
Abstract
To understand the biological relationships among various molecules, it is necessary to define the cellular expression patterns of multiple genes and gene products. Relatively simple methods for performing multi-label immunohistochemical detection are available. However, there is a paucity of techniques for dual immunohistochemical (IHC) and mRNA in situ hybridization (ISH) detection. The recent development of improved non-radioactive detection systems and simplified ISH protocols has prompted us to develop a tyramide signal amplification method for sequential multi-label fluorescent ISH and IHC detection in either frozen or paraffin-embedded tissue sections. We used this method to examine the relationship between glial cell line-derived neurotrophic factor receptor α2 (GFRα2) mRNA expression and IHC localization of its co-receptor Ret in the trigeminal ganglion of postnatal Day 0 mice. We found that approximately 70% of Ret-immunoreactive neurons possessed GFRα2 mRNA and virtually all GFRα2-expressing neurons contained Ret-immunoreactive protein. Finally, we used paraformaldehyde-fixed, paraffin-embedded sections and a monoclonal antibody against neuron-specific nuclear antigen (NeuN) to demonstrate the neuronal specificity of GFRα2 mRNA expression in adult mouse brain. This multi-labeling technique should be applicable to a wide variety of tissues, antibodies, and probes, providing a relatively rapid and simple means to compare mRNA and protein localization.
Keywords
I
Historically, ISH was developed using radiolabeled probes (Baldino and Lewis 1989; Baldino et al. 1994). Although sensitive, the use of radioactive isotopes is associated with several intrinsic and extrinsic difficulties, e.g., poor spatial resolution of signal, long exposure times, health risks associated with handling radioactive material, and high costs of radioactive material disposal. For these reasons, nonradioactive ISH techniques have gained popularity and provide solutions to many of the problems associated with radioactive probes (Lewis et al. 1988; Baldino and Lewis 1989; Baldino et al. 1994). However, most of these nonradioactive ISH methods lack the sensitivity of radioactive techniques, limiting their usefulness to relatively abundant mRNAs and to single probe detection. The recent development of the tyramide signal amplification (TSA; NEN Life Science Products, Boston, MA) method has dramatically increased the sensitivity of nonradioactive ISH detection and offers several advantages over alkaline phosphatase (AP)-based localization procedures. TSA is based on the horseradish peroxidase (HRP)-catalyzed deposition of labeled tyramine molecules at sites of probe binding (Bobrow et al. 1989, 1991, 992). In contrast to typical AP substrates, which precipitate diffusely at sites of AP enzyme activity, tyramine is converted by HRP into a highly reactive oxidized intermediate which binds rapidly and covalently to cell-associated proteins at or near the HRP-linked probe. Therefore, TSA provides better spatial resolution of signal than AP-based methods and limits the potential interference of deposited reaction product in multi-probe detection applications. First reported in 1995, TSA has been used to detect both DNA and mRNA sequences in cells and tissue sections (Kerstens et al. 1995; Raap et al. 1995; Schmidt et al. 1995; Adler et al. 1997; van de Corput et al. 1998) and has consistently proved equal to or better than standard fluorescent or chromogenic nucleotide detection techniques (van Gijlswijk et al. 1996, 1997; Komminoth and Werner 1997). For fluorescence in situ hybridization (FISH), TSA detection may provide up to a 100-fold increase in signal compared to conventional fluorescent probes (van Gijlswijk et al. 1996; Macechko et al. 1997). Several investigators have demonstrated that TSA in situ detection of viral DNA in infected cells is as sensitive as in situ polymerase chain reaction detection and is much easier to perform (Zehbe et al. 1997; Wicdorn et al. 1999).
Although most authors have used TSA ISH to detect DNA sequences, TSA has also been used to localize mRNA expression in the nervous system. TSA has proved more sensitive than AP deposition of BCIP/NBT for detection of oxytocin mRNA in rat brain sections and for peptide hormone mRNA detection in paraffin-embedded human tissue sections (Tyler and Mayer 1998; Speel et al. 1998, 1999). Recently, TSA has been used in combination with AP to perform dual fluorescence ISH and immunohistochemistry (IHC) (Hoon et al. 1999).
In this article we describe a method for performing dual mRNA ISH and antigen IHC on either frozen or paraffin-embedded sections using fluorescent TSA techniques. We used this method to examine the relationship between GFRα2 mRNA expression and Ret protein immunoreactivity in the developing mouse nervous system and GFRα2 mRNA expression and NeuN immunoreactivity in the adult mouse brain.
Materials and Methods
Tissue Preparation
Young adult and timed-pregnant Swiss-Webster mice were purchased from Harlan Sprague Dawley (Indianapolis, IN). Postnatal Day 0 and adult mice were deeply anesthetized with methoxyflurane (Metofane; Pitman-Moore, Mundelein, IL), decapitated, and their brains placed in embedding medium (Tissue Tek OCT compound; Miles, Elkhart, IN) and frozen on dry ice in liquid chlorodifluoromethane (HistoFreeze; Fisher Scientific, Pittsburgh, PA). Alternatively, tissues were fixed for 2–4 hr in 4% paraformaldehyde at room temperature (RT) and paraffin-embedded.
Probes, Antibodies, and Reagents
GFRα2 cRNA probe was generated from a plasmid containing GFRα2 nucleotides 1002–1417 of GenBank accession number AF002701 (Golden et al. 1998). The plasmid was linearized and labeled with either digoxigenin or biotin RNA labeling kits according to the manufacturer's instructions (Boehringer Mannheim; Indianapolis, IN). We have previously demonstrated the specificity of this probe sequence for ISH detection of GFRα2 mRNA (Golden et al. 1998; Heuckeroth et al. 1999). Rabbit anti-Ret antibodies (Santa Cruz Biotechnology; Santa Cruz, CA) were used to immunolocalize the GFRα2 co-receptor Ret in postnatal Day 0 trigeminal ganglia and mouse anti-neuronal nuclei (NeuN) antibodies (Chemicon; Temecula, CA) were used to identify neurons by IHC in the adult brain.
TSA reagents were obtained from NEN Life Science Products. Direct fluorescence deposition was performed using TSA Plus Direct-Green or TSA Plus Direct-Cyanine 3. Indirect fluorescence deposition was performed with TSA Plus DNP (HRP) followed by TSA Plus Direct-Cyanine 3.
Dual GFRα2 ISH and Ret IHC Detection on Frozen Sections
Sixteen-μm-thick coronal sections of postnatal Day 0 mouse brain were cut, air-dried, and fixed in 4% paraformaldehyde in PBS (10 mM phosphate, pH 7.2) for 30 min at 25C. All steps were performed at RT unless otherwise indicated. Sections were washed three times for 5 min each in PBS and incubated for 30 min in 0.1% active DEPC (in PBS). Sections were again washed in PBS and endogenous peroxidase activity was inhibited by incubation in 0.3% H2O2 (in PBS) for 30 min. Sections were then washed in PBS and incubated in 5 × SSC (0.75 M NaCl, 0.75 M Na-citrate) for 15 min. Tissue sections were then prehybridized for 2 hr at 58C in prehybridization buffer (5 × SSC, 50% formamide, pH to 7.5 with HCl, 50 μg/ml salmon sperm DNA). Slides were then incubated with digoxigenin-labeled probe (100–500 ng/ml of probe in hybridization buffer) for 12–24 hr at 58C. Posthybridization washes were performed with 2 × SSC and 0.1 × SSC (each for 1 hr at 65C) and sections were then washed in PBS and incubated for 30 min in PBS-BB (PBS containing 1.0% bovine serum albumin, 0.2% powdered skim milk, and 0.3% Triton X-100). Digoxigenin-labeled GFRα2 probe was localized with HRP-conjugated sheep anti-digoxigenin antibodies (Roche Molecular; Indianapolis, IN) diluted 1:1000 in PBS-BB and incubated for 1 hr followed by PBS washes and TSA Plus Direct-Cyanine 3 deposition according to the manufacturer's protocol (NEN Life Science Products). If only single ISH detection was performed, slides were then washed in PBS, nuclei labeled with Hoechst 33258 (0.2 μg/ml; Sigma, St Louis, MO), sections mounted in PBS:glycerol (1:1), and viewed with a Zeiss Axioskop microscope equipped with epifluorescence.
For dual GFRα2 ISH and Ret IHC localization, residual HRP activity from the initial TSA reaction described above was destroyed by incubation in 0.3% H2O2 in PBS for 10 min. Sections were then washed in PBS and nonspecific antibody binding was inhibited by a 30-min incubation in PBS-BB. Rabbit anti-Ret antibodies (diluted 1:50 in PBS-BB) were incubated on the sections for 12–24 hr at 4C (alternatively, for 1 hr at RT) followed by PBS washes and HRP-conjugated donkey anti-rabbit antiserum (Jackson Immunoresearch Laboratories; West Grove, PA) diluted 1:1000 in PBS-BB for 1 hr. Sections were again washed in PBS and reacted with TSA Plus Direct-Green according to the manufacturer's protocol (NEN Life Science Products). Sections were then incubated with Hoechst 33258, mounted in PBS:glycerol, and coverslipped.
Dual GFRα2 ISH and NeuN IHC Detection on Paraffin-embedded Sections
Four-μm-thick paraffin-embedded brain sections were deparaffinized in xylene and rehydrated in isopropanol and water. Heat-induced epitope retrieval was then performed for 20 min in 0.01 M citrate buffer, pH 6.0. After a 20-min cool-down period, sections were washed in PBS and treated twice for 15 minutes each in 0.1% active DEPC in PBS. The remaining steps of the ISH detection protocol were identical to those described above for frozen sections. Localization of NeuN immunoreactivity (mouse anti-NeuN, 1:2000) was also performed as described above for Ret immunodetection.
Results
GFRα2 ISH and Ret IHC
On the basis of our previous ISH studies utilizing radiolabeled GFRα2 cRNA (Golden et al. 1998), we focused our analysis on GFRα2 expression in the trigeminal ganglia of postnatal Day 0 mice. Strong GFRα2 mRNA signal was localized to trigeminal neurons with TSA Plus Direct-Cyanine 3 detection (Figure 1A). Only weak or no signal was observed when the GFRα2 cRNA probe was omitted or when irrelevant digoxigenin-labeled cRNA probes were used in the procedure (Figure 1B). Increased GFRα2 mRNA signal intensity and/or optimal probe dilution could be achieved by extending the TSA Plus Direct-Cyanine 3 reaction to 1–2 hr or by performing TSA Plus DNP (HRP) followed by TSA Plus Direct-Cyanine 3 (data not shown).
The tissue preparation method used for mRNA ISH was compatible with localization of Ret protein immunoreactivity in the trigeminal ganglion (Figure 1C). No reactivity was observed when primary antiserum was omitted from the procedure (data not shown). To determine the interrelationship between GFRα2 mRNA and Ret protein expression in the trigeminal ganglion, we performed sequential GFRα2 mRNA ISH and Ret IHC using TSA Plus Direct-Cyanine 3 and TSA Plus Direct-Green. Both GFRα2 mRNA (Figure 1D) and Ret immunoreactivity (Figure 1E) were observed in a subset of trigeminal ganglion cells. Dual exposure of labeled sections revealed that virtually all GFRα2 mRNA-positive cells (111 of 112 cells evaluated) possessed Ret immunoreactivity but that approximately 30% of Ret-immunoreactive cells (50 of 161 cells evaluated) lacked detectable GFRα2 mRNA (Figure 1F). In control experiments, replacement of the rabbit anti-Ret antiserum with normal rabbit serum after GFRα2 ISH resulted in no specific labeling or co-localization with the GFRα2 mRNA signal, demonstrating the specificity of the Ret IHC localization.
GFRα2 ISH and NeuN IHC
Digoxigenin- or biotin-conjugated GFRα2 cRNA probes were used to detect GFRα2 mRNA in para-formaldehyde-fixed, paraffin-embedded adult mouse brain sections. Consistent with our previous study utilizing 33P-labeled cRNA probes (Golden et al. 1998), GFRα2 mRNA was readily detected in many cell populations throughout the mouse central nervous system (Figure 2A). Weak or no reactivity was observed when the GFRα2 cRNA was omitted or when irrelevant biotin or digoxigenin labeled cRNAs were substituted (Fig. 2B). Particularly intense GFRα2 reactivity was observed in the neocortex and hypothalamus. GFRα2 mRNA signal appeared localized to neurons on the basis of anatomic distribution and the nuclear features of positive cells. However, to unequivocally identify GFRα2 mRNA positive cells as neurons, we performed dual GFRα2 ISH and NeuN IHC localization. NeuN immunoreactivity is present in the vast majority of neurons in the brain and is absent in non-neuronal cell populations (Mullen et al. 1992; Wolf et al. 1996). NeuN immunoreactivity was readily detected in paraformaldehyde-fixed, paraffin-embedded mouse brain sections using TSA Plus Direct-Green (Figure 2C). Sequential detection of GFRα2 mRNA (Figure 2D) and NeuN immunoreactivity (Figure 2E) revealed neuron-specific GFRα2 mRNA expression (Figure 2F).
Discussion
In this article we describe a relatively rapid and simple method for dual fluorescent ISH and IHC detection using TSA Plus reagents and either digoxigenin- or biotin-labeled cRNA probes. This method proved successful for at least three reasons. First, because proteinase K is not required during the ISH procedure (Braissant and Wahli 1998), protein antigenicity is preserved for subsequent IHC detection. In several previously reported methods, dual mRNA ISH and antigen immunolocalization required that IHC be performed before ISH (Shivers et al. 1986; Couwenhoven et al. 1990; Hrabovszky et al. 1995). This necessitates that the IHC protocol be performed under RNase-free conditions to preserve the mRNA for subsequent detection. This adds considerably to the difficulty of the procedure. Second, the TSA reaction generates a detectable product that is covalently linked at or near sites of probe binding (Bobrow et al. 1989, 1991, 1992). Therefore, unlike precipitating reaction products, there is limited dispersion of signal and there is no diffusion of reaction product from sites of deposition during subsequent detection steps. Third, the TSA Plus reagents have significantly increased sensitivity compared to the first generation of TSA conjugates, making them more suitable for mRNA ISH detection.
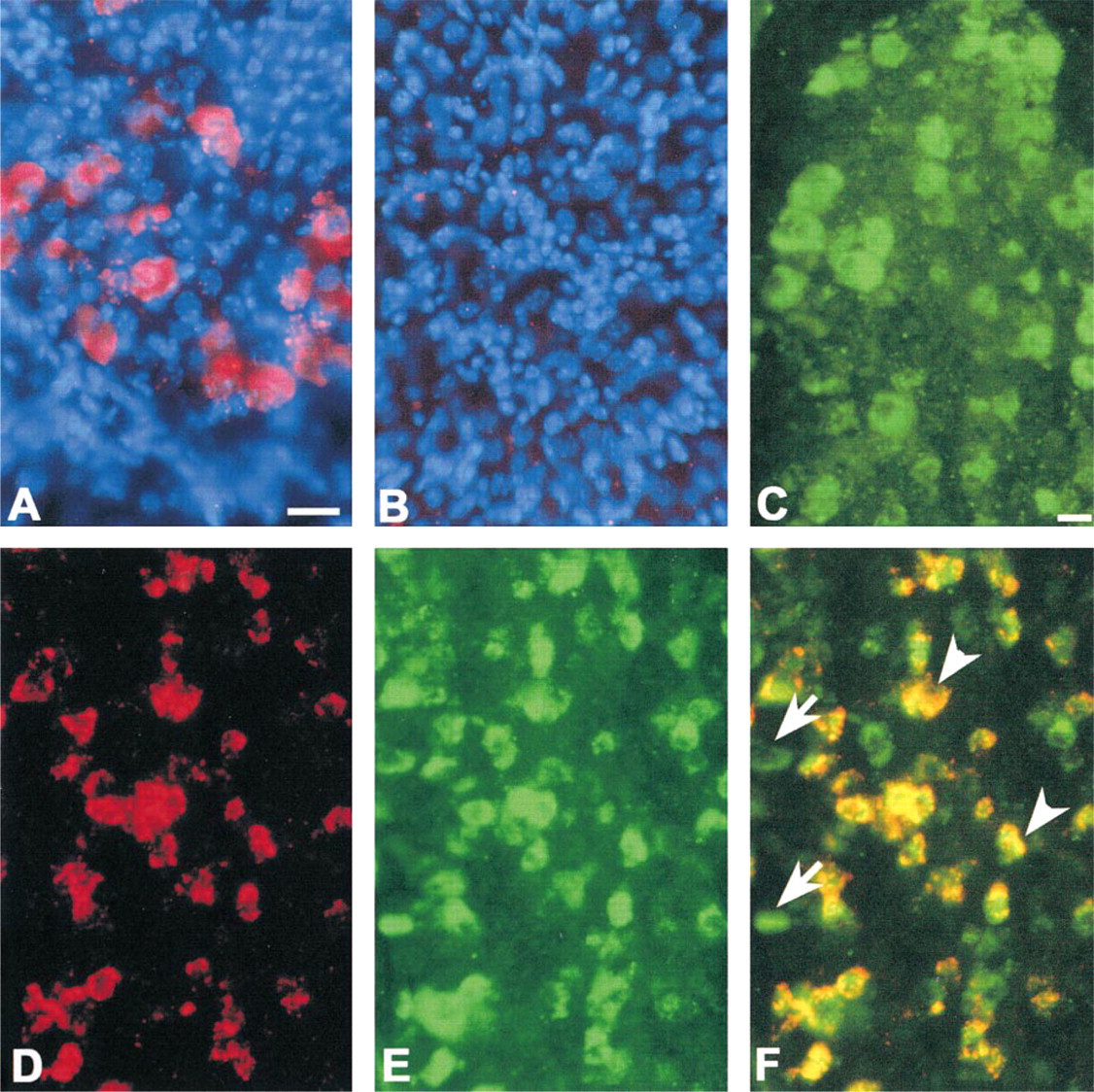
Localization of GFRα2 mRNA and Ret immunoreactivity in frozen sections of mouse trigeminal ganglion. (
The dual fluorescent ISH and IHC detection protocol was used to compare the expression patterns of GFRα2 mRNA and Ret immunoreactivity in the postnatal Day 0 mouse trigeminal ganglion and GFRα2 mRNA and NeuN immunoreactivity in adult mouse brain. GFRα2 and Ret form a receptor complex that preferentially binds neurturin (NRTN), a member of the glial cell line-derived neurotrophic factor (GDNF) family of ligands (GFLs) (Lin et al. 1993; Durbec et al. 1996; Kotzbauer et al. 1996; Trupp et al. 1996; Milbrandt et al. 1998). Because there are multiple GFLs and GFRαs, an analysis of their expression patterns and that of the co-receptor Ret may shed light on the in vivo relationships between these molecules and provide clues to their physiological function. Our analysis of the trigeminal ganglion indicated that virtually all GFRα2 mRNA-expressing neurons possessed Ret- immunoreactivity but that approximately 30% of the Ret immunoreactive neurons lacked GFRα2 mRNA. These findings are consistent with our previous studies demonstrating that GFRα2 and Ret form a functional receptor for GFLs, particularly for NRTN, and that other members of the GFRα family, GFRα1 and GFRα3, are also expressed in the trigeminal ganglion (Baloh et al. 1998; Golden et al. 1998). It appears likely that GFRα1 and/or GFRα3 are co-expressed with Ret in the trigeminal neurons lacking GFRα2. Additional multi-labeling studies are required to directly test this hypothesis. In the adult mouse brain, GFRα2 mRNA was found in many neuronal populations, as previously described (Golden et al. 1998). The neuronal specificity of GFRα2 expression was directly demonstrated by co-localization of GFRα2 mRNA and NeuN immunoreactivity in single cells.
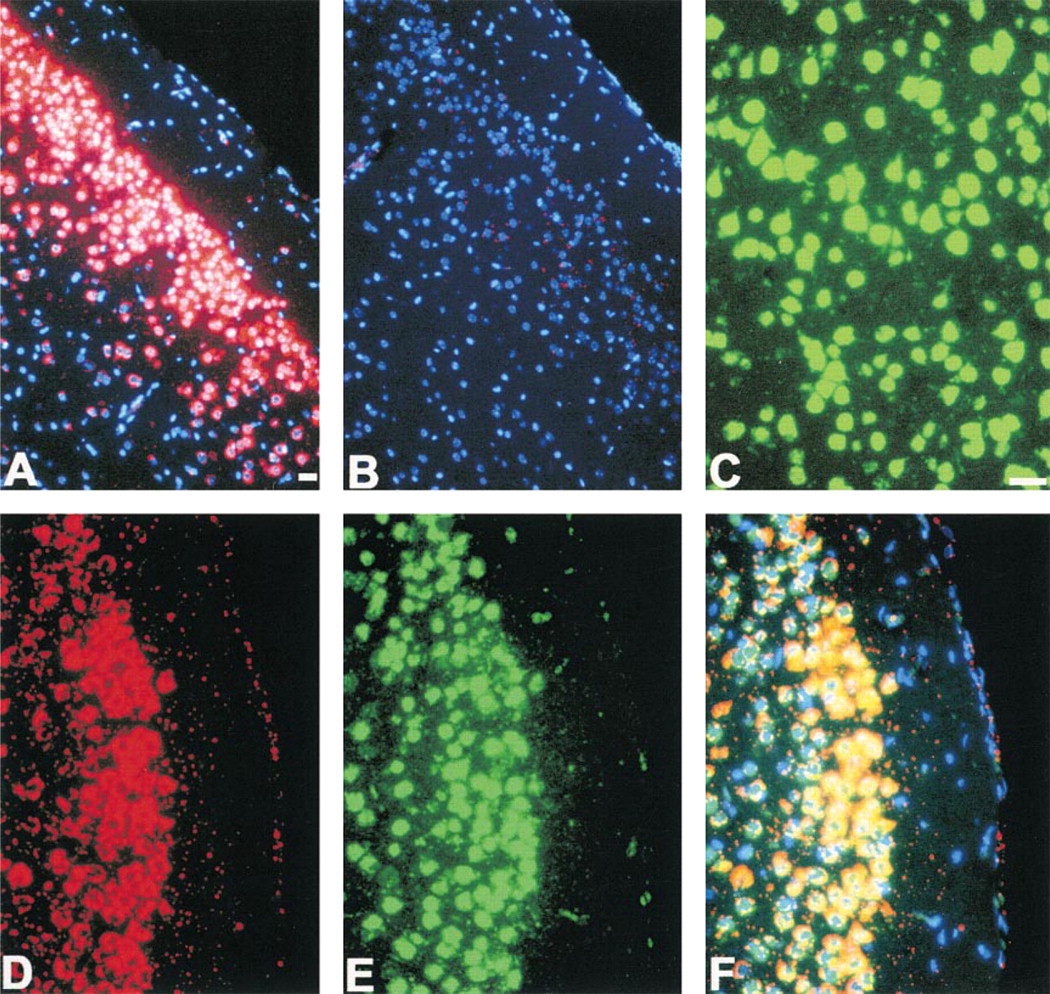
Neuronal localization of GFRα2 mRNA in paraformaldehyde-fixed, paraffin-embedded mouse brain sections. (
In this work, we used either digoxigenin- or biotin-labeled cRNA probes to detect GFRα2 mRNA. However, we have also used HRP-conjugated oligonucleotides and a similar protocol to detect nestin mRNA in the developing mouse nervous system (data not shown). Our observations and the published literature suggest both advantages and disadvantages of oligonucleotide cDNA probes vs cRNA probes for mRNA ISH. Oligonucleotides are relatively inexpensive and can be designed to hybridize with virtually any available mRNA sequence, obviating the need to generate or obtain plasmids containing the targeted gene sequence. Their small size, typically 25–50 nucleotides, facilitates tissue penetration and decreases or eliminates the requirement for tissue disruption before probe hybridization. However, DNA–RNA hybrids are less stable than RNA–RNA hybrids, specificity of binding is less stringent, and overall sensitivity is poor, necessitating the use of mixtures or “cocktails” of oligonucleotide sequences to achieve adequate detection (Trembleau and Bloom 1995). Similarly, the use of HRP-conjugated probes vs digoxigenin- or biotin-labeled probes and subsequent localization of hybrids with HRP-conjugated digoxigenin antibodies or strep-tavidin has both advantages and disadvantages. In a previous report, HRP-labeled oligonucleotides were used in combination with TSA for rapid detection of multiple genomic sequences in metaphase chromosome spreads (van Gijlswijk et al. 1996). Because HRP was conjugated directly to the probe, there was no need for “secondary” immunological reagents, greatly decreasing the time necessary to perform the procedure and eliminating all background labeling associated with hapten and hapten detection. The major drawback to HRP-conjugated oligonucleotide probes for mRNA ISH detection is nonspecific hybridization signal. HRP activity is sensitive to both high temperatures and formamide, necessitating the use of more complex hybridization buffer, decreased hybridization temperatures and increased stringency of posthybridization washes to achieve adequate signal-to-noise ratios (van Gijlswijk et al. 1996).
In total, we have described a rapid, sensitive method for dual fluorescent mRNA in situ hybridization and immunohistochemistry using TSA Plus detection reagents. Additional studies are required to determine the applicability of this method to other probes, cells, and tissues.
Footnotes
Acknowledgements
Supported in part by NEN Life Science Products and by National Institutes of Health grants NS35107 and NS35404. AUZ received fellowship support from the McDonnell Center for Cellular and Molecular Neurobiology.
We thank Cecelia Latham for technical assistance, Angela Schmeckebier for secretarial support, and Dr Mark Bobrow (NEN Life Science Products) for expert advice and insights on tyramide signal amplification.