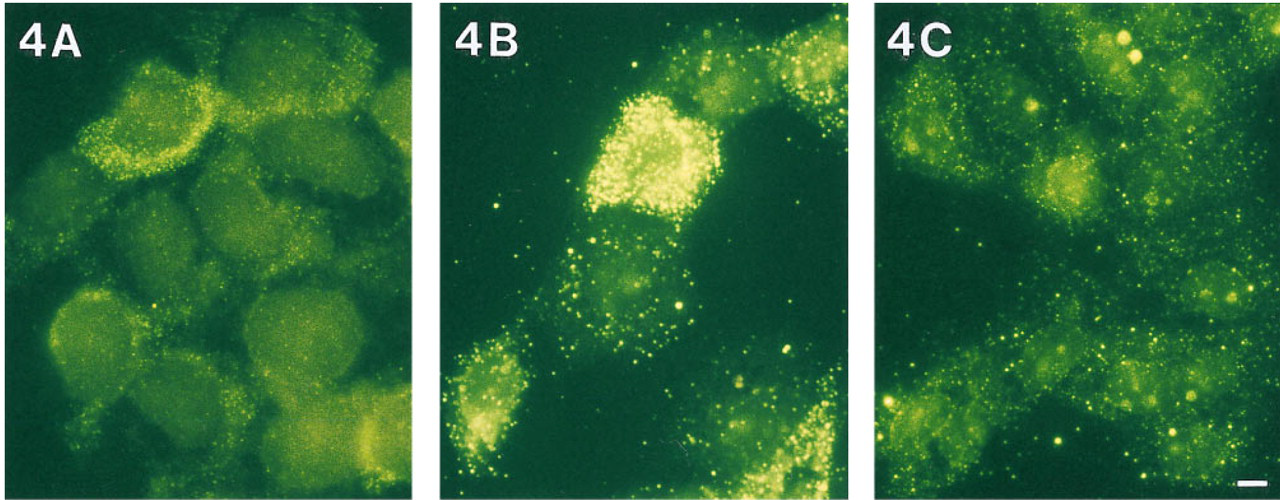
Detection of IL-1b mRNA in untreated 5637 cells using digoxigenin-labeled cDNA probes. Detection was performed using either conventional immunocytochemistry (
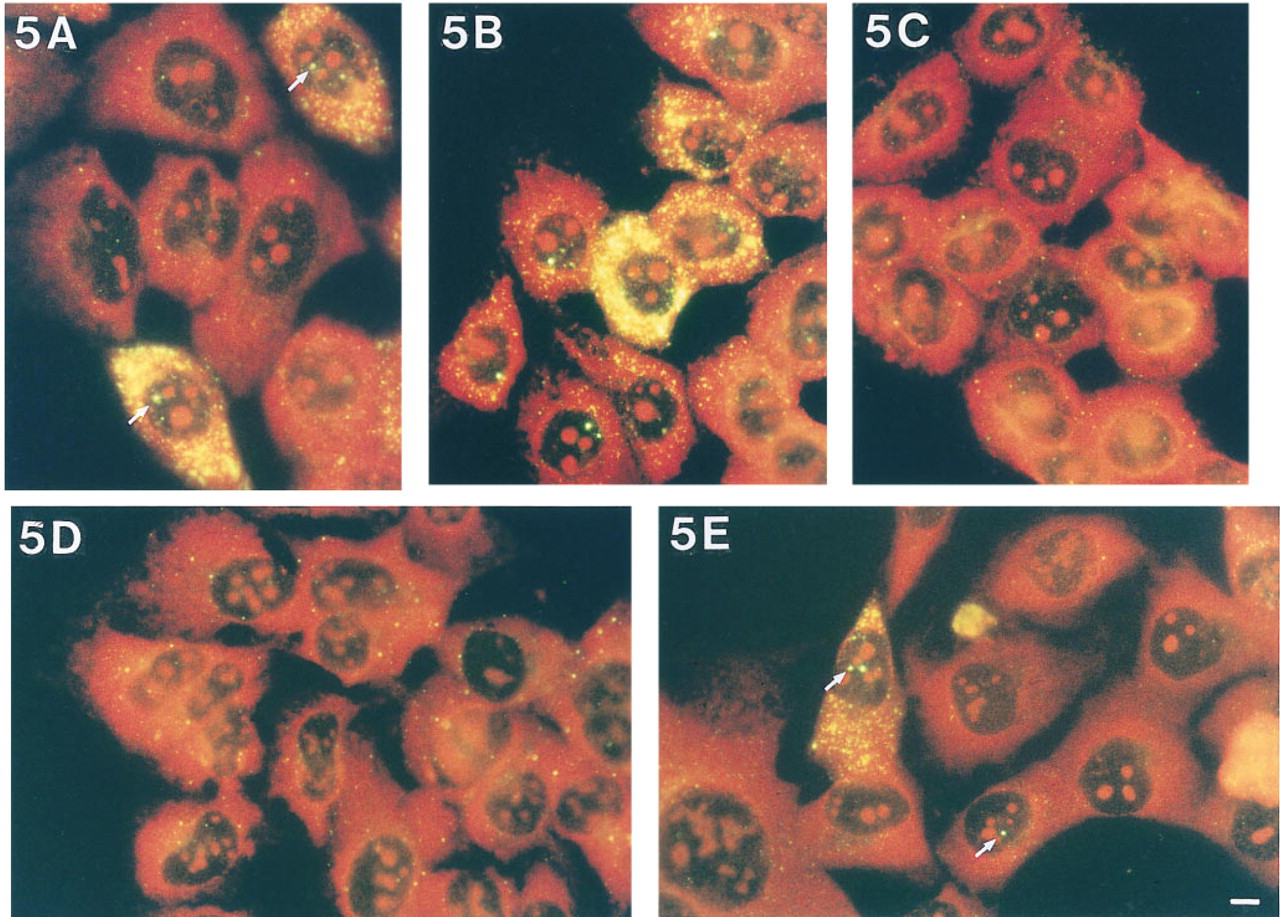
mRNA-FISH performed with a single IL-1b- or IL-8 HRP-labeled ODN probe followed by biotin TSA plus detection showed heterogeneous mRNA expression patterns (yellow signals) for IL-1b (
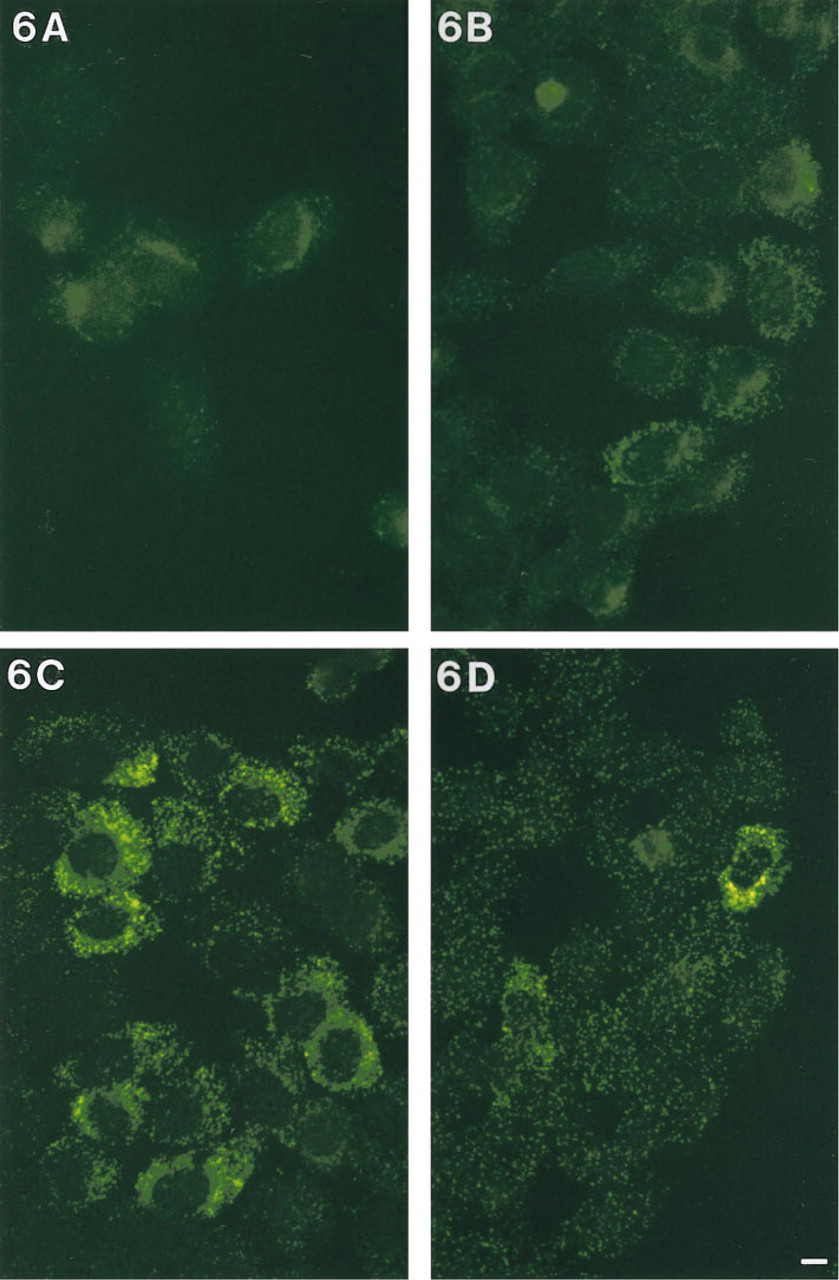
Detection of G-CSF mRNA in untreated 5637 cells using one (
Discussion
To tackle the problem of low-abundance transcript detection by mRNA-FISH, we extensively explored the potential of ODN-HRP probes and TSA in RNA-FISH, hypothesizing that the use of small probes would reduce nonspecific probe binding and that TSA would compensate for the loss of signal generation capacity associated with the use of small probes (van Gijlswijk et al. 1996). The results support this hypothesis. TSA indeed compensates for loss of signal generation capacity when a switch is made from cDNAs (425-1024 nucleotides) to ODNs (30-mers), and hybridization noise is reduced considerably. The second important gain in noise reduction was obtained by directly conjugating the TSA-driving enzyme (horseradish peroxidase) to the ODN probe. In this way the TSA detection can be applied immediately after hybridization. Therefore, the primary HRP-conjugated antibody layer that introduces the most prominent antibody-related background with haptenized probes can be omitted. The reduction of nonspecific hybridization and nonspecific antibody binding resulted in an increase in contrast, enabling us to visualize cytokine mRNAs that could not or could only marginally be detected with Northern hybridization and RT-PCR.
Semiquantitative RNA-FISH interpretation of untreated and CHX-treated 5637 and U2OS cells using single or cocktail ODN-HRP probes followed by biotin TSA plus detection developed at 37C a
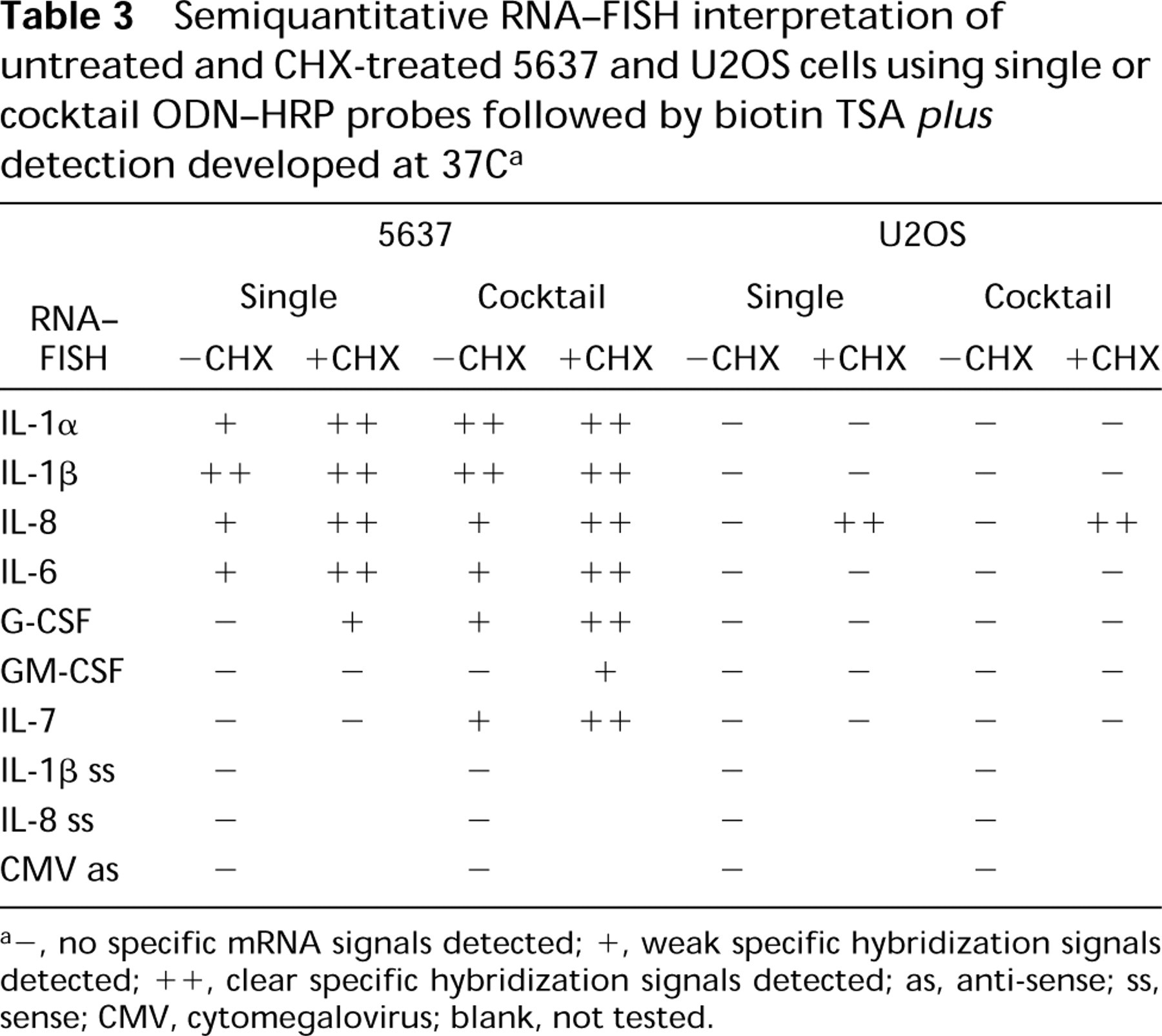
-, no specific mRNA signals detected; +, weak specific hybridization signals detected; ++, clear specific hybridization signals detected; as, anti-sense; ss, sense; CMV, cytomegalovirus; blank, not tested.
We selected the 5637 cell cytokine expression model in the anticipation that on a cellular basis interleukin expression would be fairly homogeneous. It apparently is not. This complicates correlation of the FISH data with the semiquantitative Northern hybridization and RT-PCR results and precludes determination of the sensitivity of the novel RNA-FISH methodology in terms of copy number per cell, although the ODN-HRP/TSA FISH methodology has led to mRNA detection that was not possible with other FISH approaches.
The heterogeneous expression patterns of the cytokine mRNAs indicate that a subpopulation of cells in the clonal 5637 cell line is transcriptionally active within a certain time frame. This is also reflected in the number of cells showing nuclear RNA signals. The minority of cells showed only one or two nuclear spots in the absence of a cytoplasmic RNA signal. Apparently these cells had activated a gene just before fixation of the cells. Another population of cells revealed, in addition to the nuclear RNA signals, a cytoplasmic signal, indicating that these cells were already transcriptionally active for some time before fixation. Finally, the largest population of cytokine mRNApositive cells revealed cytoplasmic signals only, suggesting that these cells had turned off their cytokine gene transcription shortly before fixation.
The mechanisms underlying gene expression are complex. The expression of some genes is strictly correlated with a phase of the cell cycle, whereas the expression of others is strictly regulated in development. In addition to transcription factors involved in regulation of gene transcription, long-range chromatin interactions may play a role in regulation of gene expression. For example, studies of mRNA hybridization patterns of individual cells have been shown that the order of transcriptional activity of globulin genes is regulated in time and is dependent on the presence of a locus control region (Wijgerde et al. 1995). This example may illustrate that detailed analysis of in situ mRNA hybridization patterns can provide important information about regulation of gene expression. The methodology presented here is likely to contribute significantly to gene expression studies at the single-cell level.
As observed in RT-PCR experiments, U2OS cells showed very low expression of IL-6, GM-CSF, and IL-7. However, these low RNA abundances could not be detected with our FISH methodology, indicating that more sensitive in situ transcript detection is needed. An obvious way to increase sensitivity is to apply repeated TSA rounds. However, this leads to unacceptably high levels of background signal (results not shown), but it also shows that with TSA strategies more sensitive transcript detection should be feasible if noise can be further reduced.
Recently, several new probe designs have been reported that may be of use in reaching the aim of further reduction of nonspecific probe binding in FISH. Nilsson et al. (1994) introduced so-called padlock probes to improve discrimination of single-base mismatches. Padlock probes are oligonucleotides with two probe sequences 20 nucleotides long at the 39 and 59 ends and a 30 to 40-mer spacer between them that carries hapten molecules for detection purposes. The two probe sequences are chosen such that, in the hybrid molecule, the 39 and 59 ends are juxtaposed. A ligation step then covalently links the two probe sequences and closes the padlock probe. Two features are of interest with respect to the uniqueness of hybridization of padlock probes: the juxtaposition of the two probe sequences of one padlock molecule requires two hybridization events, and the formation of a circular molecule allows extreme stringency conditions in the wash step. Therefore, any haphazard hybridization of a single probe sequence can be eliminated in the ligation and washing steps. After application of the padlock methodology in Southern hybridizations, it was successfully applied in DNA-FISH, including the detection of single-base mismatches of centromeric repeat sequences (Nilsson et al. 1997). Although providing superb specificity of hybridization, the sensitivity of the padlock methodology is still an issue. Signal amplification by TSA has indeed been suggested as a means to improve the sensitivity of padlock detection (Lizardi and Ward 1997).
Use of polypeptide nucleic acid (PNA) probes has also been advocated recently to improve specificity of hybridization. PNAs form more stable hybrids with DNA and RNA sequences than DNA or RNA probes do. In addition, PNAs have greater discriminatory power because of the greater differences in Tm of perfectly matching and single-base mismatching PNA.DNA or PNA.RNA duplexes (8-20C) (Egholm et al. 1993). A few recent studies describe the use of PNA probes in FISH to high copy number targets. Lansdorp et al. (1996) used PNAs to detect highly repetitive telomere sequences in metaphase spreads and interphase nuclei, and Thisted et al. (1996) and Taneja (1998) used PNA probes to detect, respectively, immunoglobulin-k lightchain mRNA in lymphoid tissue sections and transcripts containing CTG triplet repeats in human myotonic dystrophy cells. In combination with TSA, PNAs may allow low copy number mRNA detection as well.
In conclusion, we developed and explored a novel RNA-FISH technology, based on ODNs and HRP-mediated tyramide signal amplification, that permits detection of transcripts in situ that are not or are only marginally detectable by other means. This ODN-HRP/TSA mRNA-FISH approach is therefore expected to find broad application in basic cell biology and molecular pathology research.
Footnotes
Acknowledgments
Supported in part by the Dutch Science Organization, Area Medical Sciences (MW-NWO) project no. 900-543-109 and by NEN Life Science Products, Boston, MA. CMH was supported by FWF, Austria, no. J1481-MED.