
Abstract
Here we show that ultrathin cryosections of placental tissue can be used as a substrate in immunofluorescence experiments. A high degree of spatial resolution can be achieved in these preparations because there is essentially no out-of-focus fluorescence. Therefore, immunofluorescence microscopy using ultrathin cryosections provides a very useful method for determining the precise subcellular localization of antigens in tissues. In addition, ultrathin cryosections of placenta also serve as a substrate for correlative immunofluorescence and immunoelectron microscopy using FluoroNanogold as the detection system. In correlative microscopy, the exact same structures in the same ultrathin section were observed by both fluorescence and electron microscopy. Using a particle counting procedure and electron microscopy, we compared the labeling obtained with colloidal gold and FluoroNanogold and found a higher number of particles with silver-enhanced FluoroNanogold than with colloidal gold.
Keywords
I
In many studies, determination of which cells in a population (e.g., a tissue section) are positive for a particular antigen will suffice. However, in other cases more precise determination of the distribution of an antigen in a particular cell may be required. In simple tissue culture systems, relatively precise localization can be accomplished with immunofluorescence microscopy (IFM) methods, and very precise localization can be achieved with immunoelectron microscopy (IEM). In organized tissues, precise localization of antigens can be achieved by IEM. However, precise localization of antigens in tissues by IF methods may be problematic. Conventionally, tissues are sectioned at 5–15-μm thickness with a cryostat before IFM localization. Such sections can be examined with a conventional fluorescence microscope, in which case the fluorescence from the entire volume of the section will be observed. The out-of-focus fluorescence that may be in the section leads to image degradation and loss of resolution. Alternatively, the sections can be examined with a confocal microscope, in which case optical sectioning permits examination of a restricted volume of the cryostat section. This limits most out-of-focus fluorescence and yields enhanced resolution compared to conventional fluorescence microscopy (e.g., Inoué 1995; Pawley 1995). Moreover, the tissue can be sectioned with an ultracryomicrotome to yield ultrathin cryosections (in the 50–100-nm range). These sections can be used for fluorescence and electron microscopy (Takizawa et al. 1998). The use of ultrathin cryosections further eliminates the out-of-focus fluorescence because the sections are very thin (Pombo et al. 1999; Robinson et al. 2001); they are thinner than the z-dimension of an optical section in a confocal microscope (i.e., 200–500-nm range) (Majlof and Forsgren 1993; Zucker and Price 2001). Ultrathin cryosections can also be used for correlative fluorescence and electron microscopy, in which the exact same structures are imaged, further adding to the utility of these sections (Pombo et al. 1999; Robinson et al. 2001). Correlative IFM and IEM are not practical with conventional cryostat sections.
We have described methods for correlative fluorescence and electron microscopy of isolated neutrophils (Takizawa et al. 1998). We have now modified and improved the methodology so that it is more applicable to the study of intact tissues (Takizawa and Robinson 2003). Here we demonstrate the value of using ultrathin cryosections of tissue for IFM and for correlative IFM and IEM, and review some of our previous results.
Materials and Methods
Reagents
Alexa Fluor 488- and 594-labeled goat anti-mouse and goat anti-chicken antibodies, and goat anti-rabbit antibodies, 4',6-diamidino-2-phenylindole (DAPI), as well as the Pro-Long antifade kit were obtained from Molecular Probes (Eugene, OR). Alexa Fluor 594-labeled streptavidin Fluoro-Nanogold (FNG) was from Nanoprobes (Yaphank, NY). Biotin-labeled goat anti-chicken F(ab)'2 and goat anti-chicken 4-nm colloidal gold antibodies were from Jackson ImmunoResearch (West Grove, PA). Fetal bovine serum was obtained from Invitrogen (Carlsbad, CA). Gelatin was obtained from Sigma Chemical (St Louis, MO). All immunological reagents were handled in accordance with the manufacturers’ recommendations and were used within the expiration date for each product. A murine monoclonal antibody (MAb) to early endosome antigen 1 (EEA 1) was obtained from BD Transduction Laboratories (San Diego, CA). An anti-peptide antibody specific for caveolin-1α (CAV-1α) was generated in chickens. The immunogen was amino acids 3–14 from the N-terminal region of the human CAV-1α molecule with the amino acid sequence GGKYVDSEGHLY. Characterization of this antibody has been reported (Robinson and Vandré 2001; Lyden et al. 2002).
Processing Ultrathin Cryosections for Immunocytochemistry
Human full-term placentae were used as the model tissue and were obtained from the Ohio State University tissue procurement service according to a protocol approved by the Ohio State University Human Subjects Institutional Review Board. Tissue samples from cesarean deliveries were processed for fixation as rapidly as possible (no more than 15 min after delivery). Three placentae were used in this study.
Tissue was cut into small pieces and fixed in 4% paraformaldehyde (freshly prepared) in 0.1 M sodium cacodylate buffer, pH 7.4, containing 5% sucrose for 2 hr at room temperature (RT). The samples were washed and embedded in 10% gelatin in the same buffer. The solidified gelatin was cut into smaller pieces and then cryoprotected by infiltration with 2.3 M sucrose in 0.1 M sodium cacodylate (pH 7.4) overnight at 4C. After sucrose infiltration, the samples were mounted on specimen pins designed to fit a Reichert cryoultramicrotome, frozen in liquid nitrogen, and stored in liquid nitrogen until they were sectioned.
Ultrathin cryosections were cut with a Reichert Ultracut E equipped with an FC 4D cryounit. Sections (100-nm thickness or less) were cut on a Cryo P diamond knife (Diatome-US; Fort Washington, PA) and were collected on droplets of 0.75% gelatin–2.0 M sucrose (Takizawa and Robinson 1994) or 1% methylcellulose-1.15 M sucrose (Liou et al. 1996) and then transferred to round glass coverslips (12-mm diameter) pre-coated with 2.0% 3-aminopropyltriethoxysilane or to formvar film-coated, carbon-stabilized, glow-discharged nickel grids. The pick-up solutions contained 0.05% sodium azide so that the cryosections could be stored as described previously until they were immunolabeled (Griffith and Posthuma 2002).
Immunofluorescence Microscopy
Ultrathin cryosections were mounted on glass coverslips when only IFM microscopy was done. Coverslips containing sections were immersed in a solution containing 1% non-fat dry milk and 5% fetal bovine serum in PBS (MFBS–PBS) for 15 min at 37C to remove the sucrose and gelatin, then washed three times in PBS and incubated in MFBS–PBS to block nonspecific protein binding sites; 0.05% sodium azide was also present. Ultrathin cryosections were then incubated with chicken anti-CAV-1α (diluted 1:500 in MFBS–PBS) and anti-EEA 1 (diluted to 10 μg/ml) for 30 min at 37C. The cryosections were rinsed in PBS four times over 15 min, immersed in MFBS–PBS, and then incubated with Alexa 488-labeled goat anti-chicken IgG and Alexa 594-labeled goat anti-mouse IgG (both diluted 1:200 in MFBS–PBS) for 30 min at 37C. After immunolabeling, the cryosections were washed five times in PBS. The sections were then incubated with DAPI (1 μg/ml) in PBS for 10 min at RT in order to visualize nuclei. The sections were then washed five times in PBS and mounted on glass microscope slides in ProLong antifade medium. Control sections received the same treatment except for omission of the primary antibody or substitution of pre-immune chicken antibody for the primary antibody.
Correlative Microscopy
Ultrathin cryosections were collected on nickel Maxtaform “finder” grids (Graticules; Tonbridge, Kent, UK) to facilitate location of specific structures when going from the optical to the electron microscope. Sections on grids were incubated in the same manner as were the 12-mm coverslips containing ultrathin cryosections. After washing in PBS and MFBS–PBS (the same as for ultrathin cryosections on cover-slips), the grids were incubated with biotin-labeled goat anti-chicken (13 μg/ml in MFBS–PBS) for 30 min at 37C. The grids were then washed in PBS for 12 min with four changes and then immersed in MFBS–PBS. The grids were next incubated with streptavidin-labeled FNG conjugated with Alexa 594 (diluted 1:50 in MFBS–PBS) for 30 min at RT. The grids were then washed in PBS for 15 minutes with five changes.
After the labeling procedure, the grids were mounted on a glass microscope slide in PBS containing 1% N-propyl gallate and 50% glycerol, pH 8.0, to retard photobleaching (Takizawa and Robinson 2000). The preparation was then overlaid with an 18-mm round glass coverslip. The cryosections were examined immediately by optical microscopy and images were collected. The locations of regions of interest on the “finder” grid were noted for relocation in the electron microscope. The temporary slide preparations were then disassembled and the grids were washed in PBS with five changes over 15 min. The ultrathin cryosections were then fixed in 2% glutaraldehyde in PBS for 30 min to further stabilize the sections and washed in distilled water for 6 min with four changes. The sides of the grids opposite the sections were dried with filter paper. The grids were then floated on drops of distilled water and subsequently on drops of 50 mM 2-[N-morpholino]ethanesulfonic acid buffer, pH 6.15, for 4 min with two changes. FluroNanogold bound to the cryosections was then subjected to a silver enhancement procedure for 3 min at RT to render them visible in the sections (Burry 1995). A positive contrast enhancement procedure that we have recently developed was used to visualize membrane profiles in the ultrathin cryosections (Takizawa et al. 2003). Grids were then examined with the electron microscope. The same regions examined by fluorescence microscopy were relocated and electron micrographs were collected.
Immunoelectron Microscopy with Colloidal Gold
Ultrathin cryosections were incubated for the localization of CAV-1α with colloidal gold as the reporter system. Sections on EM grids were incubated with the primary antibody and then washed in the same manner as previously described for IFM. The sections were then incubated with goat anti-chicken 4-nm colloidal gold diluted 1:10 in MFBS–PBS for 30 min at 37C. The sections were then washed with five changes of PBS over 15 min and then fixed in 2% glutaraldehyde for 30 min. The sections were then washed in distilled water for 6 min with four changes. They were then subjected to the same positive contrast procedure that was used for the correlative microscopy.
Microscopy
Fluorescence and differential interference contrast (DIC) images were collected with a Nikon Optiphot microscope equipped with a Photometrics Cool Snap fx CCD camera (Roper Scientific; Tucson, AZ). Images were captured with Metamorph image analysis software system (Universal Imaging; Dowingtown, PA). All fluorescence images were collected in the monochrome mode and then converted to the appropriate pseudo-color where needed. Electron microscopy was carried out with a Philips CM-12 transmission electron microscope operated at 60 kV. Figures shown in this report were compiled with Photoshop 7 software (Adobe Systems; Mountain View, CA).
Quantitative Analysis of Immunogold Labeling
Electron micrographs of CAV-1α localization that was detected with 4-nm colloidal gold or with silver-enhanced FNG were analyzed by counting particles associated with caveola-like structures. A total of 100 randomly selected caveola were scored for each type of detection system.
Results
Immunofluorescence in Ultrathin Cryosections
Ultrathin cryosections of human placenta (70–100-nm thickness) were tested as a substrate for IFM microscopy using a chicken anti-CAV-1α and an MAb directed against EEA 1 as the primary antibodies. Detection of anti-CAV-1α and anti-EEA 1 binding was achieved with goat anti-chicken Alexa 488 and goat anti-mouse Alexa 594 as the secondary antibodies (Figure 1). In ultrathin cryosections, CAV-1α was present in small punctate structures in the capillary endothelial cells and pericytes but not in the syncytiotrophoblast (STB) layer of terminal villi of the placenta. These fluorescent structures are suggestive of individual caveolae or small groups of caveolae. The distribution of EEA 1 was decidedly different. It was located primarily in vesicle-like structures in the apical portion of the STB. Relatively few EEA 1-positive structures were observed in endothelial cells or pericytes. These structures were not observed in control incubations when pre-immune antibody replaced the specific antibody for CAV-1 or when the primary antibody was omitted (CAV-1 and EEA 1) (data not shown). These results demonstrate that ultrathin cryosections of placenta serve as an excellent substrate for high-resolution IFM labeling.
Correlative Immunofluorescence and Immunoelectron Microscopy in Ultrathin Cryosections
Ultrathin cryosections were used as a substrate for correlative IFM and IEM. Ultrathin cryosections collected on EM grids were immunolabeled with the chicken anti-CAV-1α. The detection of anti-CAV-1α binding to the section was achieved with biotin-labeled goat anti-chicken antibody followed by Alexa 594 FNG–streptavidin binding (Figure 2A). The IFM distribution of CAV-1α in placenta, as determined with FNG as the reporter system, appears identical to that obtained with conventional fluorochrome-tagged secondary antibodies (compare Figures 1 and 2A). The morphology of the ultrathin cryosection on the EM grid was determined by DIC microscopy (Figure 2B). After collection of the fluorescence and DIC images, the FNG was subjected to the silver enhancement procedure to render the FNG visible at the EM level. Electron microscopy showed that caveolae were now heavily decorated with silver-enhanced gold particles (Figure 2D). These gold-labeled caveolae correspond precisely to the fluorescence signal from FNG once the fluorescence images are at the same magnification as the EM images (Figure 2C).
Comparison of the Labeling Achieved with Fluorochrome-, Colloidal Gold-, and FNG-labeled Secondary Immunoprobes
The detection of chicken anti-CAV-1α binding to ultrathin cryosections of terminal villi of human placenta was demonstrated with (a) fluorochrome-, (b) colloidal gold-, and (c) FNG-labeled secondary immunoprobes (Figure 3). As far as we can tell, all caveolae were detected with fluorochrome-labeled secondary antibodies. Colloidal gold-labeled secondary antibodies also labeled caveolae. However, in this case not all of the caveola-like structures were labeled. Moreover, the signal was relatively weak, with about two colloidal gold particles per caveolae-like structure (Table 1). When silver-enhanced FNG was the detection system, all caveola-like structures appeared to be labeled. In addition, these structures were heavily decorated with about 27 particles per caveola (Table 1).
Discussion
Immunofluorescence microscopy has been a valuable tool in experimental and applied biomedicine. It provides a high degree of sensitivity for the in situ detection of antigens while being a relatively simple technique. The adequacy of spatial resolution that can be achieved with IFM depends on the questions being addressed. In situations where the question of interest relates to which cells in a population (e.g., tissue section) are positive, spatial resolution is less important than is sensitivity of detection. In other cases, where the subcellular location of an antigen within the positive cell is of interest, then spatial resolution is as important as sensitivity of detection. In the first situation, positive cells can be distinguished from negative cells in conventional cryostat sections of tissue (e.g., 5–10-μm thickness) by IFM. However, these types of cryostat sections are not adequate for resolution of subcellular detail by IFM (e.g., Takizawa and Robinson 2003). This is due, in large part, to out-of-focus fluorescence within the volume of the cryostat section. Much of the out-of-focus fluorescence can be reduced by optically sectioning such cryostat sections using a confocal microscope in IFM (Inoué 1995; Pawley 1995). Alternatively, “semithin” cryosections (0.2–2.0 μm) have been used to reduce out-of-focus fluorescence in IFM (e.g., Tokuyasu et al. 1972; Takata et al. 1991; Ishiko et al. 1998). Each of these approaches may be adequate in given situations where the volume of the tissue sampled is appropriate; physical sections in the 0.2–0.5-μm-thickness range are equivalent to optical sections with the confocal microscope, since they are also in the 0.2–0.5-μm range in the z-dimension (Majlof and Forsgren 1993; Zucker and Price 2001). Even though there are dramatic improvements in the reduction of image degradation due to out-of-focus fluorescence in confocal microscopy or when semithin cryosections are used, one can envision situations in which these approaches are inadequate for resolving separate subcellular structures. For example, if two (or more) different types of subcellular compartments (in the 50–200-nm size range) are closely stacked in the volume of an optical section or a semithin cryosection, and if in a double- or triple-label IFM preparation each is labeled with a different color of fluorochrome, false co-localization could occur due to overlap of the fluorescence signals (e.g., Griffiths et al. 1993). The number of separate compartments, even of the same type, also may be underestimated in these conditions due to combining the fluorescent signals in the volume of the sections. To minimize the potential for false co-localization in IFM preparations, we have utilized ultrathin cryosections (50–100-nm in thickness) for IFM. We show that these ultrathin cryosections can serve as a substrate for IFM. Moreover, the potential for false co-localization is further minimized because the thickness of the sections is about the same or less than the size of the subcellular compartments of interest (i.e., 50–200 nm). Here, we have used ultrathin cryosections for the IFM localization of the marker CAV-1α to caveolae in the placenta (Rothberg et al. 1992). This is an ideal test system for the use of ultrathin cryosections because these structures are about 70 nm in diameter in placental endothelial cells (Lyden et al. 2002). Using ultrathin cryosections we can resolve single caveola with this approach. In addition, we show that ultrathin cryosections can also be used in double-label IFM preparations. A summary of the methods employed and the conceptual basis for the advantages of using ultrathin cryosections in IFM is given in Figure 4.
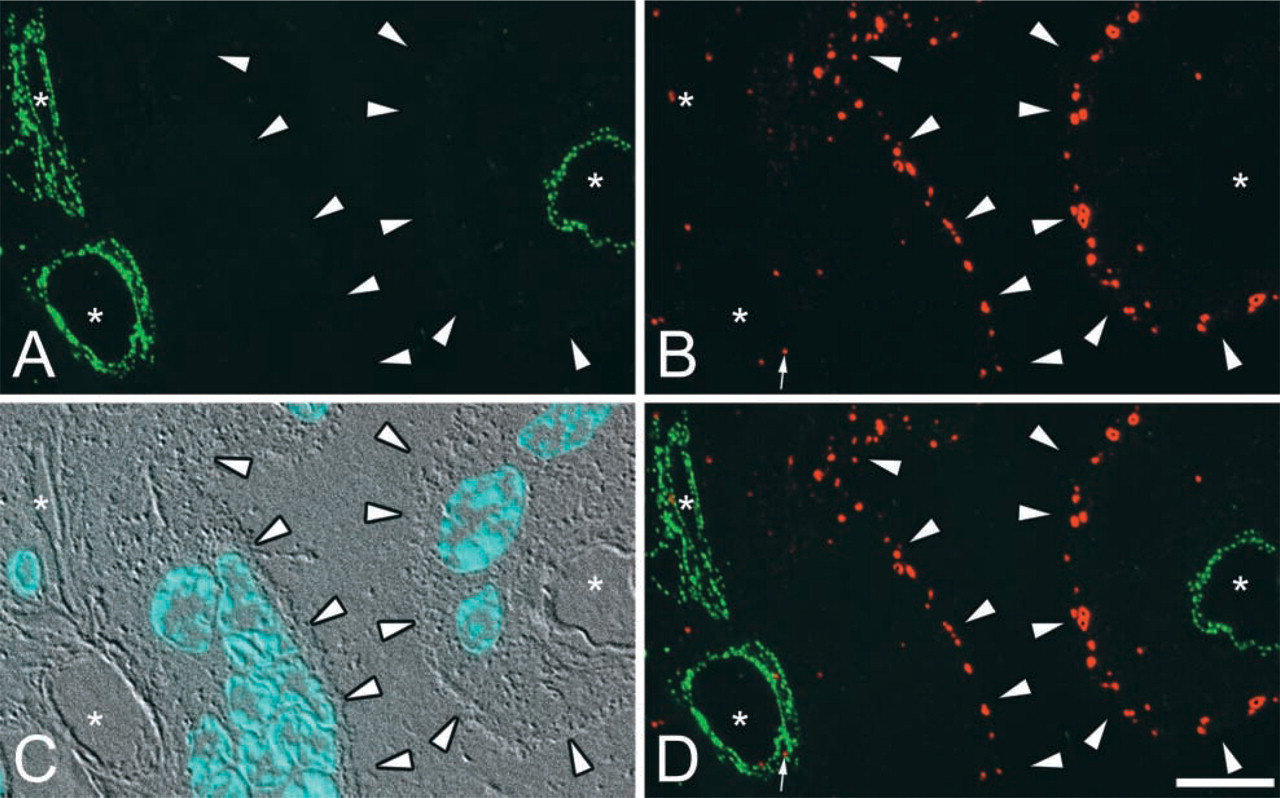
Double-label IFM detection of CAV-1α and EEA 1 on ultratrathin cryosections. (
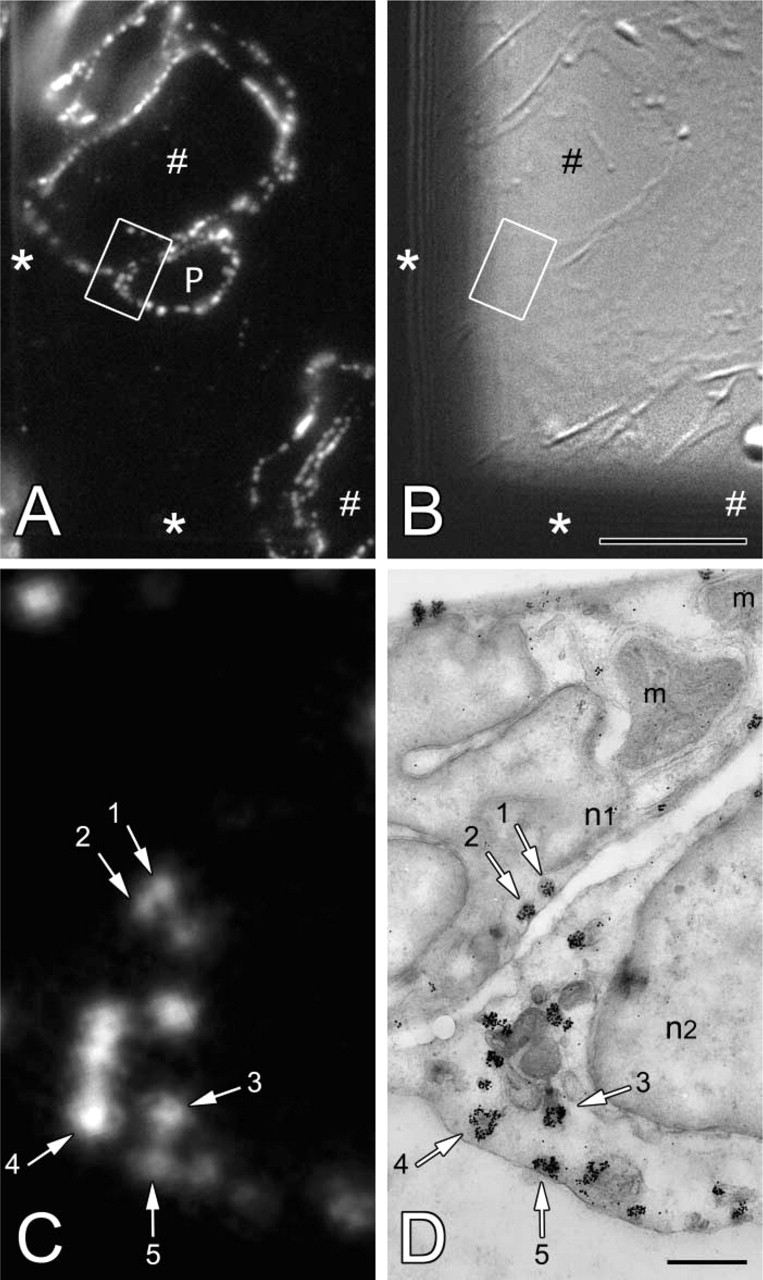
Immunocytochemical localization of CAV-1α by correlative microscopy on the same ultrathin cryosection. (
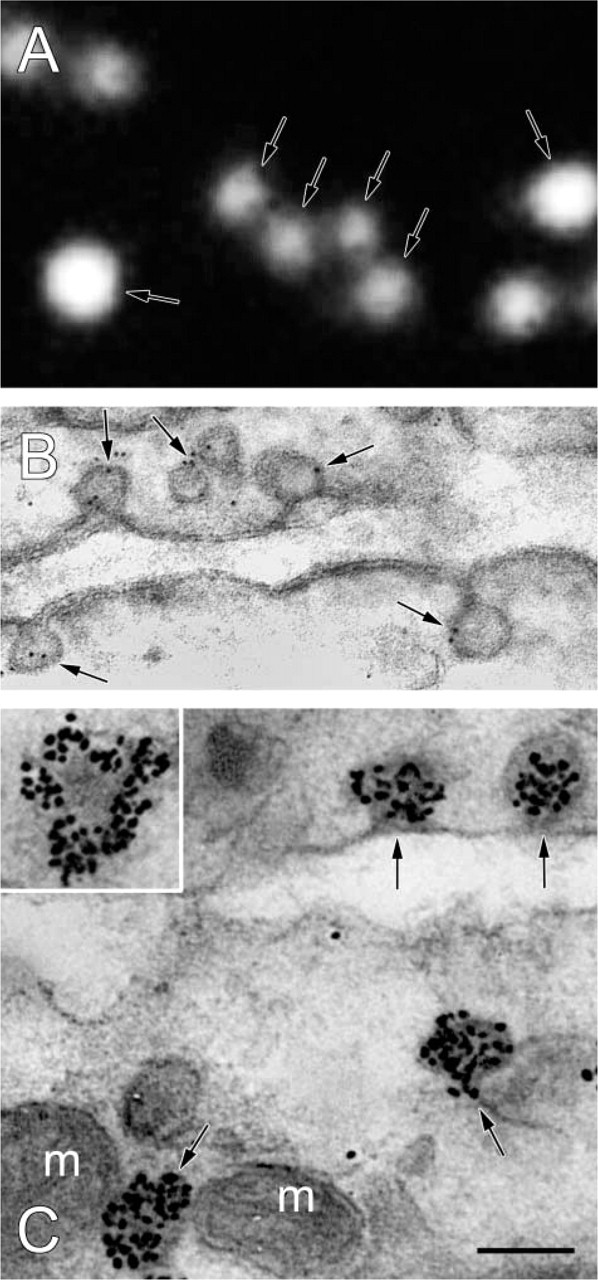
A comparison between fluorochrome-, colloidal gold-, and silver-enhanced FNG-based detection systems for localization of CAV-1α in ultrathin cryosections. (
Immunoelectron microscopy provides a very high degree of spatial resolution but is generally less sensitive than is IFM. However, IEM does provide a “reference space,” i.e., all structures are visible, not just the immunolabeled components. In IFM, on the other hand, the “reference space” is absent; in IFM, only the labeled structures are observable, and the unlabeled structures are not observed and do not contribute to the analysis (Griffiths 2001). Therefore, it is often important to carry out IEM studies to complement IFM. Correlative microscopy in which both IFM and IEM is carried out constitutes a way to combine the best attributes of each method. Increased sensitivity is obtained from IFM, while IEM provides the reference space. In past studies in which this has been done with cryosections, semithin cryosections were cut for IFM and and ultrathin cryosections were used for IEM with colloidal gold or ferritin detection of antibody binding (e.g. Tokuyasu 1980,1983; Takata et al. 1991) (see Figure 4A). Although useful, this approach has certain limitations. As noted above, semithin sections are still thick enough that structures closely stacked in the sections may contribute to false colocalization. The ultrathin section that is examined while adjacent or nearby is still not the same as the semithin section. In addition, different immunolabeling systems were used in these studies. Fluorochrome-labeled immunoprobes were used with the semithin cryosections, while colloidal gold- or ferritin-labeled immunoprobes were used for the ultrathin cryosections. This may be problematic because of the apparent differences in labeling efficiency between fluorochrome-conjugated and colloidal gold-tagged secondary immunoprobes (see Figure 3 and below). We have used a different approach for correlative IFM and IEM (see Figure 4C). In this case, the same ultrathin cryosection is used for both types of microscopy. In addition, FNG is used to detect antibody binding in both IFM and IEM. This immunoprobe combines a fluorochrome and a gold cluster compound. The former is imaged directly in the fluorescence microscopy, and the latter is imaged in the electron microscope after silver enhancement (Powell et al. 1998; Robinson et al. 2000). Thus, the same immunoprobe is used for both modes of microscopy. This point is important because it predicts that both types of microscopy will observe the same structures. This prediction was verified (see Figure 2). Thus, correlative microscopy on the same ultrathin cryosection using FNG as the detection system provides enhanced sensitivity for localization of antigens and the reference space needed to place the immunolabeling signal in the context of the cells and tissues.
Comparison of the labeling intensities for the detection of CAV-1α using 4-nm colloidal gold and silver-enhanced FluoroNanogold (FNG) as the reporter systems
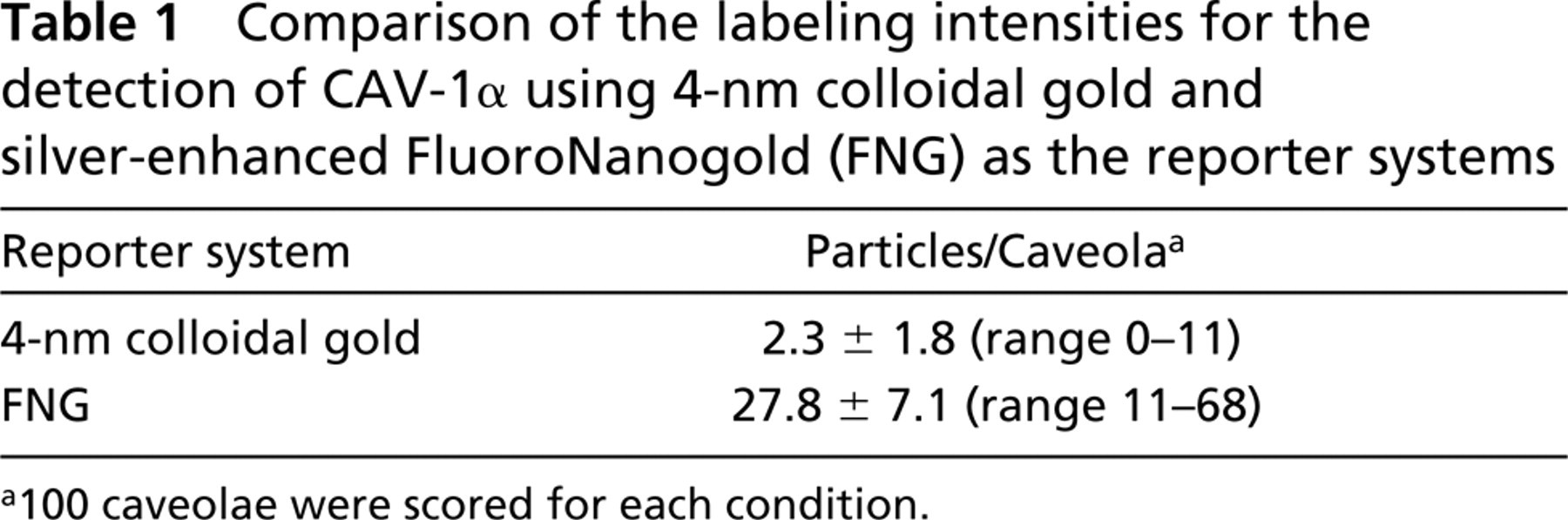
a100 caveolae were scored for each condition.
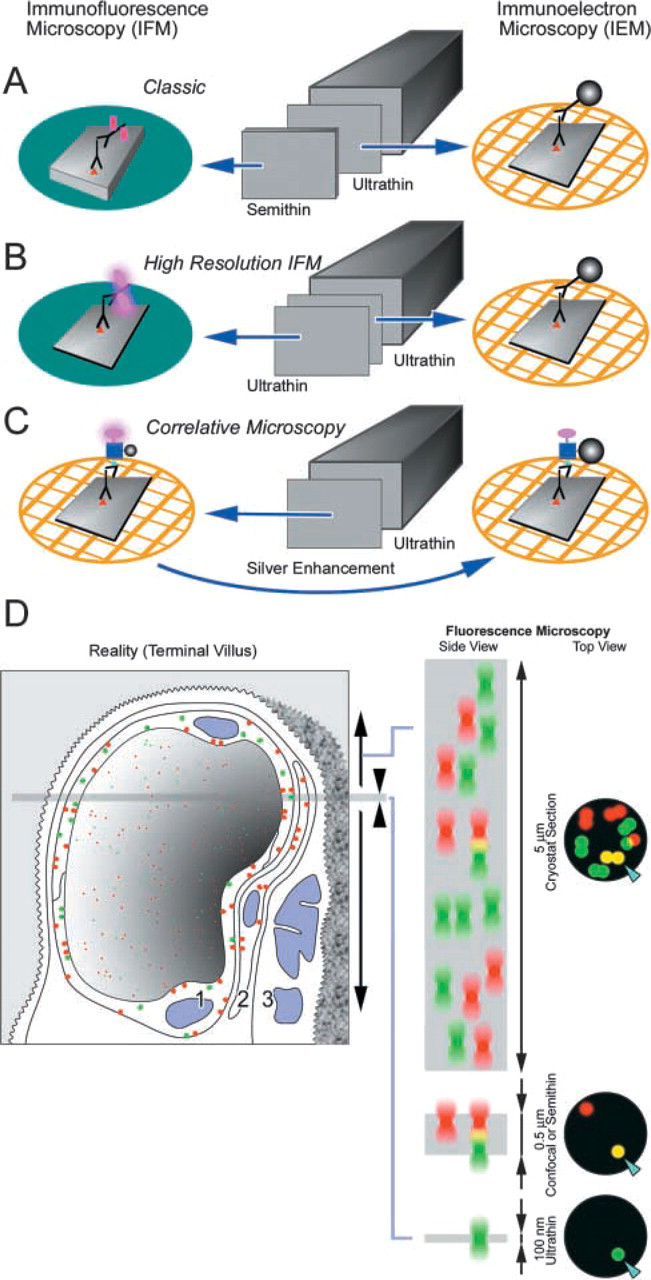
Diagram summarizing the methods employed and a model illustrating the advantages of using ultrathin cryosections for immunoncytochemistry. (
We referred above to enhanced sensitivity of detection of antigen localization in light microscopy compared to electron microscopy (i.e., fluorescence vs colloidal gold). We tested this proposal using ultrathin cryosections of placenta for the localization of CAV-1α (see Figure 3). When fluorochrome-labeled secondary antibodies were used, individual caveola were detected; the fluorescence was associated with the entire caveola. On the other hand, when 4-nm colloidal gold-labeled secondary antibodies were used, the caveola were sparsely labeled with about two colloidal gold particles per caveola. Moreover, the caveolae were not uniformly labeled using colloidal gold. Some caveola-like structures were unlabeled (not shown). Detection of CAV-1α binding to ultrathin cryosections was also achieved with FNG as the detection system. In IFM, qualitatively similar results were obtained when fluorochrome-labeled secondary antibodies were used or when FNG was used as the detection system (compare Figures 1A and 2A). After silver enhancement of FNG, the caveolae were heavily labeled. Moreover, all caveola-like structures were labeled (see Figures 2D and 3C). There were significantly more particles associated with caveolae when FNG was the detection system compared to colloidal gold. Moreover, with FNG the same structures detected by IFM were also detected by IEM. In summary, we show that ultrathin cryosections of tissue serve as an excellent substrate for IFM and for correlative IFM and IEM.