
Abstract
We tested the immunoprobe FluoroNanogold (FNG) for its utility as an immunocytochemical labeling reagent. This immunoprobe consists of a 1.4-nm gold particle to which a specific Fab' fragment and a fluorochrome are conjugated. We employed the microtubules (MTs) of human phagocytic leukocytes as a model system for testing the usefulness of FNG as a secondary antibody for immunocytochemistry. We show that these fluorescently labeled ultrasmall immunogold particles are very efficient for labeling MTs in these cells. The signal from FNG can be detected directly by fluorescence microscopy or indirectly by other modes of optical microscopy and electron microscopy, after silver-enhancement of the gold. The spatial resolution of immunolabeled MTs obtained with FNG and silver enhancement was comparable to that of conventional immunofluorescence detection. Colloidal gold (5-nm and 10-nm in diameter), on the other hand, failed to label MTs in cells prepared in a similar manner. This difference in labeling was due in large part to greater penetration of 1.4-nm gold into aldehyde-fixed cells than either 5-nm or 10-nm gold particles. The fluorescent 1.4-nm immunoprobe was shown to be an important new tool for general use in correlative microscopy.
Keywords
I
The application of immunocytochemistry in cell and developmental biology as well as in immunopathology has been extremely important in defining the distribution of molecules in both spatial and temporal domains. Two of the major reporter systems used in immunocytochemistry have been the labeling of immunoprobes with fluorescent and particulate markers. The use of fluorescent immunoprobes offers the advantages afforded by high-resolution optical and confocal microscopes for their detection as well as the availability of many fluorochromes with different spectral properties for multiple labeling. Particulate probes, especially colloidal gold, have permitted high-resolution detection of molecules at the ultrastructural level. The availability of colloidal gold particles in different sizes has allowed the detection of more than one type of molecule in the same cell.
Although colloidal gold has been invaluable for ultrastructural immunocytochemistry, several studies have shown that there is an inverse relationship between the size of colloidal gold particles and the density of immunolabeling. This correlation holds over a wide range of experimental material and conditions (e.g., Takizawa and Robinson 1994; Ghitescu and Bendayan 1990; Yokota 1988; Kehle and Herzog 1987; Lackie et al. 1985; Horisberger 1981; Slot and Geuze 1981).
The immunoprobe FluoroNanogold (FNG) consists of a 1.4-nm gold particle to which an affinity-purified Fab' fragment and a fluorochrome are conjugated. Thus, this ultrasmall immunoprobe combines the attributes of fluorescence and particulate detection systems. In a previous study we showed that 1.4-nm immunogold particles (in that case lacking the fluorochrome) penetrated into ultrathin cryosections to a greater extent than did colloidal gold particles (i.e., 1.4-nm < 5-nm < 10-nm) and that this led to enhanced immunolabeling at the ultrastructural level (Takizawa and Robinson 1994). We have now tested FNG as a secondary antibody to determine if this probe yields enhanced labeling of microtubules (MTs) in immunocytochemical preparations. MTS were selected as the target for immunolabeling because their detection requires high spatial resolution. We used human leukocytes as the model cell system because they have relatively few MTs, thereby facilitating the microscopic analysis. We show FNG to be a versatile tool that can be added to our armamentarium for immunocytochemical localizations and that this immunoprobe is an important new tool for multimodal correlative microscopy.
Materials and Methods
Reagents
Goat anti-mouse FNG and goat anti-mouse 5-nm and 10-nm colloidal gold were obtained from NanoProbes (Stony Brook, NY). Goat anti-mouse 5-nm colloidal gold was also obtained from Goldmark Biologicals (Phillipsburg, NJ). Fluorescein-labeled goat anti-mouse IgG was purchased from Cappel (Durham, NC). Murine monoclonal anti-α- and anti-β-tubulin were obtained from Amersham (Arlington Heights, IL). Normal goat serum was purchased from Jackson ImmunoResearch (West Grove, PA). Gum arabic, 2-[N-morpholino] ethanesulfonic acid, sodium borohydride, sodium thiosulfate, DAPI, dimethylsulfoxide, Triton X-100, and N-formyl-Met-Leu-Phe (fMLP) were obtained from Sigma Chemical (St Louis, MO). Glutaraldehyde, osmium tetroxide, Mowiwol, and formvar were obtained from Polysciences (Warrington, PA). Epon 812 and silver lactate were obtained from Fluka (Ronkonkoma, NY), and n-propylgallate and p-phenylenediamine were supplied by Aldrich (Milwaukee, WI). Medium 199 was obtained from Gibco BRL (Gaithersburg, MD). Fetal calf serum was supplied by Intergen (Purchase, NY). All other reagents were at least reagent grade. Stock solutions of fMLP were prepared in dimethylsulfoxide and stored at –20C.
Preparation of Cells
Human leukocytes were collected onto round glass cover-slips from finger prick blood as we have described previously (Robinson and Batten 1989). LLC-PK cells were grown on glass coverslips in Medium 199 supplemented with 3% fetal calf serum.
Cell Incubation
Leukocytes on coverslips were incubated with the chemotactic tripeptide fMLP at 10-7 M for 5 min at 37C. Cells thus treated are more favorable for microscopic analysis than untreated cells. Cells were then fixed immediately in 0.7% glutaraldehyde in PBS for 15 min. The cells were subsequently detergent-extracted in Triton X-100 and SDS and then processed for immunocytochemical localization of MTs as we have described (Ding et al. 1995). LLC-PK cells were washed in warm microtubule stabilizing buffer (PHEM buffer: 60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2, pH 6.9) to remove the serum. They were then either fixed in 0.7% glutaraldehyde in PBS for 15 min and then extracted with 0.5% Triton X-100 for 15 min or lysed in 0.5% Triton X-100 in PHEM buffer for 2 min and then fixed for 15 min with 0.7% glutaraldehyde in PBS.
Immunocytochemistry
Coverslips were incubated in sodium borohydride (1 mg/ml) in Tris-buffered saline to block free aldehydes (two changes for 15 min each at 22C) and 5% normal goat serum in PBS to block nonspecific protein binding sites (1 hr at 22C). Subsequently, cells were incubated in a mixture of monoclonal anti-α- and anti-β-tubulin as we have described (Robinson and Vandré 1995). After incubation with the primary antibodies and extensive washing, coverslips were incubated with FNG. This immunoprobe consists of a 1.4-nm gold particle conjugated with goat anti-mouse Fab' fragment and fluorescein. The FNG was diluted 1:25 over that supplied by the manufacturer in PBS containing 5% normal goat serum. Cells were incubated with the FNG for 2 hr at 22C. The coverslips were then washed at least six times in PBS. For optical microscopic analysis, we also used goat anti-mouse 5-nm and 10-nm colloidal gold particles as secondary antibodies. These antibodies were diluted 1:25 (A520 was 0.064 and 0.057, respectively) in PBS containing 5% normal goat serum. For purposes of comparison, a conventional fluorescein-labeled goat anti-mouse IgG was employed. This secondary antibody was diluted 1:50 over that supplied by the manufacturer in PBS with 5% normal goat serum. The cells were incubated and washed as described for the FNG samples. Coverslips that were to be examined directly for fluorescence were mounted in Mowiwol containing p-phenylenediamine (1 mg/ml) to retard photobleaching.
Silver Enhancement of FluoroNanogold and Colloidal Gold
Optimal visualization of 1.4-nm gold at both the optical and electron microscopic levels requires silver enhancement of the gold particles. FNG-labeled samples were incubated for silver enhancement of the gold particles for various times (30 sec to 15 min) using the procedure developed by Burry and associates. The silver enhancement process was stopped with a neutral pH fixer solution (for review see Burry 1995). Silver enhancement of colloidal gold was carried out in an identical manner for 5 min. After silver enhancement, LLC-PK cells were incubated with DAPI (0.75 μg/ml PBS for 30 min) to render chromatin visible by fluorescence microscopy. The samples were then mounted in Mowiwol lacking p-phenylenediamine.
Optical Microscopy
FNG-labeled MTs and FITC-IgG-labeled MTs were visualized by conventional epifluorescence microscopy. Silver-enhanced FNG-labeled and 5-nm and 10-nm colloidal gold-labeled samples were visualized by brightfield, phase-contrast, differential interference contrast (DIC), and epipolarization optics. A Nikon Optiphot microscope equipped with a Nikon B-2A filter set for fluorescence detection of FITC and a Nikon IGS filter set for epipolarization detection were employed for these studies. Nikon x100 phase-contrast, NA 1.4, and x100 DIC, NA 1.25 objective lenses were used. Micrographs were recorded on Kodak T-Max 400 film.
Confocal Microscopy
The samples were also examined by confocal microscopy using a BioRad MRC 600 operated in the reflectance mode. Images were collected after Kalman filtering and stored on an optical disk. The micrographs were printed to a GCC Technologies film recorder onto Kodak T-Max 100 film without additional manipulation of the images.
Electron Microscopy
Silver-enhanced FNG-labeled MTs were also examined by electron microscopy. After labeling with FNG and washing in the same manner as for optical microscopy, the cells were refixed in 2% glutaraldehyde in PBS for 15 minutes. The samples were then subjected to the silver enhancement procedure that was used for light microscopy for various times, from 30 sec to 5 min. After fixation of the developed silver particles and subsequent washing, the cells were incubated in 0.1% OsO4 (Burry et al. 1992) in PBS for 15 min. Other preparations of cells received the same treatment except for omission of the silver enhancement procedure, and other samples received the entire procedure except that they were not incubated with the primary antibody.
In additional experiments, 5-nm colloidal gold (goat anti-mouse IgG) was used as the secondary antibody for ultra-stuctural detection of MTs. The colloidal gold was diluted either 1:15 or 1:30 (A520 was 0.103 and 0.053, respectively) over that supplied by the manufacturer in PBS-goat serum as described for FNG and FITC-IgG. After a 2-hr incubation at 22C, the unbound 5-nm gold was removed by several washes in PBS. The samples were then refixed in 2% glutaraldehyde in PBS for 15 min and then washed several times in PBS. Silver enhancement of the 5-nm colloidal gold was not employed in the EM preparations. The samples were postfixed in 0.1% OsO4 for 15 min and then washed in PBS.
In all cases, the cells on glass coverslips were dehydrated and embedded in Epon as described previously (Robinson et al. 1986). Thin sections were collected on formvar-coated single-slot grids and examined with or without heavy metal contrast staining using a Philips CM-12 electron microscope.
Results
Immunocytochemical labeling of leukocyte MTs was readily achieved with FNG and conventional FITC-labeled secondary antibody using the preparation method we recently described (Ding et al. 1995). Because of the ultrasmall size of the 1.4-nm gold, we were unable to detect MTs in these same cells with the other modes of optical microscopy employed (e.g., DIC) (Figure 1). However, the FNG was very versatile because it could be visualized directly by fluorescence and also after silver enhancement by brightfield, DIC, phase contrast, and epipolarization optics (Figure 2). It should be noted that the fluorescence signal from FNG was retained, albeit diminished, after early times of silver enhancement (up to 2 min) (Figure 2), but was lost with longer silver enhancement times (data not shown). In other experiments, cells labeled with conventional FITC-tagged secondary antibodies were subjected to the silver enhancement protocol. This was to test whether the constituents of the enhancement solution caused quenching of the fluorescent signal. The fluorescence in these samples did not appear to be diminished to any great extent (data not shown). Therefore, the loss of the fluorescence signal observed in the FNG-labeled samples was due to quenching by the increased silver deposits present after longer enhancement times rather than by some interaction with components of the silver enhancement solution per se.
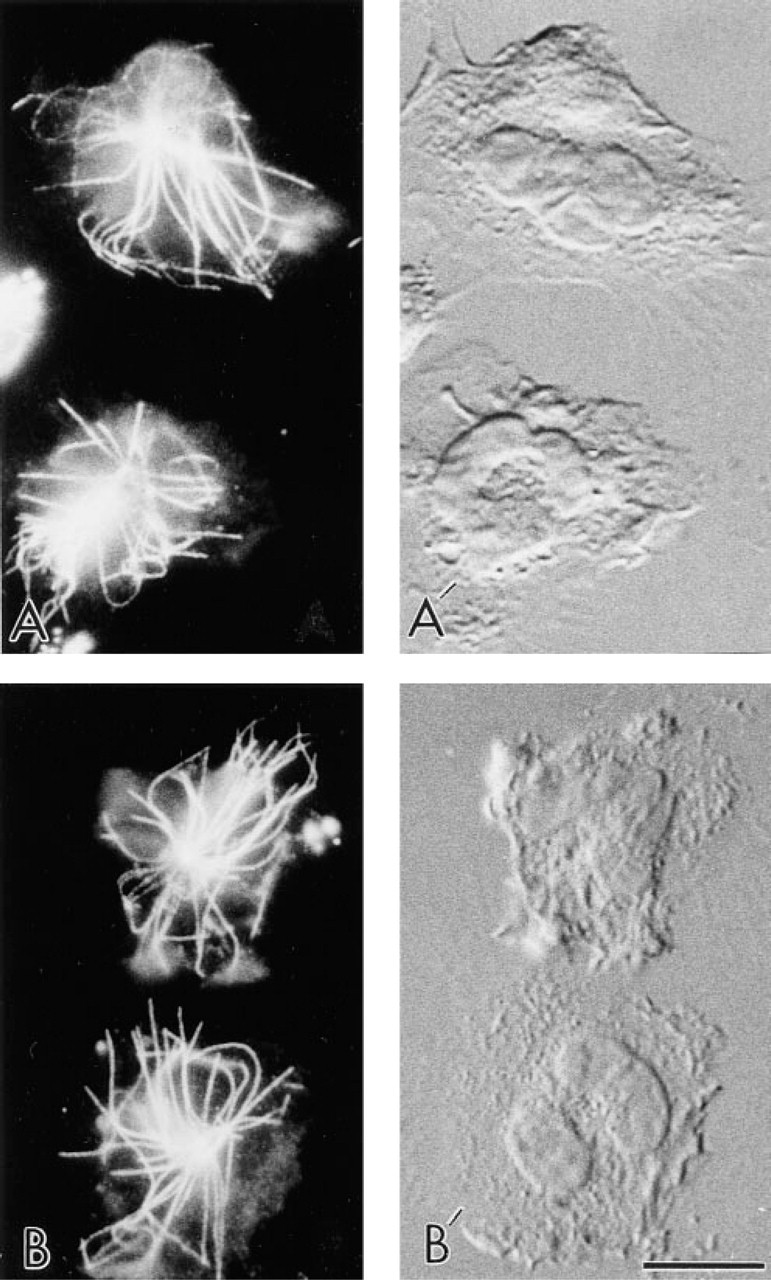
Immunofluorescence localization of MTs in human neutrophils with FNG or conventional FITC-IgG as secondary antibodies. (
The effect of silver enhancement of the 1.4-nm FNG particles was first observed by DIC optics. At the earliest times in the silver enhancement process, MTs were detected by DIC before their optimal visualization by brightfield microscopy (Figure 3).
Silver-enhanced 1.4-nm FNG particles were also visualized by confocal microscopy using the reflectance mode of operation (Figure 4) as well as the fluorescence mode (not shown). The reflectance imaging modality was particularly useful for defining regions of close association of cells with the substratum. These peripheral regions of close association with the substratum lack MTs.
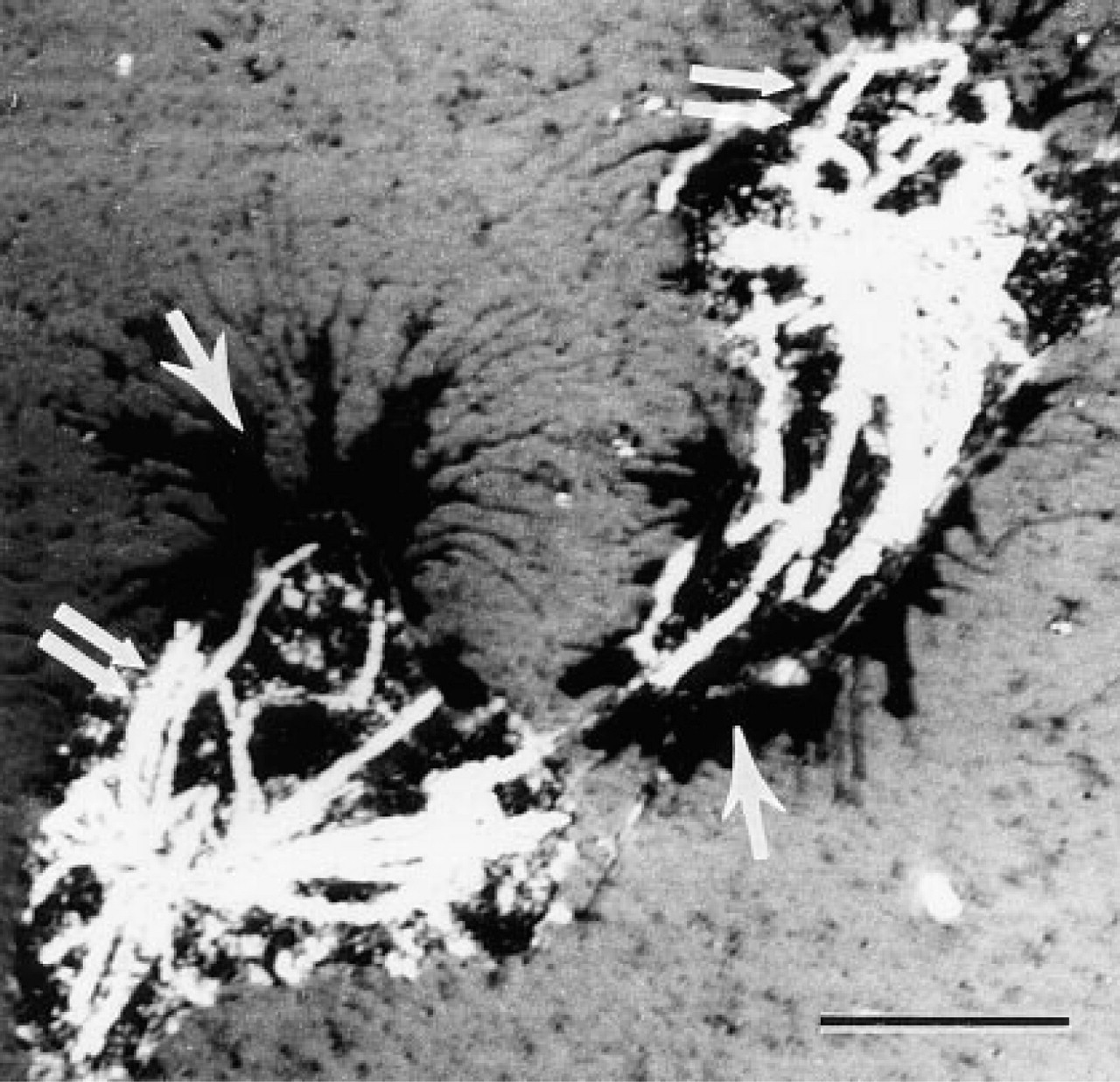
Silver-enhanced 1.4-nm FNG particles were readily detected in thin section preparations by TEM (Figure 5). The silver-enhanced particles were visualized in thin sections lacking counterstain and in thin sections that had been counterstained with heavy metals. The MTs were intensely labeled with the reaction product. MT labeling was evident by EM at the earliest silver enhancement time tested (i.e., 30 sec), even though MTs were not well visualized by brightfield optics in those samples. Little if any background detection of silver-enhanced gold particles was observed over the nucleus in these preparations. There were some silver-enhanced gold particles in the cytoplasm away from the MTs. However, this is probably specific labeling derived from unpolymerized tubulin that was not extracted from the cell during the preparative procedures. Microtubules were not detected in samples that did not receive silver enhancement or primary antibodies. On the other hand, labeling of MTs was absent at the EM level with 5-nm colloidal gold in these cells (data not shown). This is probably due to the preparative procedures employed in which the cells were fixed in glutaraldehyde and then subsequently extracted with detergents. The cytoplasm of cells prepared in this fashion may be sufficiently crosslinked to prevent uniform penetration of the 5-nm colloidal gold-IgG particles into the cells (see below).
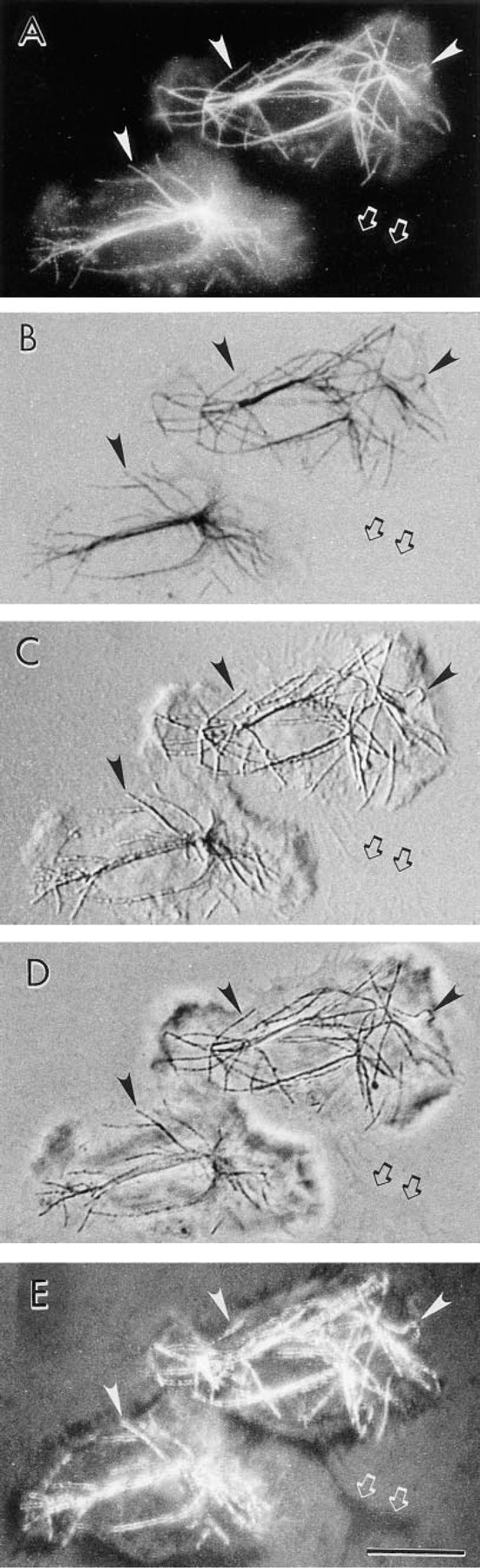
Localization of MTs in human neutrophils with FNG: detection with different imaging modalities in the optical microscope. The same two neutrophils are shown in each panel. After labeling of the cells with FNG, the sample was incubated in silver enhancement solution for 2 min. The sample was then mounted on a glass microscope slide and observed. The same individual microtubules are marked as points of reference in each panel (arrowheads). A region at which a thin cellular process is located is shown (arrows). (
Localization of MTs in human neutrophils with FNG and brief silver enhancement times. (
Localization of MTs in human neutrophils labeled with FNG and then incubated for silver enhancement. The cells were imaged with a confocal microscope operated in the reflectance mode. The MTs (double arrows) are readily apparent when observed in this manner. Areas of the cell in close association with the substratum appear black (single arrows). MTs are largely excluded from these regions of close contact. Bar = 10 μm.
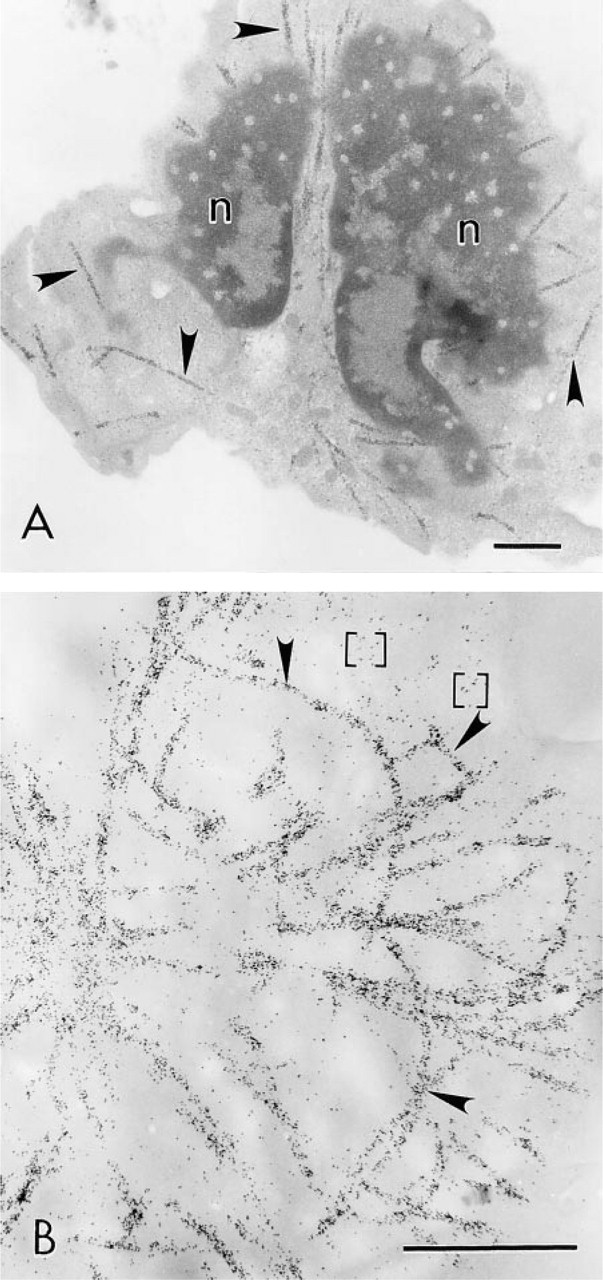
Localization of MTs in human neutrophils with FNG and silver enhancement at the electron microscopic level. (
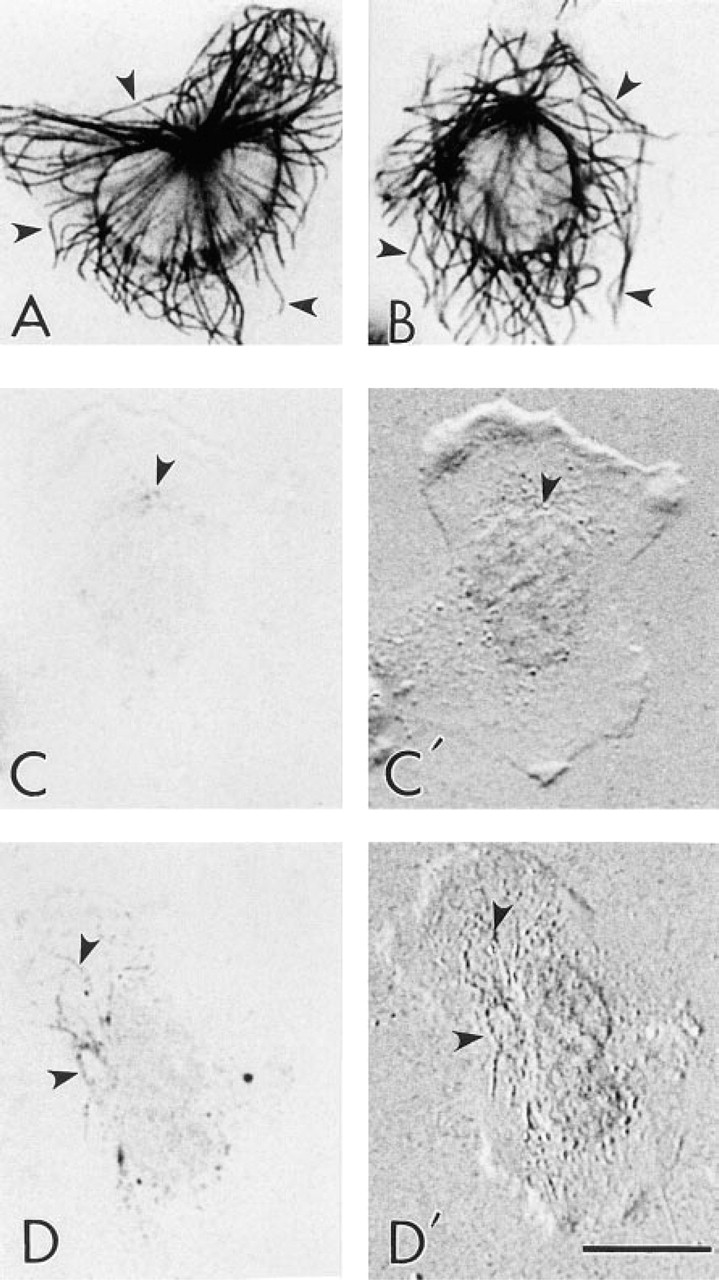
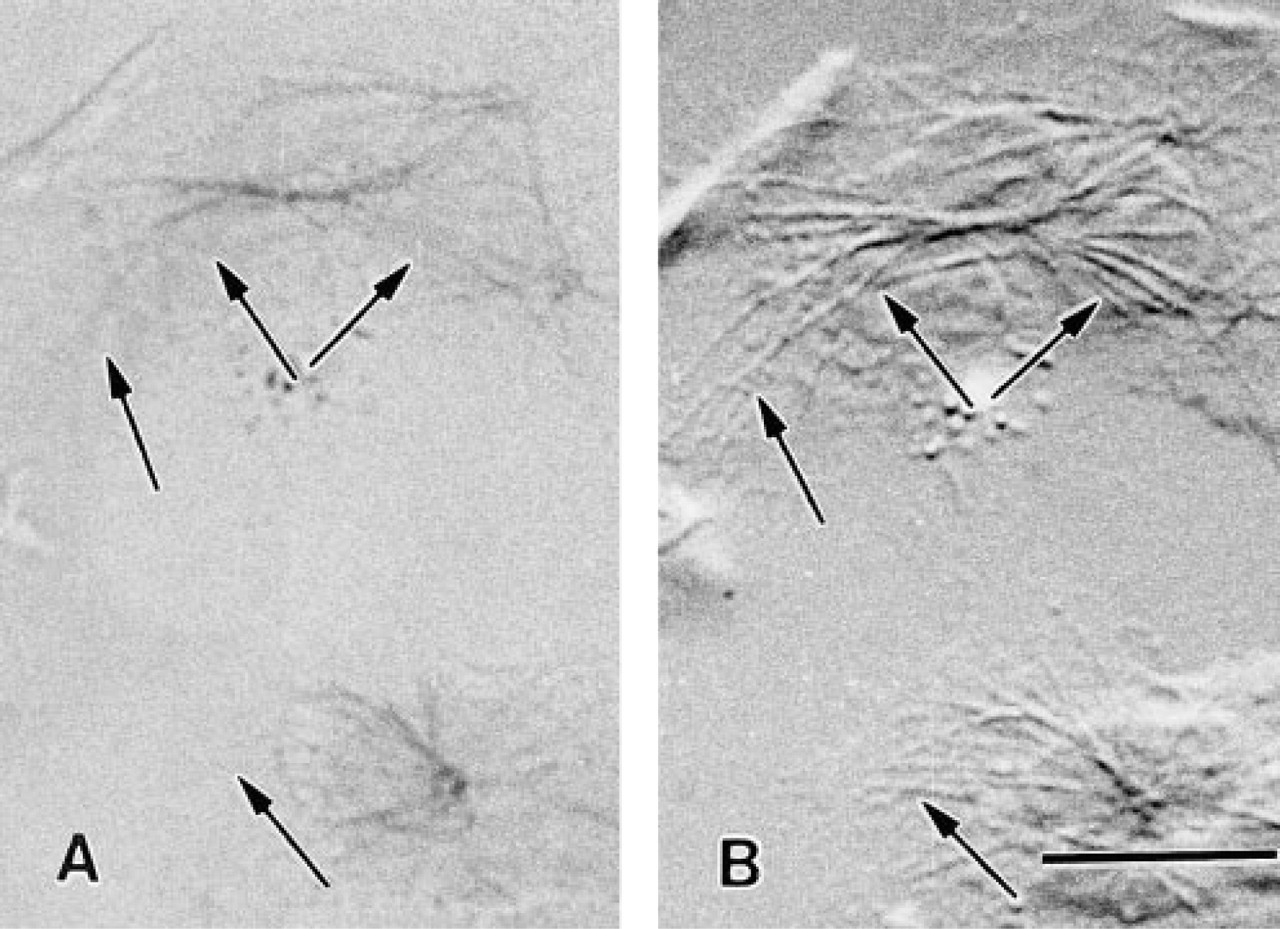
Comparison of the labeling efficiency of FNG and 5-nm immunogold particles in leukocytes at the light microscopic level. All cells are from the same cell preparation and were treated in exactly the same manner (i.e., fixation, permeabilization, primary antibody incubation, silver enhancement schedule) except for the size of the secondary immunoprobes. (
The poor labeling of leukocyte MTs achieved with the 5-nm immunogold probes at the ultrastructural level led us to a comparison of the degree of penetration of FNG to that of 5-nm colloidal gold into leukocytes. In these experiments, cells were fixed, permeabilized, exposed to primary and secondary antibodies, and incubated for silver enhancement in exactly the same manner. The only difference was in the secondary immunoprobe employed. All of the secondary antibodies were used before the expiration dates given by the manufacturers. MTs were heavily labeled after silver enhancement when FNG was used as the secondary antibody. On the other hand, when 5-nm colloidal gold served as the secondary antibody there was little if any labeling of MTs (Figure 6). As previously mentioned in conjunction with the EM results, the poor labeling of MTs achieved with 5-nm immunogold was most likely due to exclusion of the 5-nm immunoprobes from the glutaraldehyde crosslinked cytoplasm. Although there were occasional cells in these preparations (<0.5%) that showed some MT staining with 5-nm gold, they were probably damaged in some way during processing, thus allowing the colloidal gold into the cells. However, the FNG readily penetrated into the cytoplasm of leukocytes prepared in this manner, as evidenced by the labeling of the full MT array in each cell. This was an unequivocal result because examination by light microscopy allowed the examination of large numbers of cells compared to EM observation. All FNG-labeled cells displayed extensive decoration of the MTs, whereas very few cells displayed even poor MT labeling when 5-nm gold was used.
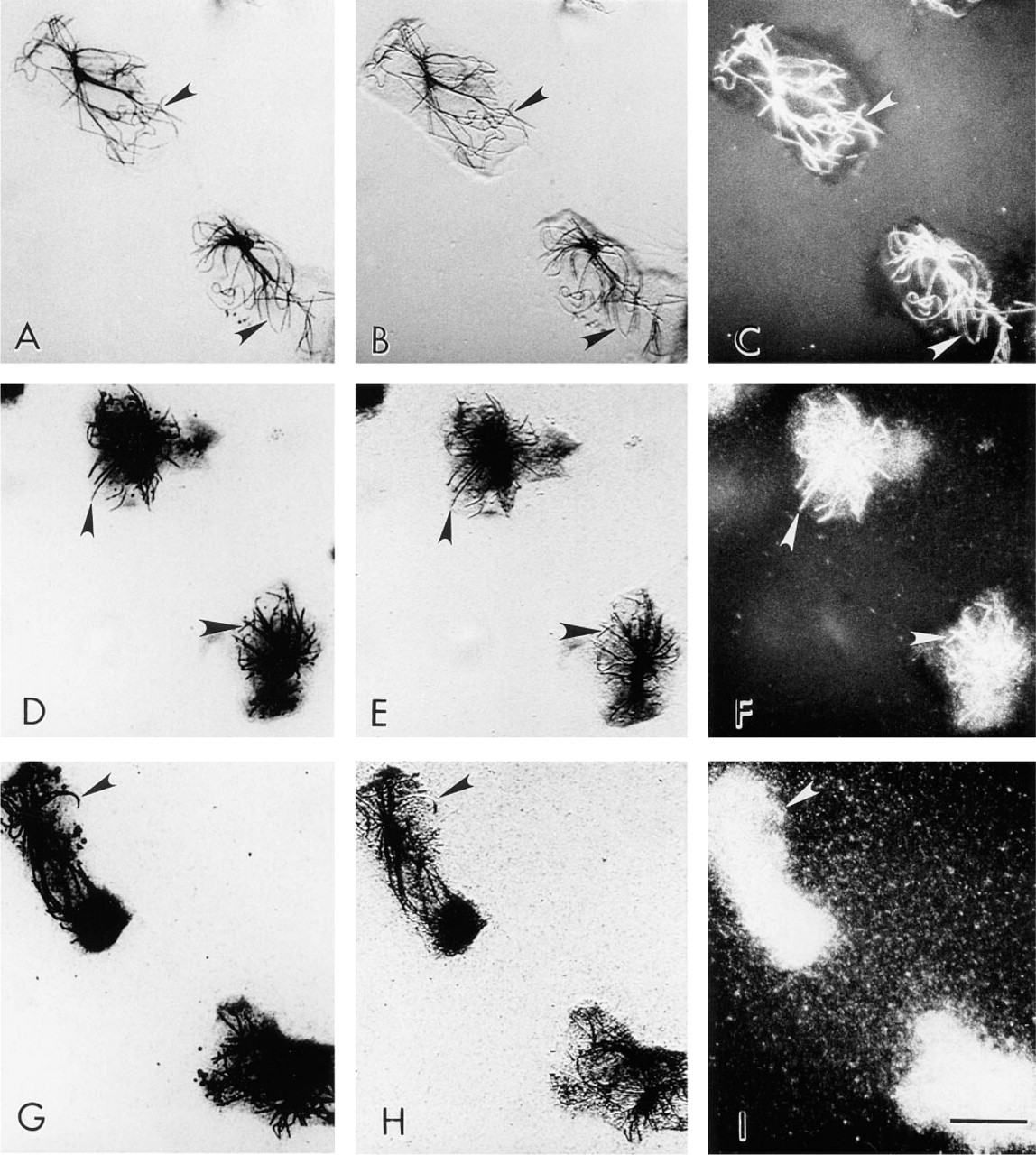
Because of the highly dynamic nature of the MTs in human phagocytic leukocytes (Ding et al. 1995; Robinson and Vandré, manuscript in preparation), we were unable to do the previous experiment in the reverse manner. That is, we could not permeabilize the leukocytes before fixation; when that is done the neutrophil MTs are difficult to detect and over half of the monocyte MTs are lost (compare Cassimeris et al. 1986 and Ding et al. 1995). To compare the ability of the FNG and the 5-nm and 10-nm colloidal gold to label MTs in the same cells that were detergent-extracted, either before or after glutaraldehyde fixation, we examined the labeling patterns in LLC-PK cells. MTs of LLC-PK cells fixed and then permeabilized were labeled with FNG, as determined by fluorescence microscopy. This labeling pattern was identical to that obtained with conventional FITC-IgG (Figures 7A and 7B). Similarly, MTs of LLC-PK cells were readily detected in samples fixed and then permeabilized before labeling with FNG and silver enhancement (Figure 7C). On the other hand, MTs were not detected when 5-nm or 10-nm colloidal gold particles were used under these conditions (Figure 7D). However, MTs of LLC-PK cells that were lysed before fixation were localized by all sizes of immunogold particles (Figures 7E and 7F). In the latter case, however, 1.4-nm and 5-nm immunogold yielded a higher labeling density than 10-nm immunogold. It is noteworthy that the silver enhancement reaction did not prevent the detection of nuclei and chromosomes with DAPI, the fluorescent DNA-binding dye (Figures 7G and 7H). These results also validated the general applicability of the FNG for immunocytochemical labeling of MTs in cells other than leukocytes. Furthermore, these results demonstrate that our lack of MT labeling in leukocytes with 5-nm gold was due to the inability of colloidal gold to penetrate into the cells rather than these specific reagents not reacting properly.
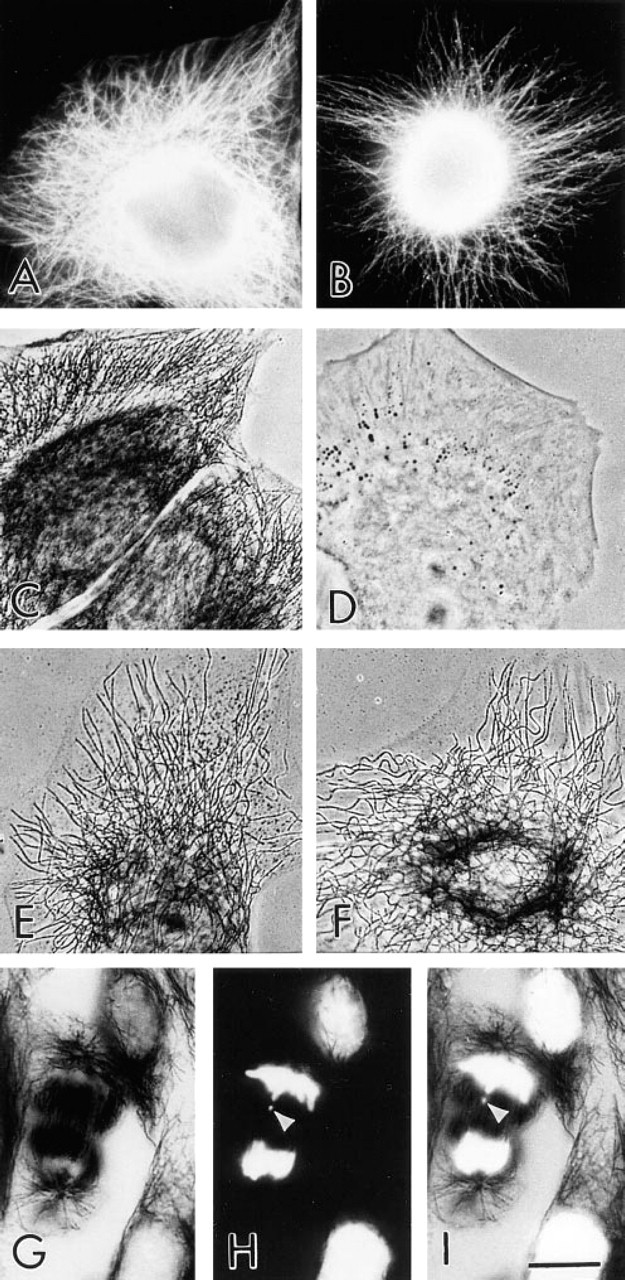
Comparison of the efficiency of FNG and colloidal gold particles for immunolabeling of MTs in LLC-PK cells at the light microscopic level: effect of the sequence of fixation and cell permeabilization. (
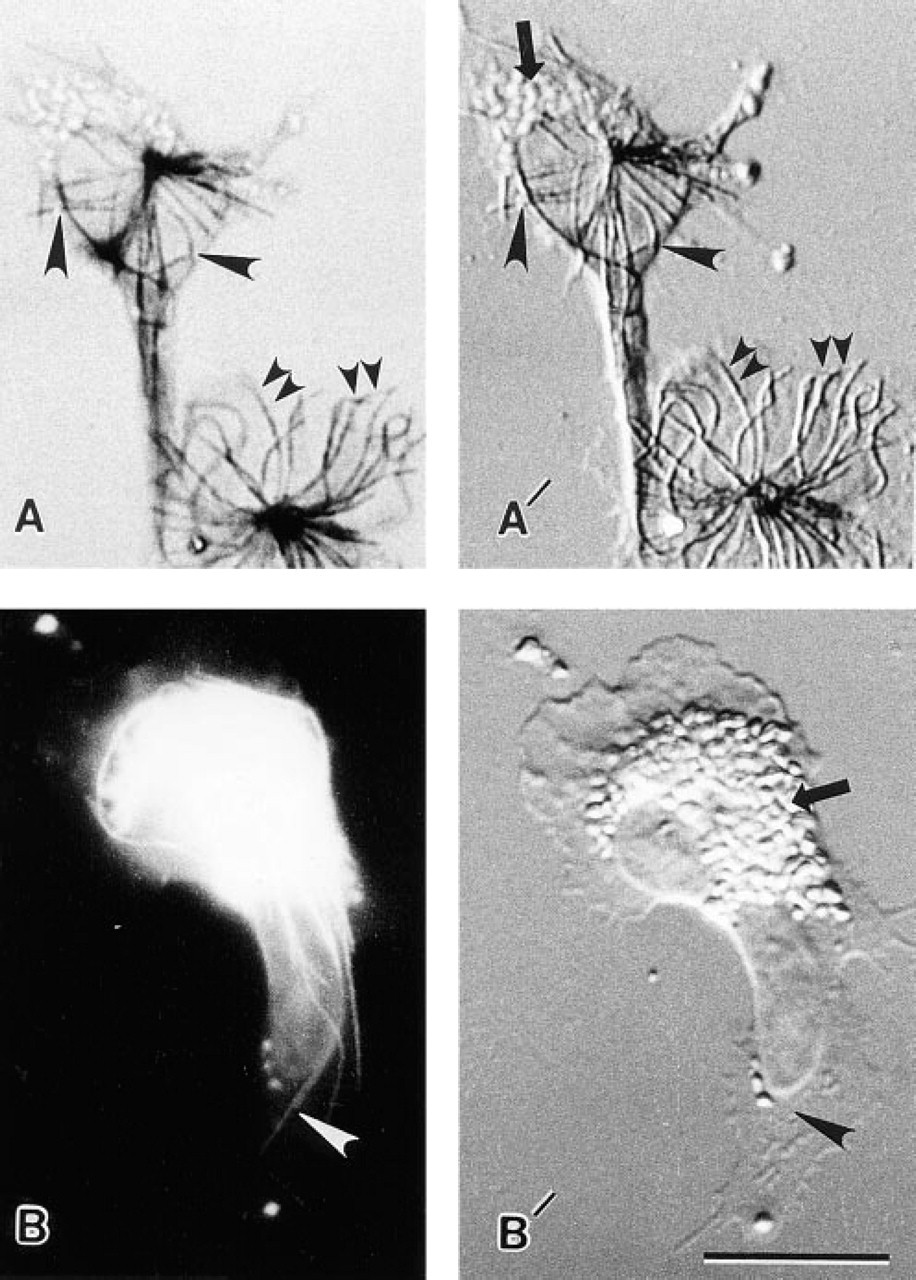
Comparison of the use of FNG with silver enhancement and FITC-IgG for detection of MTs in eosinophils. (
Effects of prolonged silver enhancement time on the localization of leukocyte MTs. (
At optimal silver enhancement times, the MTs were visualized by brightfield optics with little if any background signal. This is a very useful attribute of this approach to immunolabeling in certain situations. For example, eosinophils contain large granules that are highly autofluorescent at certain excitation and emission wavelengths (e.g., Barnes et al. 1993). The problem associated with this type of background in eosinophils can be eliminated by brightfield examination of silver-enhanced FNG-labeled MTs in eosinophils, whereas MTs in FITC-labeled cells were often obscured by the granule fluorescence (Figure 8).
The silver enhancement step is important to the optimal use of FNG at both the optical and electron microscopic levels. Comparison of Figures 2 and 3 demonstrates that the level of signal derived from the silver enhancement of FNG represents a continuum that is time dependent. It should also be recognized that at later time points (i.e., postoptimal times) the signal from silver-enhanced FNG begins to degrade until ultimately there is too much background noise to permit detection of individual MTs with any of the imaging procedures employed (Figure 9).
Discussion
Immunocytochemistry comprises a powerful collection of techniques that have been applied to many questions in biology and biomedicine. Much of the progress in immunocytochemistry has been dependent on development of reporter systems for demonstration of antibody binding to cell and tissue antigens. These reporter systems include fluorochromes, enzymes, and particulate probes. In the latter case, a seminal development in immunocytochemistry was the introduction of the iron-containing protein ferritin as an electrondense marker for electron microscopy (Singer 1959). This was followed by the introduction of colloidal gold as an electron dense immunoprobe (Faulk and Taylor 1971) and the use of gold-labeled secondary antibodies (Romano et al. 1974). Staphylococcal protein A–gold probes were subsequently introduced for indirect detection of antigen–antibody binding (Romano and Romano 1977; Roth et al. 1978). The colloidal gold immunolabeling system has been a key factor in obtaining fundamentally important information concerning cell structure. An interesting and personal review of the historical development of the colloidal gold marker system for immunocytochemistry has recently appeared (Roth 1996).
Colloidal gold was initially restricted to electron microscopy in its application to immunocytochemistry. However, colloidal gold immunolabeling has been applied to light microscopic labeling of cells (e.g., De Mey et al. 1981; Roth et al. 1980; Geoghegan et al. 1978). In these cases detection was based on observing the natural red color of colloidal gold with transmitted light. This method lacks sensitivity and requires that the antigen be present in abundance and that the colloidal gold be used in a concentrated form. The necessary step for increasing the sensitivity of the colloidal gold system so that it could be used reliably in light microscopy was provided by Holgate and co-workers (1983). They introduced the silver enhancement step, which was based on the method developed by Danscher (1981) for detection of gold in tissue sections. Immunogold–silver staining for light microscopy has been widely applied (for review see Lackie 1996). In addition to detecting the brown to black color of silver-enhanced gold by brightfield microscopy, these deposits have been detected by epipolarization and reflection contrast optics (DeWaele et al. 1988; Hoefsmit et al. 1986).
Potentially one of the most attractive avenues available for correlative microscopy is to combine fluorescence imaging with other imaging modalities (e.g., electron microscopy). Fluorescence techniques, such as immunofluorescence, display high spatial resolution and sensitivity compared to other immunocytochemical techniques routinely employed for light microscopy. The inability to correlate routinely fluorescence and electron microscopy has been a major disadvantage. Several different approaches have been employed in an effort to correlate fluorescence microscopy with other types of microscopy. An FITC–protein A–gold complex (20-nm gold) was developed for immunocytochemical labeling (Roth et al. 1980). This approach of combining colloidal gold, an immunoprobe (e.g., protein A, IgG), and a fluorochrome into the same complex has not been widely applied. Some investigators report that association of fluorochromes and proteins with colloidal gold results in diminution or complete loss of the fluorescence signal (for review see Goodman et al. 1991). Colloidal gold particles have been conjugated to fluorescent latex beads for use as a marker for retrograde transport to label neurons for examination by light and electron microscopy (Quattrochi et al. 1987). This probe, although useful for neurobiological applications, lacks the resolution and specificity required for examination of single-cell preparations. In other situations, colloidal gold and fluorescent immunoprobes have been used in the same preparation of cells (Van Dongen et al. 1985). In this case, distinctly different probes were used for double labeling (i.e., the colloidal gold immunoprobe was not fluorescently labeled).
Another approach that has been employed to correlate fluorescence microscopy and electron microscopy is fluorescence photo-oxidation of diaminobenzidine (Maranto 1982). In this method, diaminobenzidine is present in the cell or tissue section when a specific fluorochrome is excited. The diaminobenzidine becomes oxidized in the process and precipitates at the site of the fluorochrome. This reaction product can then be converted to an electron-dense product by treatment with osmium. Visualization of the products can be achieved by light and electron microscopy. This methodology has been used with several different fluorescent compounds (e.g., Lubke 1993; Schmued and Snavely 1993; Pagano et al. 1989; Sandell and Masland 1988). Deerinck and co-workers (1994) have explored the use of fluorescence photo-oxidation with eosin as a method for immunocytochemistry and in situ hybridization at the light and electron microscopic levels. Eosin was shown to have advantages for photo-oxidation of diaminobenzidine compared to fluoroscein and rhodamine.
Direct detection of ultrasmall gold (∼1-nm) at the EM level requires specialized equipment not normally available to biologists. For example, Stierhof and coworkers (1992) report that the best contrast of ultra-small gold particles, which had been used to immunolabel sections of resin-embedded bacteria, was obtained with a field emission STEM operated at 200 kV and equipped with a high-angle annular darkfield detector for collecting electrons that had undergone high-angle Rutherford forward scattering. Similarly, darkfield STEM has been used for high-resolution detection of individual 1.4-nm gold probes adsorbed to coated EM grids and for detection of anti-ferritin Fab' 1.4-nm gold probes labeling individual ferritin molecules (Hainfeld and Furuya 1992). More routine EM (i.e., TEM) observation of ultrasmall gold immunolabeling of resin sections (e.g., Burry 1995) or ultrathin cryosections (Takizawa and Robinson 1994) requires silver enhancement of the gold. In these experiments we have used the silver enhancement method described by Burry and co-workers (1992, 1995) because this method works at a higher pH (∼6.3) than other silver enhancement procedures currently available. This has the advantage of not being as perturbing to cell ultrastructure.
We show that FNG is a very useful immunoprobe. It can be detected by fluorescence microscopy and by other modes of optical and electron microscopy. For example, the labeling efficiency of samples can be examined using fluorescence microscopy before silver enhancement of the FGN for electron microscopy. This provides a means to ensure proper immunoprobe localization before completing the more extensive procedures involved in preparing the sample for electron microscopy. Retention of the fluorescence signal from FNG, even after brief periods of silver enhancement, is a highly desirable feature and will permit the combination of fluorescence and other forms of optical microscopy in the same samples after immunocytochemical labeling. As further support for this contention, we were able to combine silver enhancement of FNG for detection of MTs with transmitted light and DAPI staining for fluorescence detection of nuclei and chromosomes.
When the silver enhancement schedule was optimal there was essentially no background staining. However, it must be recognized that prolonged silver enhancement causes degradation of the immunocytochemical signal. Therefore, as a practical matter, one must determine the optimal time course for silver enhancement for different types of experimental material to achieve useful immunocytochemical localization results.
Localization of MTs with conventional immunofluorescence techniques provides excellent spatial resolution of these cytoskeletal elements. The use of FNG as a secondary antibody for detection of MTs by fluorescence microscopy or by other forms of optical microscopy after silver enhancement yields high-resolution detection of these structures equal to that obtained with conventional fluorochrome-labeled IgG probes. In certain situations, (such as with eosinophils, in which background fluorescence associated with intracellular granules is a major problem), results obtained with silver-enhanced FNG may be superior to those derived from immunofluorescence.
Labeling of MTs with colloidal gold has been achieved in studies of several cell types. However, efficient labeling of these MTs with colloidal gold-secondary immunoprobes typically requires that the cells be permeabilized with detergent before stabilization of the sample by chemical fixation (e.g., Baas and Black 1990; Mitchison et al. 1986). On the other hand, ultrasmall gold (∼1-nm) penetrates into cells fixed before permeabilization (Van de Plas and Leunissen 1993; Merdes and De Mey 1990). Similarly, we show that FNG readily penetrates into cells that are glutaralde-hyde-fixed before detergent permeabilization. Indeed, FNG penetrated as readily as FITC-IgG in both leukocytes and LLC-PK cells in our experiments. Larger colloidal gold probes (i.e., 5-nm, 10-nm) did not penetrate to any great extent into cells prepared in this manner. In addition, we verify that MTs can be labeled with 5-nm and 10-nm colloidal gold if cells are permeabilized before fixation. It should be emphasized that in the present study cells were fixed in glutaraldehyde before permeabilization. Therefore, the cytoplasm is more highly crosslinked than is routinely the case in MT preparations in other cell types. This puts constraints on probe penetration that may not be encountered in studies of MTs in other cell types. There are probably several factors involved in successful probe penetration. These include (a) probe size, (b) probe charge, (c) tertiary structure of the immunoprobe, and (d) flexibility of the probe. With the present data, one cannot determine which single factor or combination of factors accounts for good labeling with one probe and poor labeling with another. However, it is likely that probe size is a key factor leading to poor labeling with 5- and 10-nm colloidal gold immunoprobes in the leukocyte preparations. In other studies we have shown that ultrasmall gold particles penetrate further into aldehyde-fixed and cryosectioned neutrophils than do 5- or 10-nm colloidal gold particles. Furthermore, there were differences in the degree of penetration of 1.4-nm gold coupled to whole IgG compared to 1.4-nm gold coupled to Fab fragments (Takizawa and Robinson 1994). Therefore, the absolute size of the immunoprobe may be important in obtaining optimal penetration into chemically crosslinked samples.
The ability of FNG to penetrate into fixed cells far better than colloidal gold particles, along with the fact that this probe can be viewed directly by fluorescence microscopy or by other modes of microscopy after silver enhancement, makes it an ideal tool for correlative microscopy. The ability to resolve very small structures (e.g., MTs) equally well by fluorescence microscopy and by other types of microscopy after appropriate silver enhancement underlines the utility of FNG as an immunoprobe with great versatility.
In summary, we show FNG to be a versatile new immunoprobe. The utility of FNG primarily relates to its ability to be visualized directly by fluorescence microscopy and after silver enhancement by other modes of optical microscopy and electron microscopy. The ability of FNG to penetrate into aldehyde-fixed cells to a greater extent than colloidal gold particles also increases its versatility. FluoroNanogold should prove useful and find wide application in studies in which correlative microscopy is important.
Acknowledgments
Supported in part by the Ohio State University Comprehensive Cancer Center and by NIH Grant NS 31777.
We thank Jonathan Mathias for excellent technical assistance during the course of this study. We are grateful to Dr James Hainfeld for providing samples of FluoroNanogold to test and to Dr Richard Burry for sharing reagents and his technical expertise.
Footnotes
Note Added in Proof
Details of the chemical properties of FNG will be presented in a separate publication by Richard Powell, James Hainfeld, and their associates.