
Abstract
We evaluated bromodeoxyuridine (BrdU) immunohistochemistry in undecal-cified adult rat tibiae to study cell kinetics in various bone compartments: primary and secondary spongiosae, periosteum, and bone marrow. Several regimens of BrdU administration were tested (IP injections and osmotic minipumps). We compared LR White resin, methylmethacrylate, and Epon–araldite embedding, microwave irradiation for antigen retrieval, several concentrations of sodium ethoxide for deplastification, and various DNA de-naturation procedures. Paraffin-embedded decalcified tibiae and Epon-embedded bowel were used as positive controls. The best results were obtained in rats labeled with 40 mg of BrdU for 72 hr using osmotic minipumps. The procedure using a Microprobe system in Epon-embedded bone tissue with a sodium ethoxide concentration of 50% for two intervals of 20 min provided the best staining quality and tissue preservation. Labeled pre-osteoblastic cells and bone marrow cells could be counted. Epon embedding allowed preservation of tetracycline double labeling performed 1 to 5 days before sacrifice. The number of labeled pre-osteoblastic cells was correlated with the double-labeled surface area measured histomorphometrically.
T
The aim of our study was to develop an embedding procedure for immunohistochemistry and quantitative histomorphometry. We chose to evaluate osteoblast proliferation capacity, assessed by BrdU uptake, in various bone compartments (especially in primary and secondary spongiosae) and to compare these data with histodynamic parameters after double tetracycline labeling on the same undecalcified bone.
Materials and Methods
Experiment 1
Optimization of BrdU Administration and Immunohistochemistry. Male Wistar rats (IFFA CREDO; Les Oncins, France), aged 5 weeks and weighing 192 ± 8 g, were used in this study. After 5 days of acclimatization, the animals were double labeled with tetracycline (15 mg/kg b/w.) on Day 0 and with calcein (20 mg/kg bw) on Day 5 by IP injection. BrdU (Sigma; Saint-Quentin, France) was diluted in DMSO and distilled water (v/v) at a concentration of 200 mg/ml and administered according to various protocols. One rat received an IP injection 1 h before sacrifice. Another rat received 12 IP injections (i.e., 23.5 mg/kg bw/inj) over a period of 3 days. Two rats were implanted with either one or two 1003D Azlet osmotic pumps (Charles River; Saint Aubin lès Elbeuf, France), each containing 100 μl of BrdU solution (i.e., 20 or 40 mg BrdU per animal, respectively), IP for 3 days.
Epon Embedding. The embedding medium was prepared by mixing, in sequence, Embed 812 (12.6 g), Araldite 502 (11.8 g), DMP-30 (0.5 g), and DDSA (26.7 g) (TAAB; Saint-Germain en Laye, France) according to Xiang and Markel (1995).
After 24 hr of fixation in 70% ethanol at 4C, the samples were dehydrated in increasing ethanol concentrations. Tissues were infiltrated with increasing concentrations of Epon in propylene oxide at room temperature (RT) as follows: 25% Epon for 18 hr; 50% Epon for 24 hr; 75% Epon for 24 hr; and 100% Epon for 3 days. Polymerization was performed under vacuum at 60C for 5 days in fresh 100% Epon.
LR White Resin Embedding. After fixation overnight at 4C in 4% paraformaldehyde, 0.1% glutamine, 0.1 M phosphate buffer, the samples were rinsed in 0.1 M phosphate buffer and dehydrated in increasing ethanol concentrations. After dehydration, tissues were infiltrated with increasing concentrations of LRWhite resin (TAAB) in 70% ethanol at RT as follows: 25% LRWhite for 24 hr; 50% LRWhite for 24 hr; 75% LRWhite for 8 hr; and 100% LRWhite for 24 hr. Embedding was then performed in fresh 100% LRWhite for 7 days at 4C. Blocks were polymerized under vacuum at 50C for 5 days.
Paraffin Embedding. Samples were fixed in 4% paraformaldehyde, 0.1 M phosphate buffer, for 4 days at 4C. The bones were then decalcified in EDTA decalcifying solution (EDTA 2 Na: 41.3 g, 5% polyvinylpyrrolidone, NaOH 4.4 g; distilled water 1l). The decalcification endpoint was determined by specimen flexibility (after approximately 21 days). Bone embedding was performed according to a routine paraffin-embedding procedure.
Sectioning
Epon and LRWhite-embedded Specimens. Each sample was polished with a circular electrical buffing wheel (Ese 200 GTL; Escil, France). Three-μm-thick sagittal sections were cut on a sledge microtome (Polycut S; Reichert–Jung, Heidelberg, Germany) with a tungsten carbide blade (Reichert-Jung 16 cm), floated in a waterbath, and mounted on positively charged slides (ProbeOn Plus slides; Fisher Scientific, Pittsburgh, PA) adapted to the MicroProbe system (Fisher Scientific) to promote tissue structure preservation, and were dried overnight at 37C in an oven.
Serial sections were performed on Epon-embedded blocks: one for immunohistochemistry and one for histomorphometry.
Paraffin-embedded Specimens. Three-μm-thick sagittal sections were cut on a microtome (Autocut 2040; Reichert-Jung) with a disposable stainless steel blade (Reichert–Jung; Buffalo, NY), floated onto 3% albumin/warm water (42–44C), and mounted on glass slides.
BrdU Immunohistochemical Staining
Preparation of Epon-embedded Sections. Using the Microprobe system, slides were submerged in solutions of saturated sodium ethoxide diluted in 100% ethanol at different concentrations (20%, 30%, 40%, 50%, twice for 20 min, 80% for 10 min) or at a concentration of 50%, twice for 5 min, twice for 10 min, twice for 15 min, twice for 20 min to determine the best procedure to ensure optimal tissue preservation. Sodium ethoxide was used to simultaneously remove Epon and denatured DNA. The slides were then rinsed in three changes of 100% ethanol for 5 min each to eliminate sodium ethoxide, rehydrated in decreasing ethanol concentrations, and then rinsed in distilled water.
To enhance BrdU detection (Van de Kant et al. 1990; Dover and Patel 1994), some sections were treated by microwave irradiation. After rehydration, slides were incubated in a 400–ml bath of 0.1 M sodium citrate buffer, pH 6.0, which was placed in the microwave oven for 5 min at 700 W. The slides were then rinsed in distilled water and Tris, pH 7.6.
Preparation of LRWhite-embedded Sections. Slides were rehydrated with decreasing ethanol concentrations and placed in distilled water. Sections were permeabilized with 0.1% trypsin in 0.1 M HCl for 15 min at 37C, rinsed with Tris, pH 7.6, and then incubated in 4 N HCl at 56C for 30 min to denature DNA, and rinsed in 0.1 M sodium borate twice for 5 min and Tris to restore pH.
Preparation of Paraffin-embedded Sections. Slides were deparaffinized with xylene (twice for 5 min) and sections were then rehydrated and placed in distilled water. Sections were rinsed with Tris, pH 7.6, and incubated in 4 N HCl at 56C for 30 min to denature DNA, rinsed in 0.1 M sodium borate twice for 5 min and Tris to restore pH.
Staining Procedure. After each step, sections were rinsed in Tris, pH 7.6, twice for 5 min, then in Tris/BSA 1%, pH 7.6. Endogenous peroxidases were blocked with 3% H2O2 for 30 min. Sections were then incubated at RT with primary antibody (anti-BrdU; Sigma) for 1 hr. The primary antibody was revealed with a kit (Immunotech; Marseille, France) using streptavidin conjugate and DAB as chromogen. Sections were incubated with biotinylated goat anti-mouse IgG for 30 min at RT and incubated with streptavidin–peroxidase conjugate for 45 min at RT, then with DAB chromogen mixture for 20 min, and finally rinsed thoroughly in distilled water. Hematoxylin counterstaining was performed for 2 min. Sections were then rinsed with tapwater and mounted.
Evaluation. The immunostained slides were observed under light microscopy at magnifications of × 40 and × 100. The entire bone sections were observed for the localization of BrdU-labeled cells. BrdU-positive cell types were determined by observing dark brown DAB nuclear staining. Unlabeled nuclei with only blue hematoxylin staining and pale brownish nuclei were considered to be negative.
Controls. Positive controls consisted of bowel immunostaining, designed to assess correct function of the osmotic minipumps. To verify the absence of interference of our de-plastification technique with the immunostaining with another antibody, we performed the full procedure using anti-smooth muscle α-actin as primary antibody.
Three types of negative controls were designed: the complete immunohistochemistry procedure performed on bones from rats not submitted to BrdU administration and the complete procedure omitting primary antibody or using a nonimmune primary antibody on bones from BrdU labeled rats.
Experiment 2
Comparison of BrdU-labeled Pre-osteoblasts with Histodynamic Parameters. After defining the optimal procedure, six male Wistar rats were used for a 6-day experiment. Animals were double labeled with tetracycline (15 mg/kg bw) on Day 1 and with calcein (20 mg/kg bw) on Day 5 by
Processing For Bone Histomorphometry. After sacrifice, the left tibiae were excised, fixed, dehydrated in absolute acetone, embedded mineralized in methylmethacrylate, and sagitally sectioned at a thickness of 7 μm for subsequent bone histomorphometry measurements. A Leitz-TAS+ automatic image analyzer equipped with a Bosch camera connected to a Leitz Orthoplan microscope was used to determine bone surfaces (BS/TV) in the primary and secondary spongiosae. These measurements were performed on six modified Gold-ner sections. Histodynamic measurements were performed with a semiautomatic system composed of a digitizing table (Summasketch-Summagraphics) connected to a personal computer and to a Reichert Polyvar microscope equipped with a drawing system (Camera Lucida). The following parameters were measured on unstained 6-μm-thick sections under UV light: mineral apposition rate (MAR, μm/day) single- and double-labeled surface area (sLS/BS and dLS/BS, %). Left tibiae were embedded in Epon and processed according to the final procedure described earlier. Labeled osteogenic precursors, regardless of their type, were counted in primary spongiosa using a square grid. Their number was expressed in relation to the bone/cartilage area measured on the automatic image analyzer. In secondary spongiosa, labeled pre-osteoblasts were counted and their number expressed in relation to the bone perimeter. Only dark-brown nuclei were taken into account. Pre-osteoblasts were defined, as previously described (Kember 1960), as ovoid cells located between the juxtatrabecular osteoblast layer and bone marrow (Figure 1). BrdU-labeled cells/mm2 in the marrow were counted with a grid. Six-μ-thick unstained sections were prepared for histodynamic measurements under
Results
Staining specificity was confirmed, as the immunohistochemistry negative controls did not show any significant nuclear labeling. Immunostaining quality was determined by staining intensity in comparison with background staining.
Experiment 1
BrdU Regimen. No BrdU labeling was observed in the secondary spongiosa pre-osteoblasts of tibiae from rats receiving one or multiple
Continuous BrdU administration by osmotic pumps allowed labeling of proliferative pre-osteoblasts in bone. However, two osmotic pumps provided a better staining signal and an increased number of labeled cells compared to rats implanted with one pump.
BrdU Staining of Paraffin-embedded Tissues. In the small intestine, nuclei of crypt epithelial cells were BrdU labeled. BrdU labeling was seen in bone marrow cells (Figure 1A) and in chondrocytes of the proliferative zone of the growth plate. BrdU-labeled proliferating preosteoblasts were visible on the surface of trabecular bone.
BrdU Staining of LR White-embedded Tissues
Bone Specimen. As in paraffin-embedded bones, few proliferating pre-osteoblasts were labeled over the surface of trabecular bone. However, DNA denaturation by 4 N HCl at 56C for 30 min induced disruption of bone sections and morphological details were poorly preserved (Figure 1B). A degree of shrinkage and ruffling of sections was also observed during the immunostaining procedure. For these reasons, consistently good results were not obtained with this resin embedding.
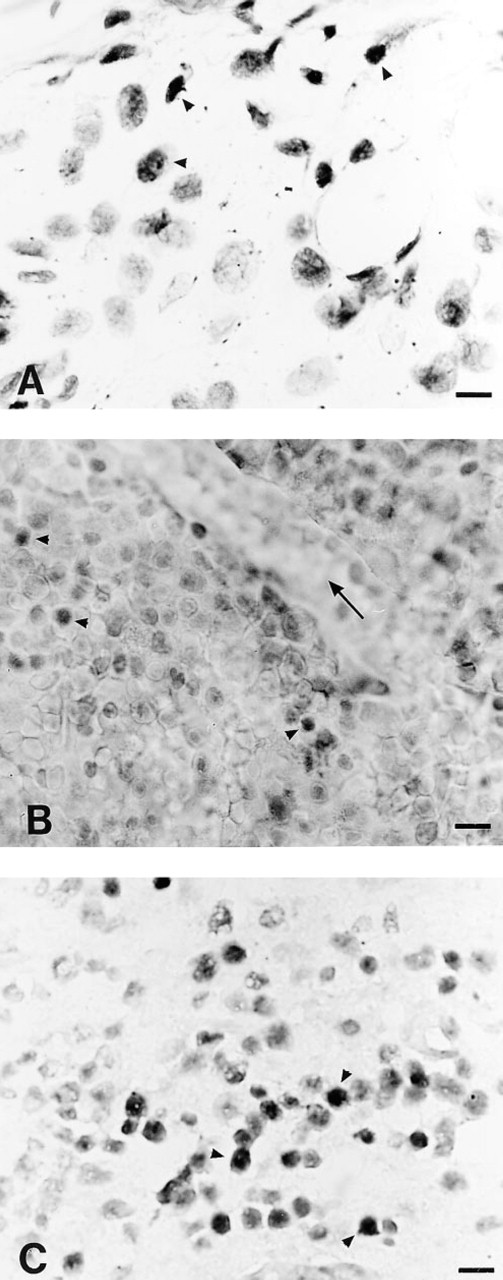
Comparison of different embedding media for BrdU immunostaining. Arrowheads, BrdU-positive cells.
BrdU Staining of Epon-embedded Tissues. BrdU staining was similar to that in paraffin-embedded small intestine. The staining result was consistent throughout the section. The sodium ethoxide concentrations that allowed complete deplastification of slides of bone sections were 50% twice for 20 min and 80% for 10 min. However, the 80% concentration did not ensure sufficient tissue preservation. When lower concentrations were used, Epon was not completely removed and BrdU-labeled cells were occasionally observed in bone marrow. The stained cells were not evenly distributed throughout the section. Deplastification with a 50% sodium ethoxide solution (twice for 20 min) combined with the use of the Microprobe system provided flat sections that remained adherent to the slides throughout the staining procedure (5 hr). In this case, BrdU uptake in bone marrow cells was comparable to that of paraffin-embedded cells (Figure 1C). Preosteo-blasts were observed in the perichondrium and periosteum (Figure 2A), close to the surface of trabecular bone in the secondary spongiosa (Figure 2C) of the tibial metaphysis. Labeled chondrocytes were found in the proliferative zone (Figure 2B) of the growth plate. Few BrdU-positive osteoclasts (Figure 2D) were observed, and only two to three labeled osteocytes were seen on each section.
The use of microwave irradiation to enhance BrdU staining induced shrinkage and damage to bone sections. Morphological details were poorly visualized and background was higher. Conventional histomorphometry was performed on these Epon-embedded sections, as the toluidine blue staining was comparable to that obtained on MMA sections (Figure 3A). Double tetracycline labeling was visible under
Experiment 2
In Epon-embedded tibiae, the number of labeled preosteoblasts per mm of bone perimeter in secondary spongiosa was 1.70 ± 0.16/mm2 and the number of osteogenic precursors in primary spongiosa was 6.77 10-6 ± 0.81/mm2 of bone/cartilage area. Approximately 20.6 ± 3.2/mm2 dividing cells were detected in bone marrow. The coefficients of variation of the measurements were 19.1% and 29.2% in secondary spongiosa and bone marrow, respectively.
Histodynamic Measurements. On Epon-embedded sections, mineral apposition rate was 3.54 ± 0.8 μm/d: the single-labeled surface (sLS/BS) = 14.2 ± 3.2% and the double-labeled surface (dLS/BS) = 29.9 ± 6.2%. With MMA, MAR was 3.22 ± 0.4 μm/d: sLS/BS was 12.1 ± 2.7% and dLS/BS was 26.3 ± 5.1%. The histo-dynamic measurements of labeled surfaces were positively correlated between MMA and EPON (slS/BS r 2 = 0.52, p<0.1; dLS/BS r 2 = 0.86, p<0.01). Labeled surfaces were higher with Epon than with MMA, but the difference was not statistically significant. This difference was probably related to the fact that Epon sections were more ruffled than MMA sections. Linear regression analysis showed a positive relationship between double-labeled surface and BrdU-labeled pre-osteoblasts in secondary spongiosa (y = 0.03 × 0.95, r = 0.87, p<0.01), demonstrating that, under physiological conditions, the mineralizing surface provides a fairly accurate estimation of pre-osteoblast proliferation.
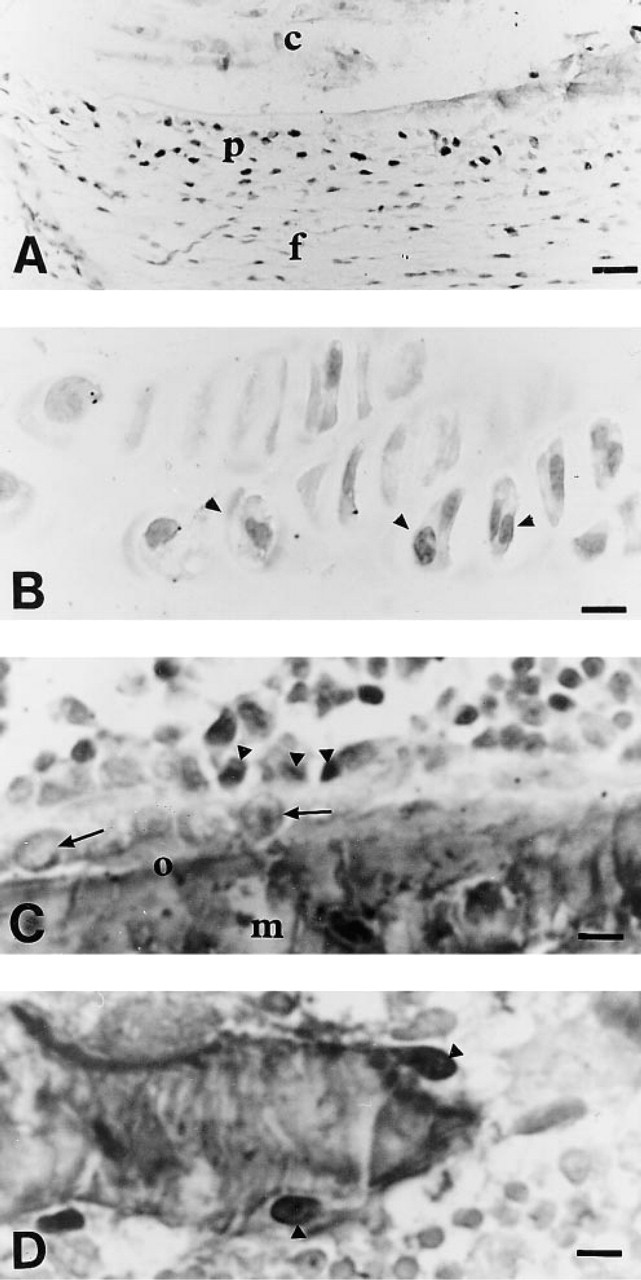
BrdU labeling in different bone areas.
Discussion
Because our aim was to perform quantitative evaluation of BrdU-labeled cells, preservation of tissue architecture and homogeneous immunolabeling throughout the section were mandatory, as emphasized by Oprins et al. (1994). Glycol-methacrylate (GMA) has been shown to provide excellent results for immunohistochemistry of non-collagenous matrix proteins in bone tissue (Groot et al. 1986; Ingram et al. 1993) and MMA for bone marrow antigens (Bernhards et al. 1992). However, as emphasized by Xiang and Markel (1995), our experiments confirmed that MMA was not a suitable embedding medium for BrdU labeling. LR White was designed to allow light and electron microscopy on the same sample and has been show to yield excellent results for BrdU staining on small sections of noncalcified tissues (Tamatani et al. 1995). It is hydrophilic resin that eliminates the need for deplastification step. However, in our hands the various DNA denaturation procedures, such as 4 N HCl and heat exposure, induced ruffling and lifting of the sections. Moreover, when visualized, BrdU staining intensity was much lower than on Epon-embedded sections.
We found that the best results in terms of tissue preservation were obtained with a sodium ethoxide concentration of 50% twice for 20 min combined with Microprobe. The use of 100% sodium ethoxide for 20 min, as recommended by Xiang and Markel (1995), allowed good deplastification but poor tissue preservation, making quantitative evaluation impossible. As shown by Apte and Puddle (1990), sodium ethoxide induced excellent DNA denaturation. Moreover, the gap capillary technique (Microprobe) improved tissue preservation and saved reagents (Frank et al. 1993b). Unlike Dover and Patel (1994) and Tischler (1995), we found that microwave section irradiation did not improve BrdU staining, but had a positive effect on cultured cells (data not shown). It also induced shrinkage of sections, leading to poor tissue preservation. These discrepancies among the results are probably related to the difference in embedding medium (paraffin vs. Epon).
The coefficient of variation in secondary spongiosa was 19.1%, which is an acceptable level of precision in view of those found in coventional quantitative bone histomorphometry (Chavassieux et al. 1985). The coefficient of variation was much higher in bone marrow (29.2%). Quantification in this compartment could probably be improved by thresholding images using an automatic image analyzer to count nuclei exhibiting the same level of brown staining.
Normal remodeling rat bone has a relatively low turnover compared to healing cortical fracture, intestine, or tumors. Unlike growth plate proliferative chondrocytes, pre-osteoblasts (Farnum and Wilsman 1993) did not exhibit labeling after a single bolus injection of BrdU. We found that the best staining of secondary spongiosa was obtained with 40 mg of BrdU per animal administered for 72 h. Kimmel and Jee (1980) found scarce positive [3H]-thymidine nuclei in bone cells 2.5 mm away from the growth plate (i.e., in the IISP) 1 h after [3H]-thymidine injection. These data demonstrated that BrdU labeling is much less sensitive than [3H]-thymidine autoradiography. However, Dobnig and Turner (1995) counted only 4% of labeled osteoblasts in rat secondary spongiosa after 1 week of [3H]-thymidine administration.
Owen (1963), in rabbit periosteum, showed that pre-osteoblasts were actively dividing cells. Similarly, in our experiment, the highest labeling was observed in periosteal pre-osteoblasts located close to the primary spongiosa. We observed scarce labeled osteocytes after a 72-h exposure to BrdU. A positive osteocyte nucleus was due to BrdU uptake by a stromal cell or pre-osteoblasts that divided at the beginning of BrdU exposure. This is in agreement with Roberts et al. (1987), who used [3H]-thymidine incorporation in the periodontal ligament as a simple model of osteogenesis. These authors estimated that osteoblast histeogenesis took approximately 60 hr (Roberts et al. 1987).
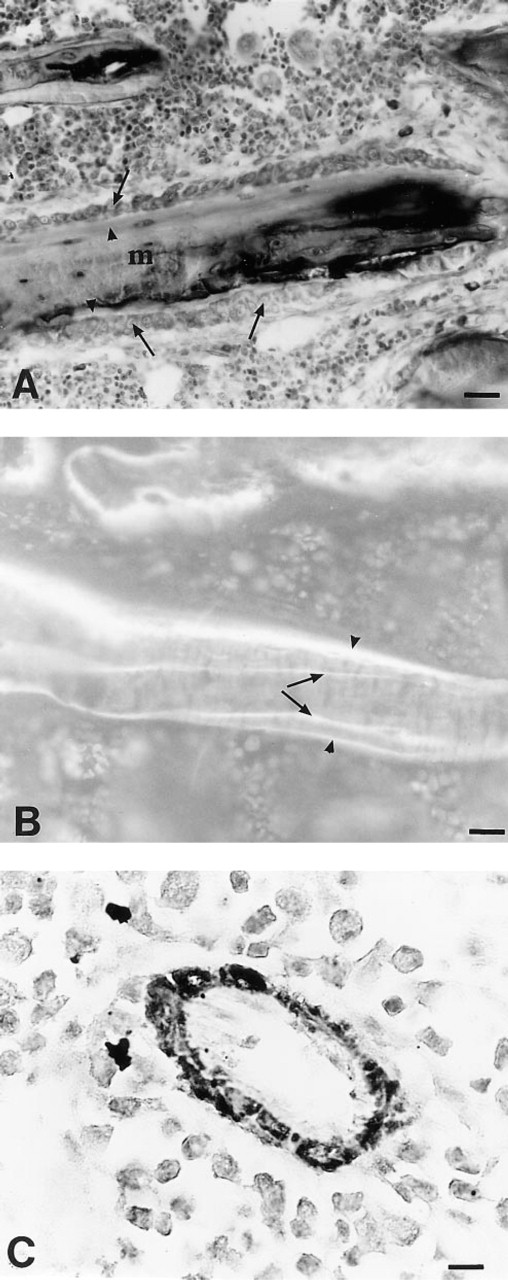
Epon-embedding allowed immunohistochemistry as well as usual strains for quantitative histomorphometry in trabecular bone.
We found a positive correlation between the number of BrdU-labeled pre-osteoblasts in the secondary spongiosa and the fluorochrome-labeled surface measured under
In conclusion, we have developed and improved a technique that allows visualization of dividing pre-osteoblasts in various compartments of adult rat metaphysis and provides reliable measurements, at least in primary and secondary spongiosa. Identification of osteoblastic precursors within the bone marrow, as performed by FACS analysis (Van Vlasselaer et al. 1994) on ex vivo samples, could provide a better understanding of the mechanisms underlying bone response to various stimuli.
Footnotes
Acknowledgements
We would like to thank Dr J. F. Mosnier for helpful discussion and advice.