
Abstract
Study of lipoxygenase expression (LOX; EC 1.13.11.12) during organogenic nodule formation in hop (Humulus lupulus var. Nugget) showed that LOXs are developmentally regulated throughout the process, suggesting their involvement in the response of internodes to wounding, nodule formation, and plantlet regeneration from these nodules. LOX activity and lipid peroxides exhibited a huge increase during the first week of culture, which may indicate a role for LOX and LOX products in response to wounding in hop, as reported for other systems. Western blotting analysis showed a de novo synthesis of LOX isoenzymes in response to wounding and the detection of three different isoenzymes. Confocal analysis of LOX immunofluorescence revealed the presence of the enzyme in cortical cells of induced internodes and in prenodular cells, mostly appearing as cytoplasmic spots. Some of them were identified as lipid bodies by cytochemical and double immunofluorescence assays, suggesting the involvement of a lipid body LOX during nodule formation. Immunogold labeling detected LOX in peroxisomes, lipid bodies, and plastids of nodular cells. Quantification of the labeling density provided statistical significance for the localization of LOX (three different isoenzymes) in the three compartments, which suggested a possible involvement of LOX in metabolic functions of these organelles during organogenic nodule formation and plantlet regeneration.
L
LOXs have several functions in plants: response to wounding stress and pathogen attack (Farmer and Ryan 1992; Rosahl 1996), mobilization of lipids for seed germination (Holtman et al. 1997; Feussner and Wasternack 2002), synthesis of cutin monomers and signal molecules (Blée and Schuber 1993; Brash 1999), and growth and development (Hildebrand 1989). The signal transduction cascades that trigger wound healing include fatty acids, which control the total period and strength of MAPK (mitogen-activated protein kinase) activity. In this way, LOXs and the oxylipin signature play a role in signaling events (Baudouin et al. 1999).
However, the exact role of LOXs in these processes often remains unclear, due in part to the presence of many LOX isoenzymes in plants (Siedow 1991). LOX isoenzymes have been reported in cytosol, chloroplasts, lipid bodies, vacuoles, microsome membranes, plasma membrane, nuclei, and mitochondria (Rosahl 1996; Hause et al. 2000).
Several compounds in the linoleate cascade from LOX, e.g., jasmonic acid, methyljasmonate, and some volatile aldehydes, have physiological effects in plants (Creelman and Mullet 1997). Jasmonic acid is derived through the octadecanoid pathway from linoleic acid, an important fatty acid constituent of membranes in most plant species. Jasmonates have attracted interest as signaling molecules activating gene expression in different plant responses, such as to wounding (Léon and Sànchez-Serrano 1999).
It has been suggested that LOXs are capable of oxygenating not only free polyunsaturated fatty acids but also complex lipid esters such as phospholipids and even biomembranes (Brash et al. 1987; Macarrone et al. 1994). During the early stages of cucumber seed germination, the lipid body LOX is induced and oxygenates, in vivo, fatty acid residues of the storage lipids (Feussner et al. 1997a). These oxygenated fatty acid residues are preferentially cleaved off by a lipase (Balkenhohl et al. 1998) and may then serve as endogenous substrates for glyoxysomal β-oxidation (Feussner et al. 1997b). Gluconeogenesis is a process that may occur in high energy-requiring processes such as morphogenesis. Previously, we reported on starch accumulation during organogenic nodule formation from Humulus lupulus var. Nugget internodes followed by its consumption during plantlet regeneration from these nodules (Fortes and Pais 2000a).
Nodular structures have been studied in several species as an additional morphogenic pathway for regeneration strategies, automated micropropagation, and genetic transformation for desirable characteristics. Hop (Humulus lupulus L.) is an economically important plant known for the production of acid resins and essential oils which are used in brewing. In spite of its economic importance, hop plants are extremely sensitive to different types of pathogens. Recently, resistance to fungi was introduced in H. lupulus var. Eroica by particle bombardment of petioles, which evolved to organogenic nodules and regenerated transgenic plantlets (Batista and Pais 2002).
Although morphogenic processes such as somatic embryogenesis and organogenic nodule formation play pivotal roles in plant biotechnology, little is known about the processes involved in their induction and development. Here we report on the expression and subcellular localization of LOXs and discuss their possible role during organogenic nodule induction and plantlet development.
Materials and Methods
Plant Material and Culture Conditions
Humulus lupulus (var. Nugget) plants were maintained under in vitro conditions and internodes were morphologically induced according to the protocol previously described (Fortes and Pais 2000a). Internodes were submitted to several incisions (wounding treatment) before inoculation. All cultures were incubated at 25 ± 2C with a 16-hr photoperiod (35 μE.m–2.sec–1) provided by cool-white Philips fluorescent tubes.
Enzyme Extraction
Extracts for the measurement of LOX activity were prepared according to Koch et al. (1992). Plant material was frozen in liquid nitrogen and stored at −80C until use. Approximately 1 g of tissue was ground in a mortar and pestle in 3–4 ml of ice-cold 0.1 M potassium phosphate buffer [pH 7, 1% (w/v) PVP, 0.1% (v/v) Triton X-100, and 0.04% (w/v) sodium metabisulfite]. The homogenate was centrifuged at 16,000 × g for 10 min at 4C and the supernatant filtered through Miracloth before precipitation for 1 hr at 4C under agitation with solid ammonium sulfate (60% saturation). The precipitate was brought to 2.5 ml with 0.1 M potassium phosphate buffer, pH 7, and then loaded onto prepared PD10 columns (Pharmacia; Uppsala, Sweden) and eluted with 3.5 ml phosphate buffer. Assays of LOX activity were carried out immediately without freezing the samples. Protein determinations were estimated by the Bradford method using BSA as standard (Bradford 1976). All the experiments were performed with a minimum of three tissue sample replicates per time point. Each assay was performed three times. Data from each assay are expressed as means ± SE.
Enzyme Assay
LOX activity was evaluated according to the method of Suurmeijer et al. (1998). Column fractions were assayed by mixing in a cuvette 5 μl of a 30 mM linoleic acid solution in methanol with 945–985 μl of 100 mM phosphate buffer (pH 6.8). The reaction was started by adding 10–50 μl of enzyme solution and the increase in absorbance at 234 nm was measured at room temperature. Enzyme activities (in ΔA min–1 mg protein–1) were calculated from the linear part of the curve. All optical measurements were performed on a spectrophotometer (UV-1603; Shimadzu, Tokyo, Japan). Controls were run with enzyme denatured by heating and alternatively without linoleic acid or without enzyme extract.
Lipid Peroxidation Analysis
The level of lipid peroxidation was measured in terms of malondialdehyde (MDA) equivalent content using thiobarbituric acid reaction (Dhindsa et al. 1981). Plant material (0.5 g) was homogenized in 0.1% (w/v) TCA and the homogenate was centrifuged for 5 min at 10,000 × g. Four ml of 20% (w/v) trichloroacetic acid (TCA) containing 0.5% (w/v) thiobarbituric acid (Merck; Darmstadt, Germany) was added to a 1-ml aliquot of supernatant. The mixture was heated at 95C for 30 min and then rapidly cooled in an ice bath. After centrifugation for 10 min at 10,000 × g, absorbance of the supernatant at 532 nm was read and the value for the nonspecific absorption at 600 nm was subtracted. The concentration of MDA equivalents or TBARS (thiobarbituric acid-reactive substances) was calculated using its extinction coefficient of 155 mm–1 cm–1 (Dhindsa et al. 1981). All experiments were repeated at least three times and with a minimum of three tissue sample replicates per time point. Data from each experiment are expressed as means ± SE.
Electrophoresis and Western Blotting
Plant material was frozen in liquid nitrogen, powdered by mortar and pestle, and further homogenized in a buffer as described by Borrel et al. (1997). It consisted of 50 mM Tris-HCl buffer (pH 8) containing 0.2 M sucrose, 10 mM NaCl, 0.1% (w/v) sodium azide, the protease inhibitors PMSF (1 mM), leupeptin, pepstatin A (each 1 μM), and 0.15 mM EDTA. The extract was centrifuged and the supernatant filtered through Miracloth and precipitated overnight with 4 volumes of acetone (Merck) at 4C. The extract was centrifuged at 15,000 × g for 15 min and the pellet was vacuum-dried, resuspended in 100 mM Tris-HCl buffer (pH 8), 10% SDS, 5% β-mercaptoethanol, and boiled for 5 min in 1/5 volume of loading buffer (50% glycerol, 0.15% BFB). The denatured proteins were subjected to electrophoresis on a gradient of SDS-polyacrylamide consisting of a 12% (w/v) acrylamide resolving gel and a 6% (w/v) stacking gel. Total proteins were estimated by the Bradford method using BSA as standard (Bradford 1976). Approximately 30 μg of protein was loaded per lane.
Protein samples and prestained standard proteins (Bio-Rad; Hercules, CA) separated by electrophoresis were transferred to Immobilon membranes (Millipore; Bedford, MA). Before immunodetection, an equal amount of total proteins was confirmed with Ponceau S (Sigma; St Louis, MO) staining. For immunodetection, the membrane was blocked in 2% powdered skimmed milk, 0.05% Tween-20 in PBS at 4C overnight. Then the membranes were incubated overnight at room temperature, with the anti-LOX antibody diluted 1:1000 in the blocking buffer, washed, and incubated for 2 hr with alkaline phosphatase-conjugated anti-rabbit IgG (Boehringer Mannheim; Mannheim, Germany), diluted 1:1000 in the blocking buffer. Finally, the proteins recognized by the antibody were revealed by treatment with a nitroblue tetrazolium, bromo-chloroindolyl-phosphate (NBT-BCIP) mixture.
Immunofluorescence Assays
Plant material at different time points was fixed overnight at 4C in 4% (w/v) paraformaldehyde in PBS, pH 7.3, and cryo-protected by immersion in graded sucrose solutions in PBS (0.1 M, 1 M, and overnight in 2.3 M). Cryoprotected samples were then embedded in tissue freezing medium (Jung; Nussloch, Germany), and sections of 20–30 μm were performed in a cryostat (Leica CM 1800; Vienna, Austria) at −20C. Sections were then placed onto aminopropyltrieth-oxysilane (Sigma)-coated slides and stored at −20C until they were used. Frozen glass slides carrying the sections were thawed at room temperature and immunolocalization was performed essentially as described by Testillano et al. (1995) with minor modifications. After permeabilization with 2% cellulase in PBS (Onozuka R-10) for 40 min, sections were washed in PBS and blocked for 5 min with 5% BSA in PBS. Alternatively, before blockage with BSA sections were pre-treated with Triton X-100 at 0.5% in PBS for 20 min. They were then incubated for 1 hr with the first antibody diluted 1:10 in 1% BSA in PBS (polyclonal antibody raised against a plastid 13-LOX, LOX-H3, from potato). This antibody was kindly provided by Dr. José Sànchez-Serrano and obtained as described by Royo et al. (1999). After rinsing steps in PBS, sections were incubated for 1 hr in darkness with the secondary antibody, anti-rabbit Alexa Fluor 488 (Molecular Probes; Leiden, The Netherlands) diluted 5:25 in Evans Blue (Sigma) to remove autofluorescence of cellulose plant cell walls. Sections were then washed, slightly, dried and mounted in Mowiol. Confocal optical sections were collected using a Bio-Rad MRC-1024 confocal scanning head mounted on a Zeiss Axiovert 135 microscope. Controls were made by replacing the first antibody with PBS or pre-immune serum. Alternatively, sections incubated with anti-LOX were further stained for 30 min with a 1-mg/ml stock of Nile Red (Sigma) in acetone diluted 5:100 in PBS. A blue laser was used for visualization of LOX immunofluorescence signal and green laser irradiation was used for Nile Red detection. Merged images of both signals were obtained.
Total Lipid Staining
Detection of total lipids was performed in cryostat sections by staining in a 0.3% solution of Sudan Black in 70% (v/v) ethyl alcohol for 20–30 min at 50C, followed by differentiation in 70% ethyl alcohol for 5 min and a rinse in distilled water.
Transmission Electron Microscopy
Samples were fixed overnight at 4C in 4% (w/v) paraformaldehyde in PBS, pH 7.3. After three washes (5 min each) in PBS, they were dehydrated in a methanol series at 4C. Then, samples were washed in pure methanol, infiltrated and embedded in Lowicryl K4M at −30C and polimerized under UV irradiation. Semithin sections were performed for preliminary histological analysis. Lowicryl ultrathin sections were placed on colodion- and carbon-coated nickel grids and used for immunogold labeling.
Immunogold Labeling
Immunogold labeling was performed essentially as previously described (Risueño and Testillano 1994). Grids carrying Lowicryl ultrathin sections were floated for some minutes on drops of double-distilled water, PBS, and 5% BSA in PBS. Then they were incubated for 1 hr at room temperature with anti-LOX polyclonal antibody diluted 1:10 in 1% BSA in PBS. After three washes in PBS, sections were incubated with a goat anti-rabbit IgG conjugated to 10-nm colloidal gold (BioCell; Cardiff, UK) diluted 1:25 in 1% BSA in PBS for 45 min. Then grids were washed first in PBS followed by double-distilled water and air-dried. Sections were counter-stained with 5% aqueous uranyl acetate for 20 min and lead citrate for 15 sec, and were observed in a JEOL 1010 electron microscope at 80 kV. Alternatively, sections were stained only with DAB for catalase detection (peroxisome localization). Immunolocalization of LOX was also performed in ultrathin sections, which were further stained with DAB. These sections were floated for some minutes (after immunolocalization) on a catalase-stabilizing buffer (cacodylate 0.1 M, sucrose 6%, NaCl 2.5%). Then sections were floated on a DAB solution (Teorell Stenghagen buffer 0.01 M, 0.20% 3,3 DAB, 0.15% H2O2) for 2 hr at 37C, washed three times with Teorell Stenghagen buffer 0.01 M (pH 10.5), postfixed for 30 min in 1% osmium tetroxide, washed in distilled water, and air-dried before electron microscopic observations.
Double Immunogold Labeling with Anti-LOX and Anti-Catalase Antibodies
Ultrathin Lowicryl sections from cryoprocessed samples were floated on drops of distilled water, PBS, and 5% BSA in PBS. They were then incubated with anti-LOX and with anti-rabbit secondary antibody conjugated with 10-nm gold particles as mentioned above. Then the sections were washed in PBS, floated on drops of glutaraldehyde 1% for 5 min to fix the labeling, washed in PBS, and floated in NH4Cl2 for 20 min to block aldehyde groups as putative sources of unspecific labeling. After washing in PBS, ultrathin sections were incubated with anti-catalase (Nishimura et al. 1983; Yamaguchi and Nishimura 1984) diluted 1:500 for 1 hr at room temperature. After washing with PBS, the sections were incubated with anti-rabbit secondary antibody conjugated with 5-nm gold particles (BioCell), diluted 1:25 in PBS for 45 min. Then the grids were washed in PBS, floated on glutaraldehyde 1% for 5 min, and washed with PBS, after which they were rinsed in distilled water and air-dried. Finally, the sections were counterstained with 5% uranyl acetate and 1% lead citrate and observed in a JEOL 1010 microscope at 80 kV.
Different controls were performed, excluding the primary antibodies, exchanging the gold particle sizes used for primary antibody visualization, and changing the order of the antibodies in the sequential incubations.
Quantitative Analysis of Gold Labeling
The density of gold labeling, expressed as the number of gold particles per μm2, was determined in different compartments in nodular cells by counting the number of gold particles over each cell compartment. The areas were estimated by a point-counting procedure using a 15 × 15-mm square grid. The minimum sample size (MSS) was determined by the progressive mean technique. All quantitative analyses were carried out using a BASIC program developed by Dr. J. Renau-Piqueras and the results represented in histograms. The statistical significance of the differences between mean values was determined by the Student's t-test, considered significant at p≤0.01.
Results
Developmental Stages Analyzed During Organogenic Nodule Induction and Plantlet Development
The morphogenic process in hop has been previously described (Fortes and Pais 2000a). Briefly, the following stages have been considered: stage zero, corresponding to internodes at the time of excision from the parent plant; 7 days after culture on induction medium in which cambial and cortical cells of internodal explants were dividing; 15 days after culture initiation, in which several prenodular structures are formed inside the calluses; 28 days after culture initiation, corresponding to nodule formation; and 45 days after culture initiation, in which plantlet regeneration occurs from organogenic nodules.
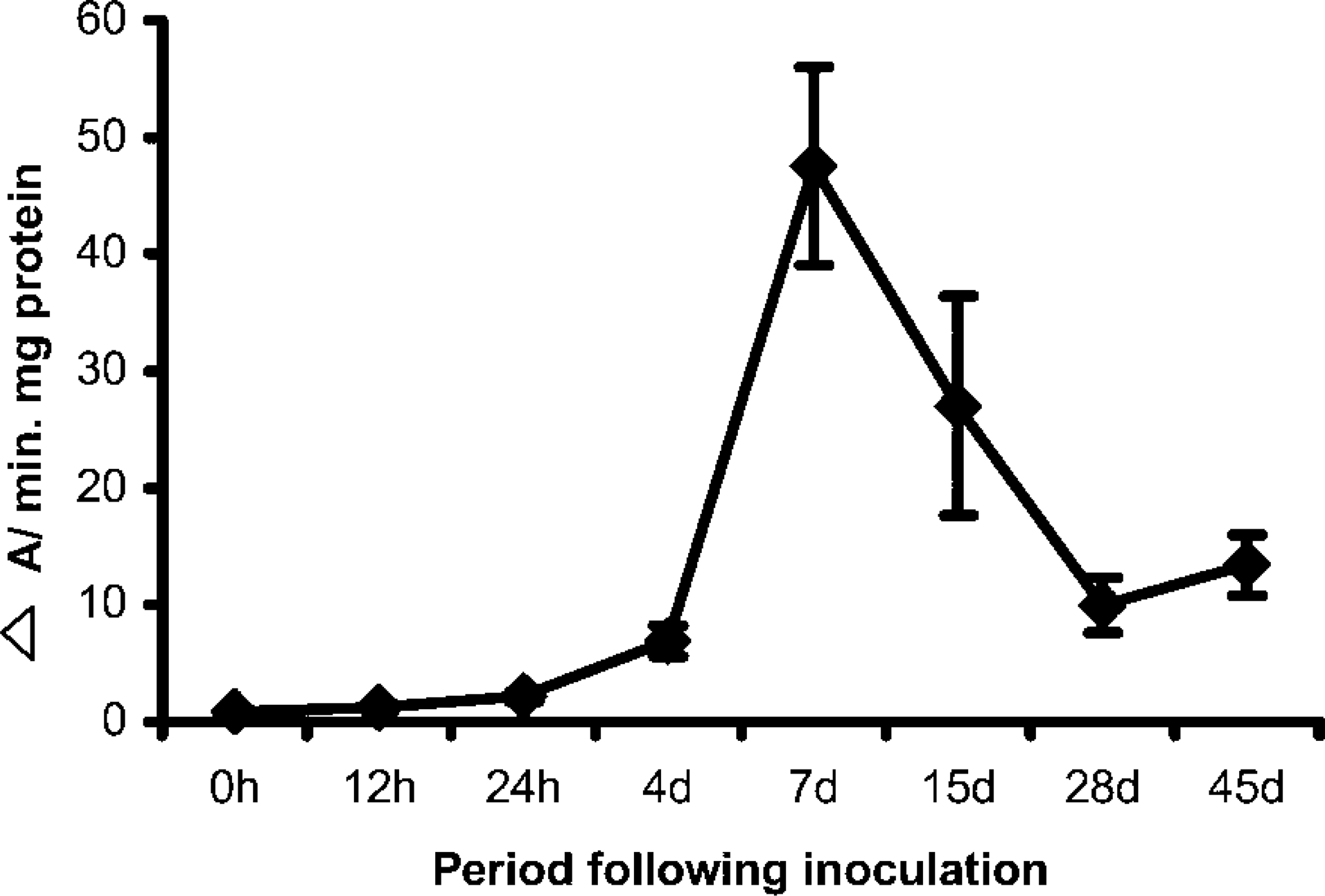
LOX activity during organogenic nodule induction and plantlet regeneration (45 days) from nodules. Results are expressed in Δ A/min mg protein for 0–24 hr and for 4–45 days. Three replicates were measured from three independent experiments.
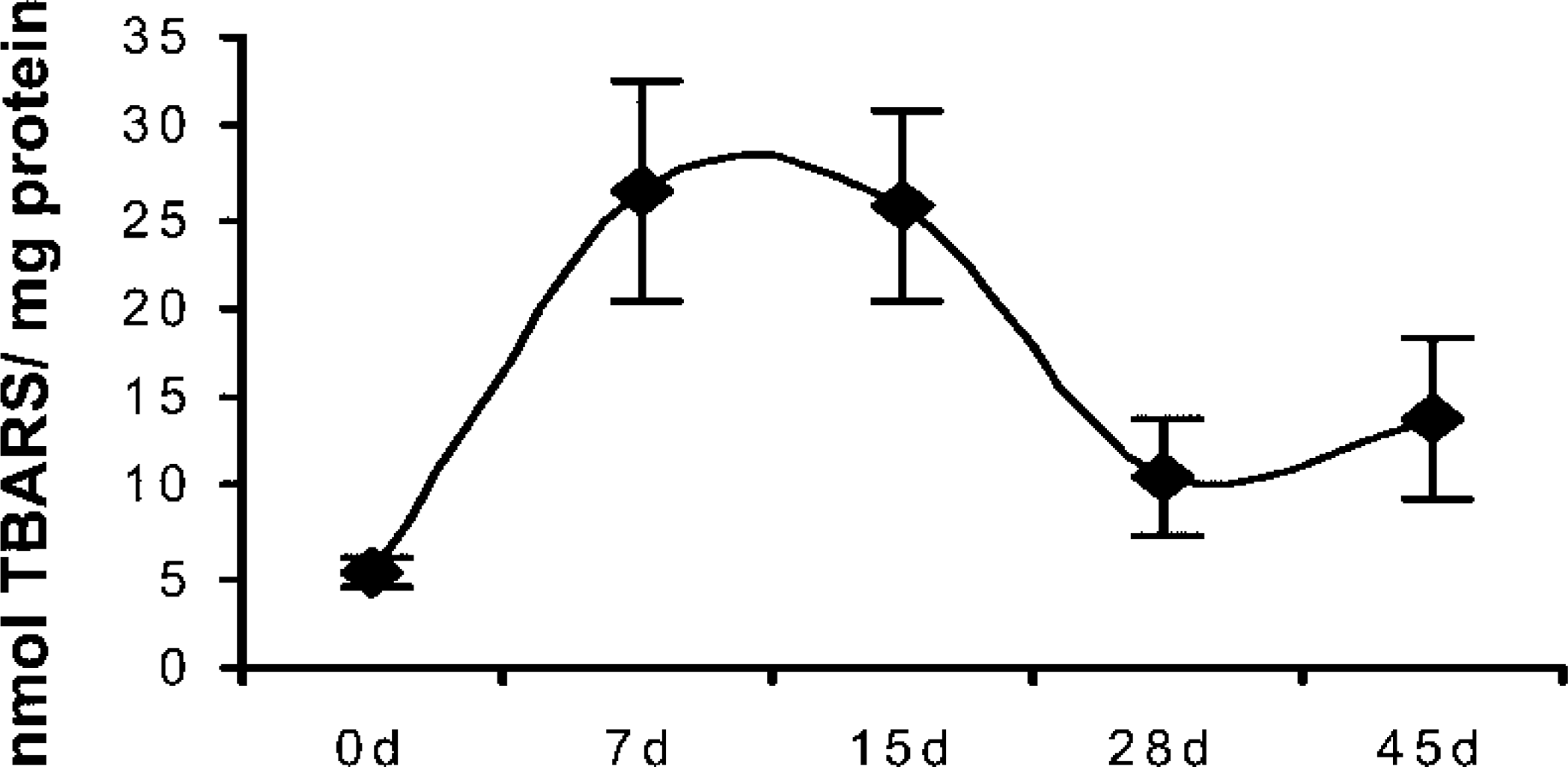
Lipid peroxide content during organogenic nodule induction and plantlet regeneration from nodules. Results are expressed in nmol TBARS/mg protein 0–45 days after induction. Three replicates were measured from three independent experiments.
LOX Activity
At day zero LOX activity was low either in extracts from the whole plant or from internodes submitted or not to wounding (0.90 ΔA/min mg protein (Figure 2). LOX activity gradually increased throughout the first week after induction, showing a maximum at day 7 (47.5 ΔA/min mg protein). An increase in LOX activity was already detected 12 hr after inoculation (Figure 1), which was confirmed by the corresponding increase in protein detected by immunoblotting (not shown). At day 15, when prenodules are being formed (Fortes and Pais 2000a), the levels of global LOX activity decreased (27.0 ΔA/min mg protein). At day 28, when nodules are evident, the global LOX activity decreased (10 ΔA/min mg protein) about five- and threefold compared to days 7 and 15, respectively (Figure 1). At day 45, when plantlets are being regenerated from nodules, LOX activity slightly increased to 13.3 ΔA/min protein.
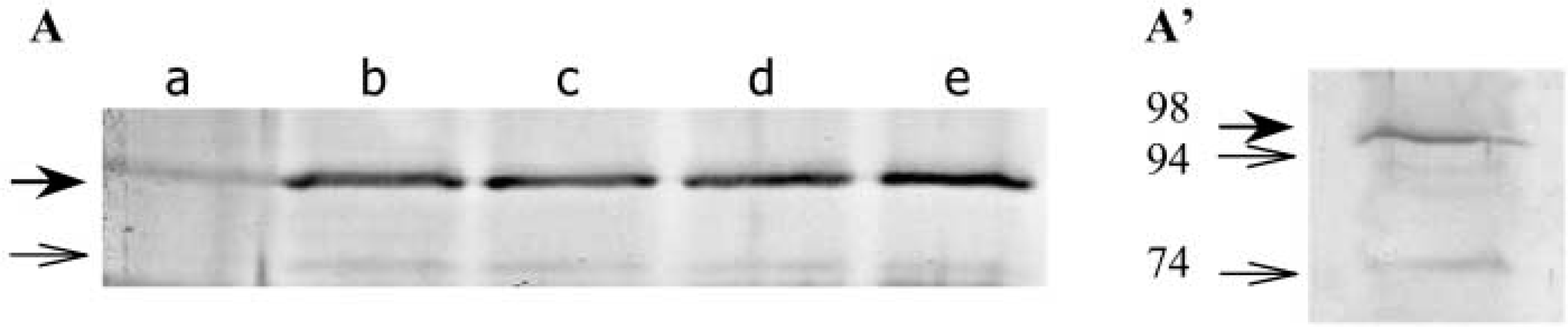
Immunoblotting analysis of LOX in internodal explants and organogenic nodules. (A) Protein extracts from different periods of culture (Lane a, 0 days; Lane b, 7 days; Lane c, 15 days; Lane d, 28 days; Lane e, 45 days) after SDS-PAGE and immunoblotted with polyclonal antisera for LOX. Thirty μg of protein were loaded per lane. (
Lipid Peroxides
Content of LOX (Figure 2) was low at stage zero (5.3 nmol TBARS/mg protein). At day 7 LOX increased to 26.4 nmol TBARS/mg protein. At day 15, when prenodules were formed, LOX content was still high (25.5 nmol TBARS/mg protein). Nodule formation occurring 28 days after induction was accompanied by a decrease in LOX content (10.5 nmol TBARS/mg protein). When plantlet regeneration occurred from these nodules, a slight increase in LOX was observed (13.7 nmol TBARS/mg protein), in parallel with the increase of LOX activity during the same period (Figure 1).
Immunoblotting
LOX protein was barely detected by Western blotting at day zero (Figure 3A). After 7 days in culture, LOX protein was detected in the immunoblot. The levels of LOX isoenzymes evaluated by Western blotting using a polyclonal antibody were constant until plantlet regeneration from organogenic nodules took place (Figure 3A). Extensive running of a gel enabled the separation of two bands corresponding to different isoenzymes with a molecular mass between 94 and 98 kD (Figure 3A′). A minor band with a lower molecular mass of approximately 74 kD was also present (Figures 3A and 3A′).
LOX Immunofluorescence
Cellular localization of LOX at different time points during culture was analyzed by immunofluorescence with LOX antibodies and observed under a confocal laser scanning microscope.
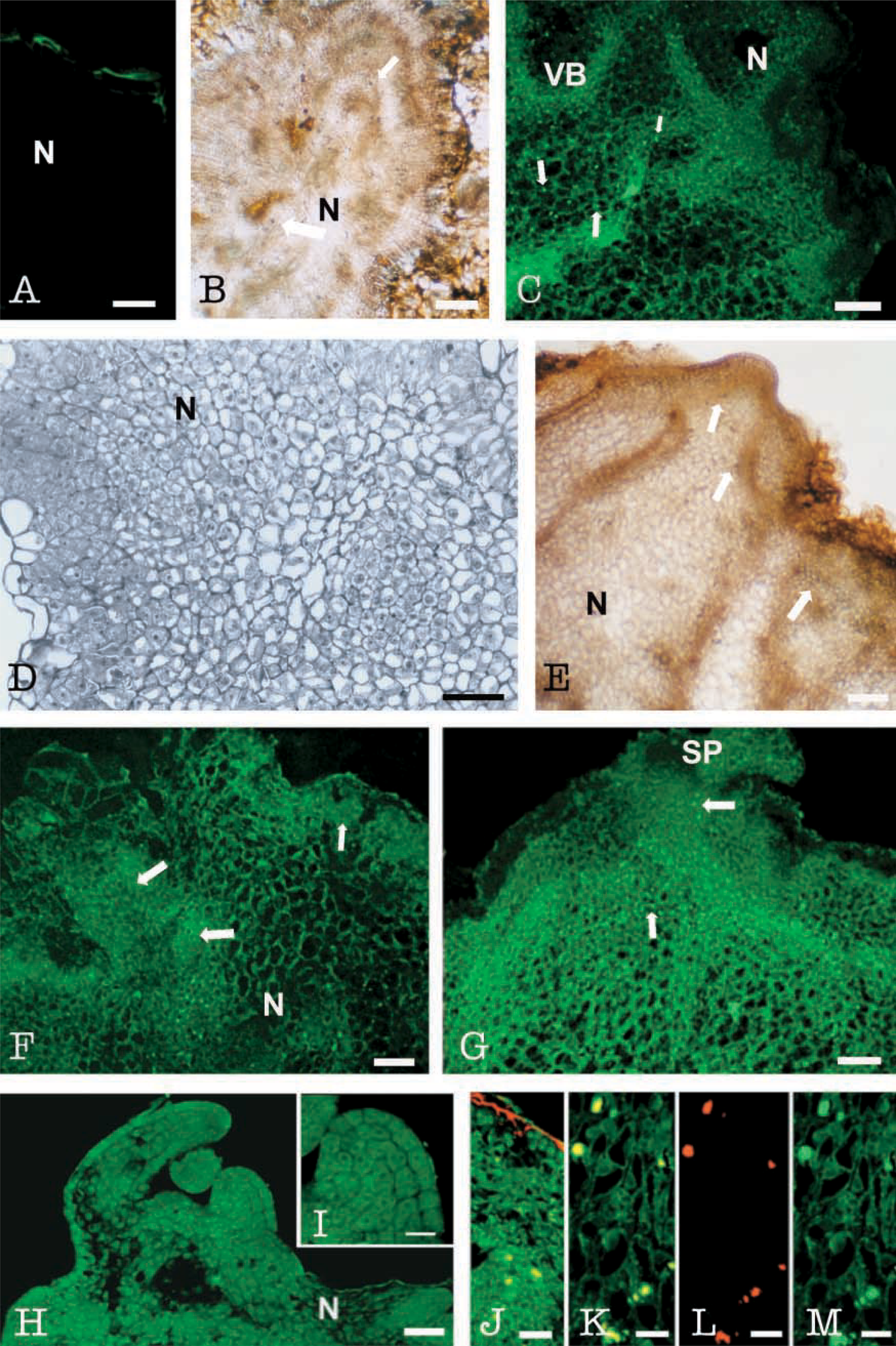
At stage zero, cross-sections of internodal explants showed a typical dicotyledon stem structure (Figure 4A). LOX immunofluorescence was low at this stage and was restricted to cambial, cortical, and subepidermal cells. Cortical and subepidermal cells showed a low signal, respectively, in cytoplasm and in chloroplasts (Figure 4B). The large vacuoles and cell walls appeared dark. Controls omitting the first antibody and with the preimmune serum did not show fluorescent signal (Figure 4D) except at the vascular cells, which displayed unspecific autofluorescence in all samples (Figure 4B).
After 7 days in culture, cell division occurred in both cambial and cortical cells, and groups of proliferating small cells with dense cytoplasm and small vacuoles were observed in these areas (Figure 4C). Highly vacuolated calli cells appeared at the periphery (Figure 4C). An intense immunofluorescence signal was found in these groups of proliferative cortical cells (Figure 4G), whereas vacuolated peripheral cells exhibited very low signal (Figure 4G). Control with preimmune serum showed autofluorescence only in the sclerenchyma cells, probably due to accumulation of cutin in their cell walls occurring at this stage (Fortes et al. 2002), and in the vascular cells (Figure 4D).
At day 15, prenodules were formed and prenodular cells displayed a high LOX immunofluorescence signal, whereas surrounding vacuolated cells showed much lower fluorescence intensity (Figure 4H). Some bright spots (arrows in Figure 4H) were found in prenodular cells surrounding vascular bundles. Sudan Black staining revealed positive dark spots of similar size, shape, and location, interpreted as lipid bodies (Figures 4E and 4F).
At day 28, initial stages of nodule formation were observed. Positive LOX immunofluorescence was found in regions surrounding the vascular bundles (Figure 5C, arrows). Peripheral areas with highly vacuolated cells displayed much lower LOX signal (Figure 5C). Sections stained with Sudan Black showed preferential dark contrast in regions similar to those with high LOX signal (Figure 5B). As in previous developmental stages, some spots were observed in these regions exhibiting high LOX signal (arrows in Figure 5C) and dark staining of lipids (arrows in Figure 5B). Controls with preimmune serum appeared dark, showing a thin autofluorescent layer only at the periphery (Figure 5A), probably corresponding to the cutin layer covering the organogenic nodules (Fortes and Pais 2002a,b).
After 45 days in culture, meristematic cells appeared at the periphery of the nodules (Figure 5D). Along with growth of the nodules, positive LOX immunofluorescence was mostly observed in the meristematic areas (Figure 5F and 5G, arrows) and shoot primordia (Figures 5G-5I). Nodular regions outside the shoot primordium, i.e., at the level of vascular bundles, showed a huge accumulation of LOX protein in globular spots (Figure 5G arrows). Sudan Black staining for lipids also revealed the presence of lipid bodies as discrete dark spots in nodular cells (Figures 5B and 5E, arrows). Staining of lipids was performed with Nile Red over some sections subjected to LOX immunofluorescence. Confocal analysis of the double fluorescence signals, red for the Nile Red (Figure 5L) and green for the LOX (Figure 5M), revealed co-localization as yellow signal (Figures 5J and 5K) on discrete spots of nodular cells.
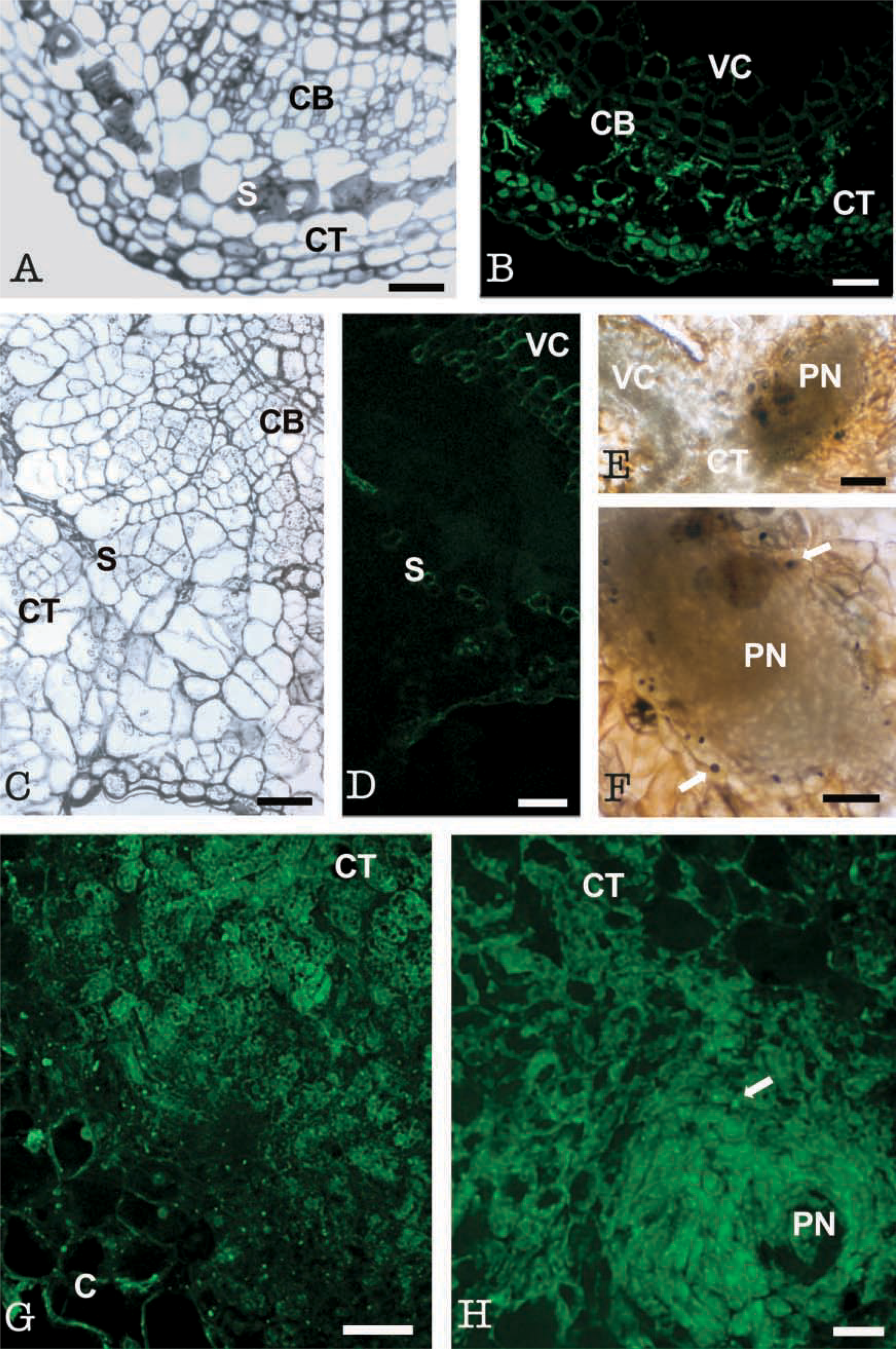
Tissue-specific expression of LOX in internodes at the time of excision from the parent plant and after 7 and 15 days of culture. (
LOX Immunoelectron Microscopy
To study the subcellular localization of LOX, immunogold labeling at the transmission electron microscopy level was performed on ultrathin Lowicryl sections from samples at different stages.
At stage zero, very few gold particles were found on cytoplasmic regions of cortical cells (Figure 6A). At later stages, labeling was found in dividing cambial and cortical cells, prenodular, and nodular cells, showing a similar pattern of distribution on different subcellular compartments. Controls with the preimmune serum showed no gold particles in any subcellular compartment (not shown).
After immunoelectron microscopy with anti-LOX antibodies, gold particles appeared in various subcellular compartments, the labeling in cytoplasm and nucleus being very sparce (Figures 6–9). No significant labeling was observed on cell walls (Figures 7B, 7C, and 8C) and vacuoles (Figure 7A). Labeling was found on lipid bodies (Figures 6B, 7A, and 7B). Gold particles were also found on plastids, specifically on chloroplasts (Figure 6C), in which labeling was preferentially localized in the stroma.
In nodular cells, many gold particles appeared in the peroxisomes (Figures 7B, 7C, 8, and 9). The labeling was mostly localized on the nucleoid, and some particles were also found on the matrix and membrane of these organelles (Figures 7B, 7C, and 8). Frequently, a lipid body was observed close to a peroxisome (Figure 7B). To cytochemically characterize the peroxisomes, a DAB reaction was performed on some sections after the immunogold labeling. Positive catalase reaction identified peroxisomes (Figured 9A and 9B), which also appeared labeled with anti-LOX (Figure 9B). A decrease in the intensity of the gold labeling was observed after catalase cytochemistry (compare Figures 9B and 8), probably due to a loss of gold particles during the cytochemical reaction.
Immunogold labeling with anti-catalase antibodies was performed to confirm the identification of peroxisomes (Figures 9C and 9D). Gold particles highly decorated peroxisomes in cytoplasm of prenodular and nodular cells; no significant labeling appeared in other structures (Figures 9C and 9D). The presence of LOX in peroxisomes was further revealed by double immunogold labeling using anti-LOX and anti-catalase antibodies revealed by gold particles of different sizes (Figure 9E). Co-localization of both LOX and catalase antigens ocurred on peroxisomes, which showed many dispersed gold particles of 10 and 5 nm (Figure 9E).
Controls exchanging the gold particle sizes used for primary antibody and changing the order of the antibodies in the sequential incubations provided similar co-localization results. Controls excluding the primary antibodies did not show the presence of gold particles.
Quantitative Analysis of the Immunogold Labeling
To assess the differences in labeling density among different subcellular compartments, quantification of the gold labeling density as number of gold particles per unit area was performed in nodular cells. Results of the mean values are summarized in the histogram of Figure 10.
The highest labeling density was obtained for peroxisomes (65.02 ± 8.80), followed by lipid bodies (30.26 ± 14.30) and plastids (20.52 ± 6.30). Mean values in cytoplasm and vacuoles were very low and not statistically significant, representing a low level of background (Figure 10).
Tissue-specific expression of LOX in nodules before and after plantlet regeneration. (
Discussion
Widespread application of gene transfer techniques for crop improvement cannot be successfully achieved if the processes leading to morphogenesis are not well understood, enabling a controlled induction of morphogenesis.
LOX expression was studied during induction and development of organogenic nodules (ONs) derived from hop internodes. Wounding applied to internodes for ON induction led to LOX activation. LOX activity was low at day zero. Immunolocalization studies carried out on ultrathin sections of control explants (day zero) showed very low levels of LOX in cytoplasm and lipid bodies. After 7 days in culture, explants showed a huge increase in LOX activity. Although the activity measurements reported can also account for auto-oxidation products, the increase in LOX activity was also suggested by de novo synthesis of LOX protein, as revealed by Western blotting, and by immunolocalization experiments. In addition, the majority of polyenoic fatty acid derivatives arise from the action of LOX (reviewed by Feussner and Wasternack 2002). For the same period (7 days after internode inoculation), explants induced on medium without growth regulators exhibited LOX activity levels that are half of those observed for explants induced on medium with growth regulators (Fortes and Pais 2000b). The expression of LOX at early stages may be partially due to auxin treatment. Auxin has been considered to induce LOXs (Wang et al. 1999). In fact, explants cultured for 7 days on medium containing only 6-benzylaminopurin (BAP) showed less LOX activity than in the presence of both BAP and IAA (Fortes and Pais 2000b).
One branch of the LOX pathway involves 13-LOX activity and leads to the formation of 13-hydroperoxy linoleic and linolenic acids. Linolenic acid is a precursor for the plant growth regulators traumatin and jasmonic acid. Traumatin was identified as a compound responsible for inducing callus growth after wound healing (Vick 1993). It may be that the high LOX activity detected 7 days after induction will lead to increased synthesis of traumatin. At this stage, callus formation from internodes occurred initially in areas submitted to wounding. For some plant species it has been proposed that, upon wounding, a systemic signal (peptide systemin) would trigger the release of fatty acids from cellular lipids and its subsequent conversion to jasmonic acid (Ryan 2000). Jasmonates have been considered as key signaling molecules in the transduction of wound stress events triggering the expression of defense-related genes (Wasternack and Parthier 1997). Recently, an increase in jasmonic acid was detected 24 hr after induction (Fortes et al. 2003). In hops, it has been shown the increase of reactive oxygen species (ROS) after internode wounding, a stress applied for induction of organogenic nodules (Fortes and Pais 1999). To signal the activation of wound-related pathways (Bate and Rothstein 1998), plants may also use C6 volatiles, whose biosynthesis involves LOX and HPL.
In prenodules, the amount of LOX protein was higher than in cortical cells. At this stage prenodular cells are proliferating to give rise to nodules. Nodular areas with high meristematic activity showed increased amounts of LOX, which suggests its role in shoot meristem formation. LOX has been suggested to be involved in growth and development (Hildebrand 1989). In pea, LOX distribution has been associated with cells in which expansion and growth are occurring, accounting for its role in pod growth and development (Rodríguez-Concepción et al. 1996). LOX has been implicated in tissue hydration (Hildebrand 1989), in cell elongation (Rodríguez-Concepción and Beltrán 1995), and in nitrogen storage (Tranbarger et al. 1991). Chateigner et al. (1999) have correlated the expression of LOX-g with axis growth resumption in pea seedlings and have suggested that this isoform could contribute to cell elongation during axis growth by remodeling the cell membrane phospholipid content. The spatial and temporal localization of LOX in prenodules and organogenic nodules suggests a role of LOX both in nodule formation and in plantlet regeneration from these nodules. Developmental programs, such as germination and root nodule formation, may require oxylipins as signal molecules that regulate processes of growth and metabolism (Porta and Rocha-Sosa 2002).
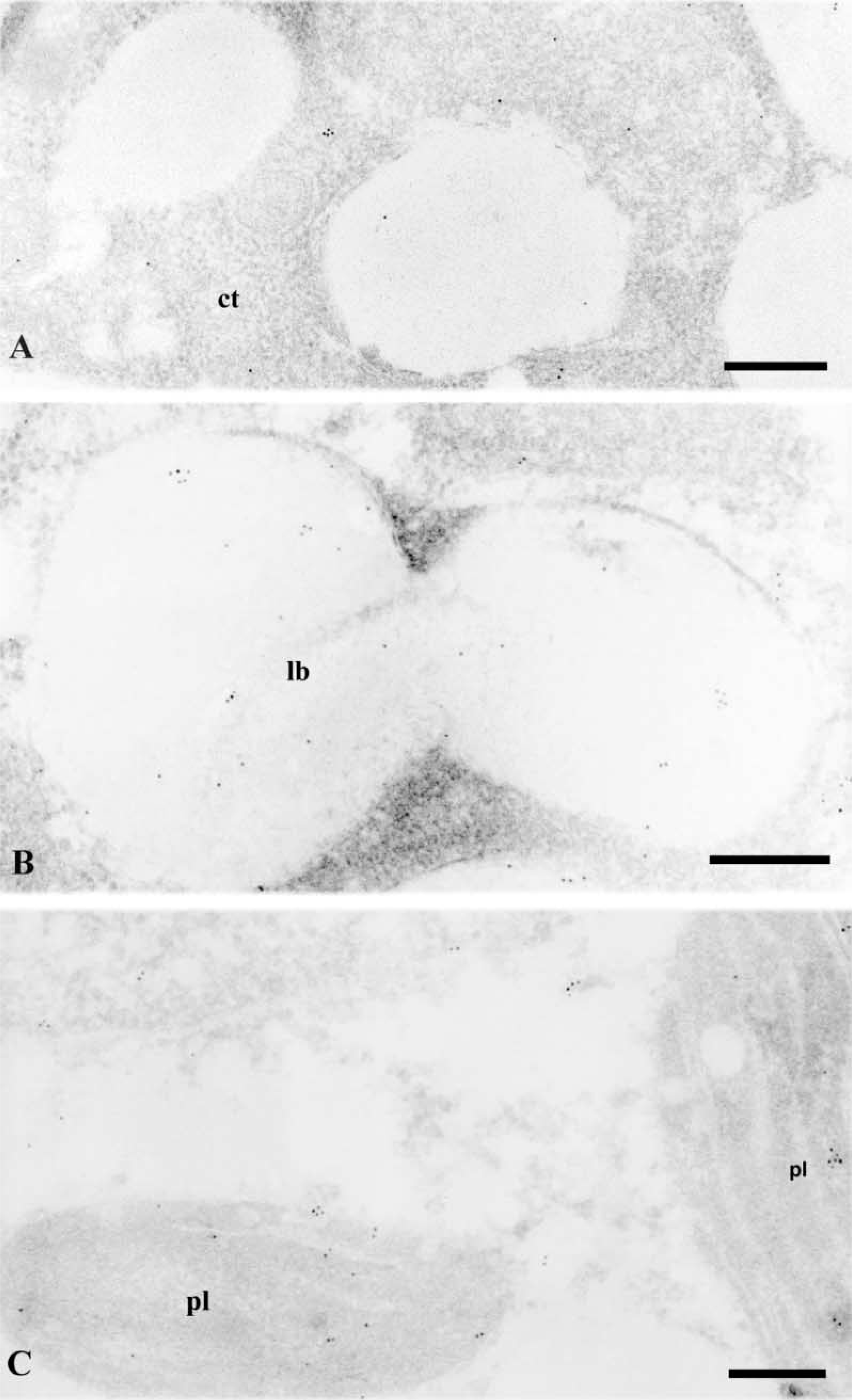
Anti-LOX immunogold labeling in hop internodes at diferent culture times. (
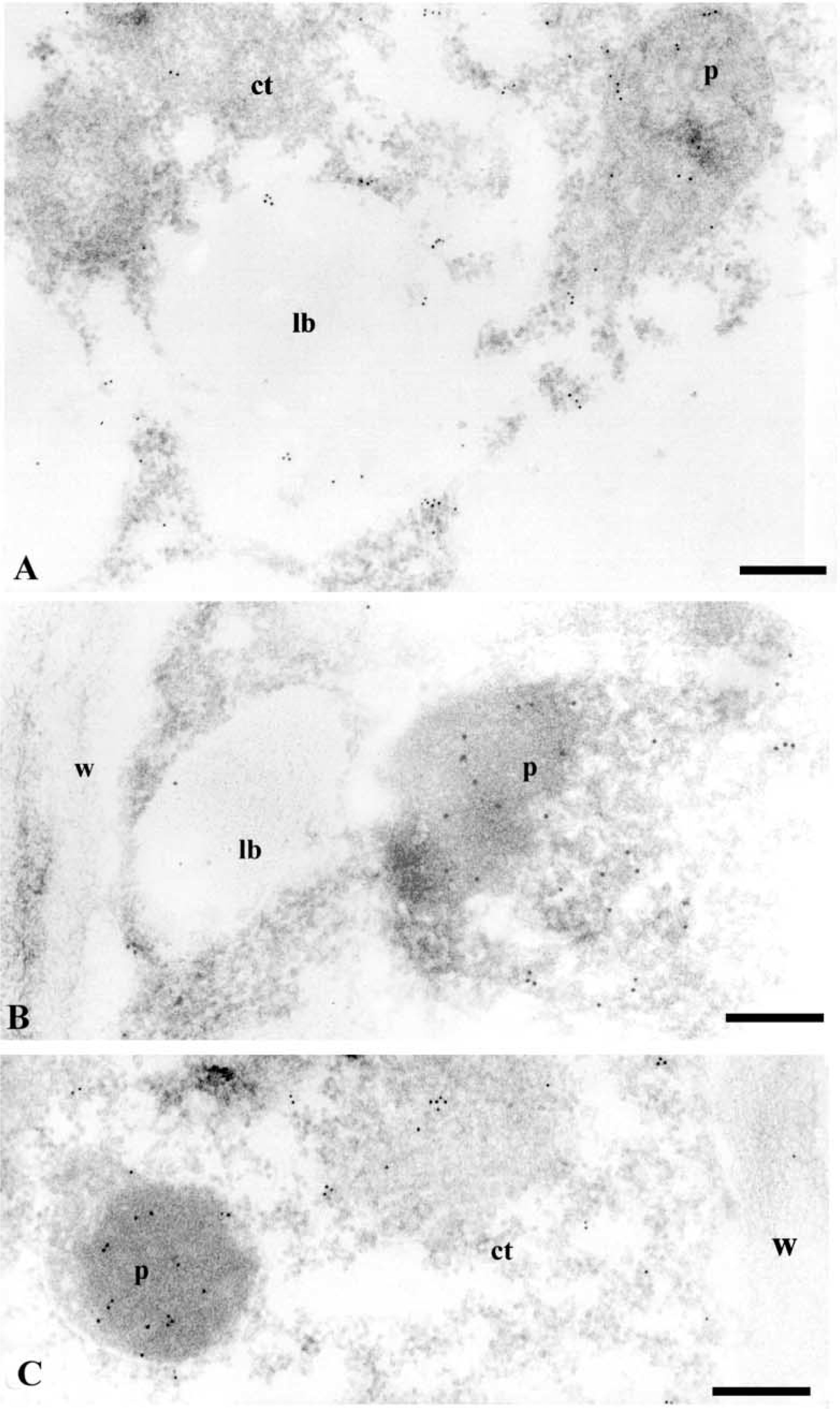
Anti-LOX immunogold labeling on specific organelles of nodular cells. (
In hop organogenic nodules, LOX was particularly abundant in cells surrounding vascular bundles. This high-fluorescence LOX signal may be related to the active cell division occurring in these regions. Recently, LOX was shown to be involved in the control of potato tuber development, a morphogenic process (Kolomiets et al. 2001). In this morphogenic system, Lox1 class transcripts were shown to accumulate in the vascular tissue of the perimedullar region, the site of the most active cell growth during tuber enlargement (Kolomiets et al. 2001).
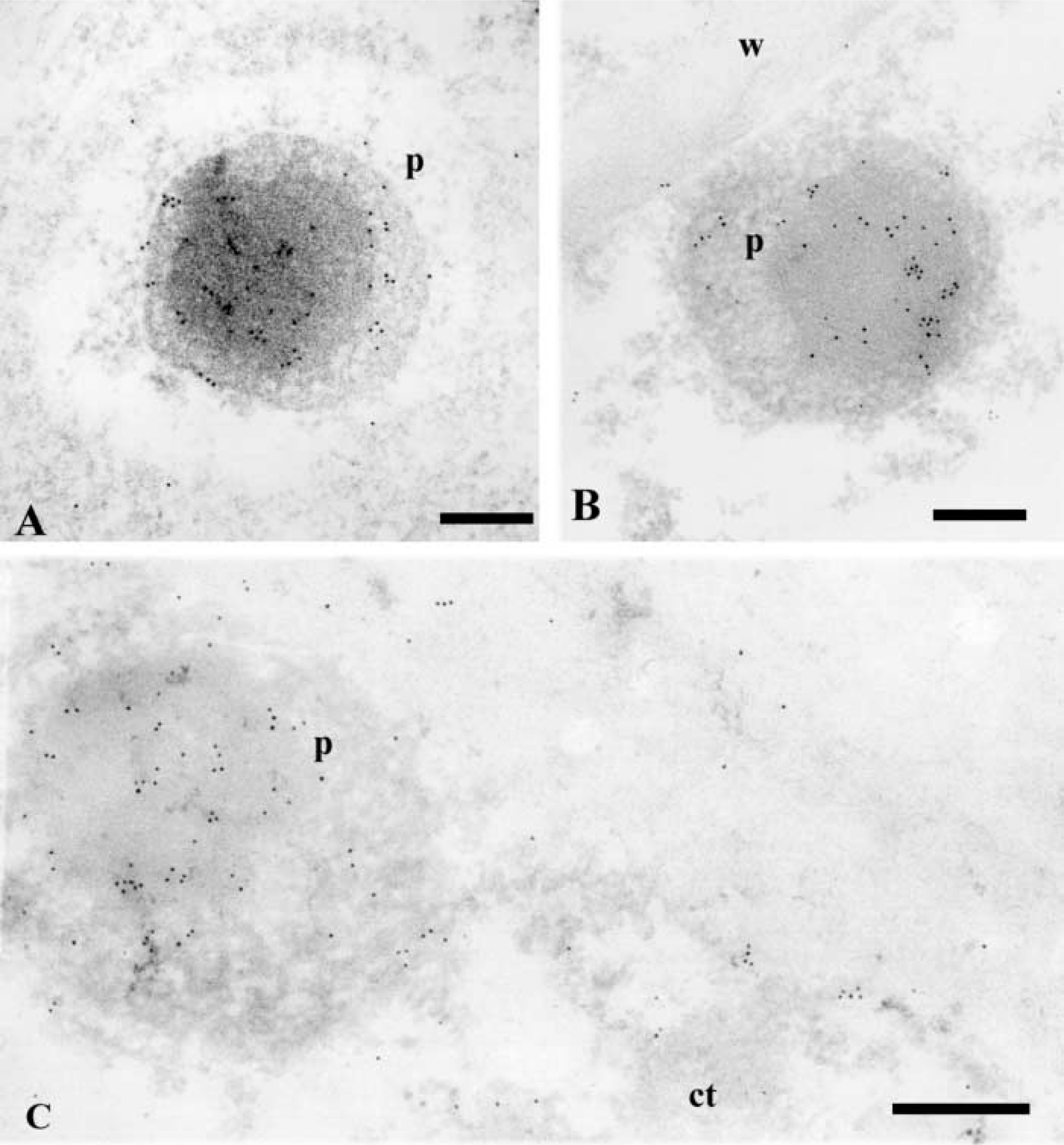
Anti-LOX immunogold labeling on peroxisomes of nodular cells. (
Our results suggest that several LOX isoenzymes with different subcellular localizations may be expressed throughout the morphogenic process. Western blotting revealed three bands, which is in accordance with the detection of LOX by immunogold labeling in peroxisomes, lipid bodies, and plastids. This localization of LOX isoenzymes in different cellular compartments may be related to different lipid substrates and downstream activities processing their hydroperoxide products. In plants, LOX-derived hydroperoxy fatty acids are metabolized through four major pathways involving enzymes such as the hydroperoxide lyase (HPL), allene oxide synthase (AOS), and peroxygenase (POX) and divinyl ether synthase (Feussner and Wasternack 2002).
In prenodular cells, LOX is localized in lipid bodies. This suggests the involvement of LOX in the mobilization of storage lipids into sugars to be used as a respiratory substrate in this high energy-requiring process of nodule formation. These lipids may be used to produce sugars via gluconeogenesis. Previous results showed a starch accumulation/mobilization cycle during ON formation (Fortes and Pais 2000a). According to Creelman and Mullet (1997), induction of vegetative storage protein synthesis (mediated by jasmonic acid) under conditions of high sugar accumulation creates a sink for carbon and nitrogen and releases phosphate from sugar phosphate pools for further carbon fixation.
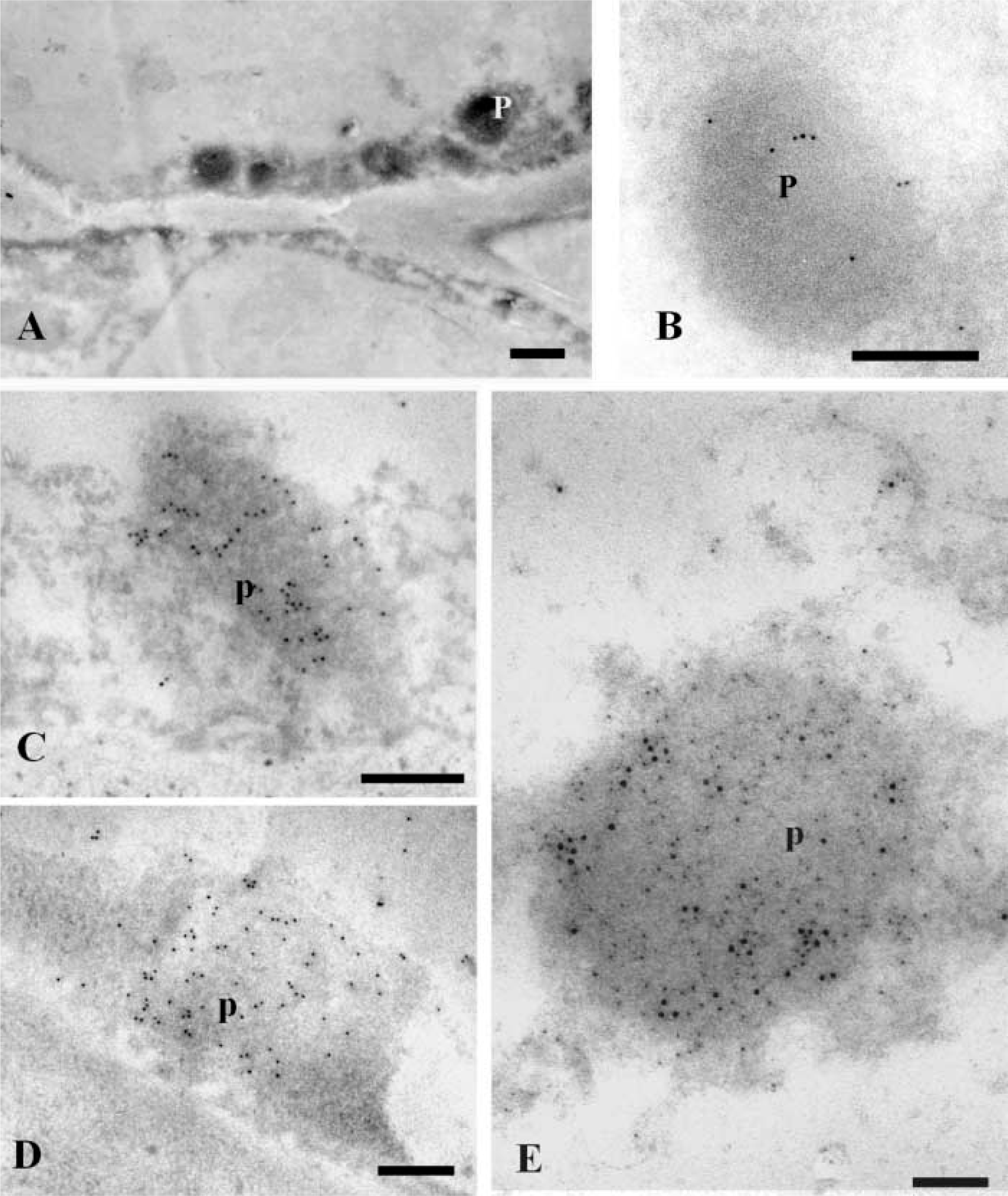
DAB cytochemistry for catalase combined with anti-LOX immunogold labeling and double anti-LOX-anti-catalase immunogold labeling in nodular cells. (
The presence of LOX in plastids may be related to jasmonic acid synthesis (León and Sánchez-Serrano 1999) or may be involved in the degradation of thylakoid membranes during the transition from chloroplast to amiloplast.
Cells from the periphery of hop's organogenic nodules showed co-localization of LOX fluorescent spots with lipid bodies. Nodular areas in which plant regeneration was taking place showed a huge accumulation of lipid bodies in which LOX was detected by TEM. These results may suggest the involvement of LOX in the mobilization of storage lipids, providing an additional energetic source for plantlet regeneration through gluconeogenesis. This breakdown occurs partially in peroxisomes, which contain fatty acid β-oxidation and the glyoxylate cycle enzymes (Landolt and Matile 1990). In barley and in cucumber, the LOXs oxygenate storage triacylglycerols and this oxygenation process seems to initiate the mobilization of the storage lipids (Feussner et al. 1997a,b). In germinating cucumber seeds, a special isoform of 13-LOX was found in lipid storage organelles (lipid bodies). This isoform oxygenates the storage triacylglycerols rather than free linoleic acid released by the action of a lipid hydrolyzing enzyme. Lipases that appear to act mostly on oxygenated ester lipids over nonoxygenated lipids have been described for microsomal membranes of several plants (Stahl et al. 1995). These oxygenated lipids would then be reduced to enter β-oxidation in peroxisomes. LOX in peroxisomes may play a role in preparing lipids for the action of lipases. Alternatively, they may be acting on fatty acids released by lipase actions on lipid bodies, as suggested by the intimate association between lipid bodies and peroxisomes.
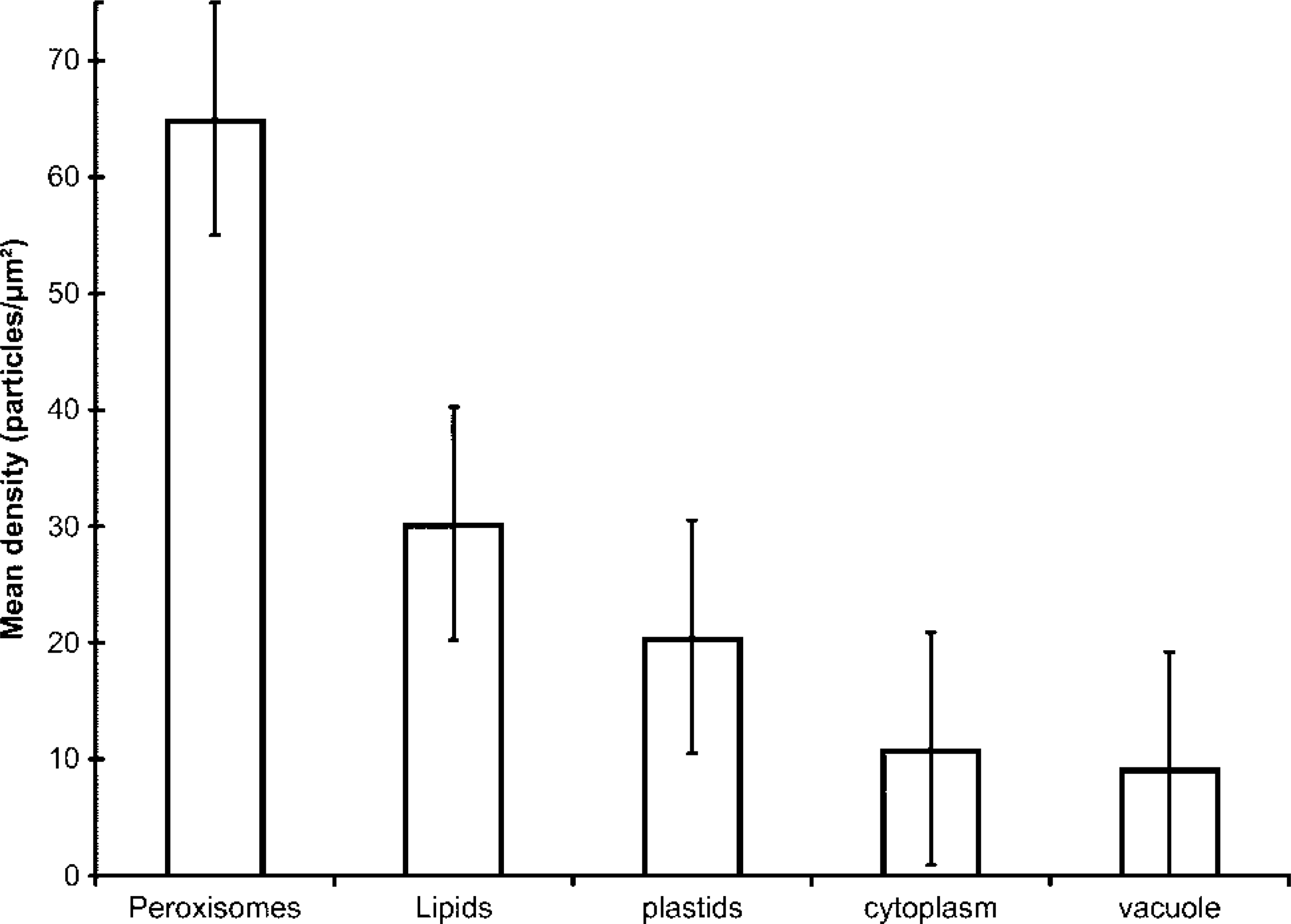
Histogram representing the labeling density after anti-LOX immunolocalization in different cellular compartments of nodular cells. The peroxisomes showed the highest labeling density, followed by lipids and plastids. The mean densities in cytoplasm and vacuoles were very low and not statistically significant, representing background.
To our knowledge, this is the first report mentioning the localization of LOX in peroxisomes of plant cells. Previously, Yokota et al. (2001) reported the involvement of a 15-LOX with a molecular mass of 72 kD, in the programmed degradation of peroxisomes in normal rat liver. This was in accordance with the results of van Leyen et al. (1998), which suggest a role for LOX in programmed organelle degradation. However, these authors have localized LOX in peroxisomal membranes, whereas our results indicate localization, in both the peroxisomal matrix and the membrane. This suggests that LOX in peroxisomes may play an additional physiological role in this morphogenic process of organogenic nodule formation. It may be that nodular cells that entered programmed cell death, so that nodules separate into “daughter nodules” (Fortes and Pais 2000a), initially showed LOX in peroxisomes as a way to degrade their membranes and induce the leakage of their contents. Nodule separation appears to be initiated by the formation of a necrotic layer at the future region of nodule separation (Fortes and Pais 2000a), which accumulates reactive oxygen species (Fortes and Pais 1999). Yokota et al. (2001) showed a selective occurrence of membrane disruption, which implies that only some peroxisomes were selected to be degraded. Recently, del Río et al. (2002) suggested that peroxisomes could have a function in plant cells as a source of signal molecules such as nitric oxide, superoxide radicals, and hydrogen peroxide. Low levels of nitric oxide and ROS are involved in many physiological processes, including growth and development (del Río et al. 2002, and references therein). It may be that LOX activity in peroxisomes is acting as a source of ROS. The important pool of new metabolites available in peroxisomes can have an effect on metabolic pathways in other cell compartments.
Changes in the distribution and abundance of LOX during development and in different tissues and cellular compartments is due in part to the expression of different members of the Lox gene family (Creelman and Mullet 1997). Our results suggest the involvement of a Lox multigene family in different metabolic pathways leading to nodule formation and to plantlet regeneration from these nodules in hops. Recent work carried out using reverse transcriptase-PCR with degenerated primers has revealed the presence of three different cDNA fragments coding for lipoxygenases (unpublished results). To accomplish a more comprehensive study, obtaining isoenzymes-specific probes will bring insights into protein dynamics and localization of the different isoenzymes and information concerning which isoenzymes are active and which LOX pathway-derived products are being produced at different stages of the morphogenic process.
Footnotes
Acknowledgements
Supported by the Portuguese Foundation for Science and Technology, by a PhD grant to Ana M. Fortes (PRAXIS XXI/BD/19536/99). This work was also supported by the Portuguese Project PCII/1999/BME/32734, by the Portuguese-Spanish Joint project Acc. Int. HP 2000–0065, and by the Spanish Projects DGESIC PB 98–0678 and CAM 076/0054/2000.
We wish to thank Dr José Sànchez-Serrano (Department of Plant Molecular Genetics, CNB, Madrid, Spain) for kindly providing the anti-LOX antibody. We also thank Dr Rui Malhó (Centro de Biotecnologia Vegetal, FCUL, Lisbon) for critically reading the manuscript and Ms Angeles Ollacarizqueta (CIB, CSIC, Spain) for technical assistance in confocal microscopy.