
Abstract
We developed an ELISA-based method for rapid selection of optimal blocking agents to be used in antigen quantification by immunogold labeling electron microscopy. Casein, skim milk, BSA from two sources, acetylated BSA, fish skin gelatin, horse serum, and goat serum were tested for their ability to block nonspecific binding of antibody to recombinant Vitreoscilla hemoglobin (VHb) antigen expressed in Escherichia coli cells by ELISA and the results were confirmed by quantitative immunogold labeling transmission electron microscopy (TEM). Ability to minimize NSB was also evaluated by dot-blot and Western blotting methods. The results demonstrated that ELISA was most accurate in predicting the most efficient blocking agent for TEM. Existing methods could not provide an accurate picture of the ability of various reagents to suppress background labeling. The sensitivity of detection of antigens by immunoelectron microscopy depends on the assay procedure being optimized to obtain the highest possible signal along with as low a background (noise) as possible. Our study indicated that an ELISA-based evaluation of various blocking agents could help in the rapid selection and optimization of a suitable protocol for immunogold localization and quantification of antigens by TEM.
Keywords
I
Materials and Methods
VHb Antigen and Anti-VHb Antibody
E. coli DH5α cells harboring the plasmid PUC8:15 expressing VHb protein (Dikshit and Webster, 1988) were used as a source of antigen. Cells from the stationary phase were harvested, washed with Dulbecco's PBS, and processed either for electron microscopy or sonicated in PBS to prepare lysate. Cell lysate was centrifuged at 12,000 × g and the clear supernatant was used for ELISA, dot-blotting, and Western blotting analysis. Polyclonal antiserum against VHb protein (anti-VHb) was raised in rabbit as described previously (Joshi et al. 1998). Before use the serum was pre-adsorbed with the antigen -ve cell extract (lysate of cells not expressing VHb, i.e., containing only the vector and lacking the gene coding for VHb) of E. coli. The pre-adsorption step was performed to minimize antibody molecules in serum that crossreact with cell proteins other than the target antigen.
ELISA of VHb Antigen
Fifty μl of lysate containing 2.5 μg of protein (Bradford 1976) was coated into every third and fourth column of eight wells of 96-well ELISA plates (TARSON) overnight at 4C. Antigen containing lysate, adsorbed onto the plastic well, represented an artificial immunospecimen. Previous studies in our laboratory have shown that under these conditions the plastic surface of ELISA wells that is immersed in lysate solution is fully saturated with adsorbed protein. To the wells in first and second set of columns was added (a) 50 μl PBS and (b) 50 μl of antigen -ve cell extract containing 2.5 μg of protein. The former served as control for the evaluation of antibody binding to clear plastic and the latter as an antigen-negative artificial immunospecimen control for each set. Then the wells of each set of four columns were blocked by filling them completely with different blocking agents constituted in PBS containing 0.01% Tween-20 (polyoxyethylenesorbitan monolaurate; Sigma Chemicals, St. Louis, MO) (PBST) for 1 hour at room temperature (RT). The various blocking agents used are listed in Table 1. Wells were washed thrice with PBST and 50 μl of rabbit anti-VHb diluted 1:1000 in PBST was added to each well in the first three sets of columns and incubated at RT for 1 hr. To wells of the fourth column was added normal rabbit serum (NRS) instead of rabbit anti-VHb, diluted to the same protein concentration as the test antibody. After three washes with PBST, 50 μl of goat anti-rabbit IgG HRP conjugate (Sigma) diluted 1:40,000 in PBST was added as second antibody to each well and allowed to react for 1 hr at RT. The reaction product was developed with 3,3′ 5,5′-tetramethyl-benzidine liquid substrate system for ELISA (Bangalore Genie; Bangalore, India), for 30 min and was terminated by addition of 50 μl of 0.1 M H2SO4 to each well. The resultant yellow color, which was in linear range, was read at 450 nm through an ELISA reader that had been blanked with distilled water (Bos et al. 1981). The absorbance of a corresponding strip of eight wells without lysate or with control antigen-negative cell lysate was taken to represent the binding to clear plastic/antigen-negative control, respectively. The OD in wells in which NRS replaced anti-VHb represented sented the nonspecific binding by control serum. Comparison between sets of different blocking agents was done by the Wilcoxon rank sum test.
Blocking agents used
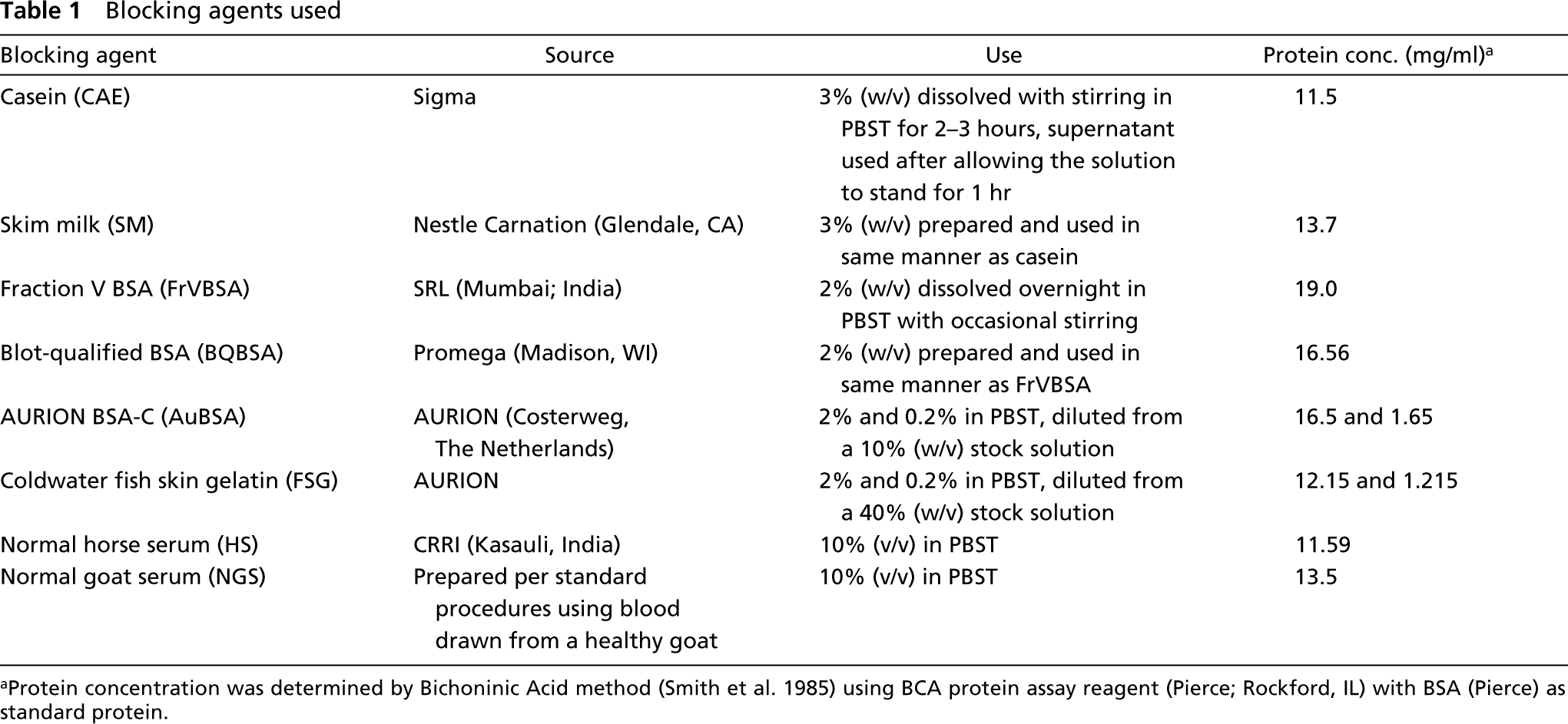
Protein concentration was determined by Bichoninic Acid method (Smith et al. 1985) using BCA protein assay reagent (Pierce; Rockford, IL) with BSA (Pierce) as standard protein.
Dot-blotting of Antigen
Five microliters of cell lysate containing 20 μg of total protein was spotted onto pieces of nitrocellulose paper (Advanced Microdevices; Ambala, India) and allowed to dry for a few minutes under an infrared lamp. The spots were placed into individual wells of a 12-well cell culture plate and blocked for 2 hr by immersion in 2 ml of various blocking agents accompanied by continuous rocking on a shaker. Then the spots were washed three times with PBST and incubated in 1 ml of rabbit anti-VHb, diluted 1:500 in PBST, for 1 hr at RT. After three washes with PBST, goat anti-rabbit IgG HRP conjugate (Sigma) diluted 1:16,000 in PBST was added as second antibody and allowed to react further for 1 hr at RT, with the wells covered with aluminum foil. After washing as before, the reaction product was developed using 3,3′ 5,5′-tetramethylbenzidine for localization of HRP (Bangalore Genie) for 15 min. The reaction was stopped by washing the spots with distilled water. In two parallel sets of control experiments; (a) the antigen containing cell lysate was replaced with antigen-negative cell lysate and (b) rabbit anti-VHb antibody was replaced with NRS.
Western Blotting Analysis
Western blotting of cell lysate was carried out according to the procedure of Paiement and Roy (1988) using 10 μg of protein loaded per lane. After electrophoresis and transfer to nitrocellulose sheets, individual lanes were cut out and blocked in various blocking agents before being probed with antibody in a manner similar to that followed for the dotblotting protocol, with the exception that all incubations were carried out in individual test tubes using 10 ml of solution for each step. The blots were developed for 15 min in 3,3′ 5,5′-tetramethylbenzidine for localization of HRP and the reaction was terminated by several washes in distilled water.
Processing of Cells for TEM
E. coli cells were fixed for 30 min in 1% glutaraldehyde (EM Sciences; Ft Washington, PA) + 4% paraformaldehyde (Polysciences; Warrington, PA) constituted in PBS, dehydrated in alcohol, and processed for embedding in LR White resin (EM Sciences) at 60C for 24 hr. Ultrathin sections were cut, flattened with xylene vapor, collected on nickel grids, processed for immunogold labeling, stained with 2% aqueous uranyl acetate, and observed in a JEOL 1200 EX II TEM.
Immunogold Labeling Method
Sections on grids were first blocked for 30 min by floating on drops of PBS containing different blocking agents along with 0.01% Tween-20, followed by washing on drops of PBST (five changes, 5 min each). This was followed by incubation overnight (16–18 hr) at 4C with rabbit anti-VHb antibody diluted 1:500. All antibody dilutions were made in respective blocking buffers that had been diluted tenfold with PBS. Then the grids were washed and bound antibodies were localized by incubating the sections for 1 hr on goat anti-rabbit gold conjugate (10 nm; Sigma) diluted 1:20. Finally, grids were washed on drops of PBST and water before being stained with 2% aqueous uranyl acetate. All incubations, except for the primary antibody step, were carried out at RT.
Controls Used
The specificity of immunogold labeling was judged by (a) comparing the probe density on the cells with that on the adjacent plastic resin, (b) comparing the probe density on the cells from which the first antibody had been omitted, (c) comparing the probe density on sections of antigen-negative cells that had been labeled in a similar manner as test sections and, finally, (d) by substituting the rabbit anti-VHb antiserum with NRS.

Dot-blotting assay of antigen-positive cell lysate using different blocking agents. (A) CAE; (B) SM; (C) NGS; (D) HS; (E) FrVBSA; (F) BQBSA; (G) 0.2% FSG; (H) 0.2% AuBSA; (I) 2% FSG; (J) 2% AuBSA. Note the higher level of background staining in BQBSA-blocked panel. The rest of the blots show similar levels of background.

Western blotting assay of antigen-positive cell lysate using different blocking agents. (A) CAE; (B) SM; (C) NGS; (D) HS; (E) FrVBSA; (F) BQBSA; (G) 0.2% FSG; (H) 0.2% AuBSA; (I) 2% FSG; (J) 2% AuBSA. Arrowhead indicates the position of test antigen. Note the presence of minor bands in some of the lanes (arrows), indicating less effective suppression of background.
Quantitation of Probe Density
The resultant probe density, using test and control serum, was quantified from random micrographs of cell sections as described previously (Ramandeep et al. 2000, 2001). Briefly, prints of negatives were made and overlaid with transparencies. Cell outlines were traced, cutout and weighed (Pearse 1972; James 1996). The gold particles in each cell and surrounding clear plastic were counted manually by point counting. The area of each cell was computed by comparison with the weight of an area corresponding to 100 μm2 cut out from the same transparency sheet. To calculate the probe density, 42 (minimum) to 111 (maximum) individual cells were counted for each test and control sample. Total clear plastic area was computed (usually larger than the total area occupied by cells) and the number of gold particles bound there in were also counted. At least seven or eight sections were used for analysis in each case.
Analysis
The total probe density on cells and clear plastic was calculated for the test and control of each sample processed. Densities were expressed as the number of gold particles/μm2. Comparisons between samples were done by two-tailed t-test.
Results
Evaluation of Blocking Agents by Dot-blotting Assay The efficiency of various blocking agents in suppressing nonspecific background labeling was first tested by dot-blotting assay. When anti-VHb antiserum was used to probe antigen +ve lysate, spotted onto nitrocellulose membrane, a uniform level of test as well as background signal was obtained in almost all cases (Figure 1). When antigen -ve cell lysate (-ve immuno-specimen)-spotted membranes were used, no appreciable signal was observed in any of the samples. Similar negative signal was observed when NRS was used in place of anti-VHb antiserum (data not shown).
Evaluation of Blocking Agents Using Western Blotting
Results of Western blotting of antigen +ve cell lysate are presented in Figures 2A–2J. A prominent band representing the antigen was seen with all blocking reagents, along with some additional faint bands, especially when blocking agents other than casein were used. When similar experiments were carried out using antigen -ve cell lysate or where anti-VHb antiserum was replaced with NRS, no significant binding was observed (data not shown).
Evaluation of Blocking Efficacy Using ELISA
ELISA was used to evaluate the blocking efficiency of various proteins in suppressing nonspecific labeling. Results of +ve signal and background binding are presented in Table 2. When anti-VHb antiserum was used for probing wells coated with Ag+ve E. coli cell lysate (positive immunospecimen) almost uniform results (OD 0.3–0.4) were obtained. In experiments using Ag -ve E. coli cell lysate (negative immunospecimen), signals lower than the corresponding Ag+ve cell lysate coated wells were obtained in all cases (p<0.01). In this case, wells blocked with CAE demonstrated the lowest OD (0.05) followed by SM (0.08). Both of these values were significantly less than (p<0.05) the corresponding OD values of wells blocked with other blocking agents in which the absorbance ranged from 0.1 to 0.16. On carrying out ELISA of only clear plastic (uncoated) wells that had been subjected to blocking, once again CAE and SM-blocked wells gave lowest signal (OD = 0.005 and 0.006, respectively). These values are significantly lower (p<0.05) than those obtained with all other blocking agents. The rest of the blocking agents evaluated gave OD values ranging from 0.01 to 0.06.
For antigen +ve E. coli cell lysate coated wells probed with NRS, the lowest signal was again observed in wells blocked with CAE (OD = 0.04). This was significantly lower than those obtained in other cases. CAE was followed by SM, which had a slightly higher signal (OD = 0.06).
ELISA results of absorbance in wells (artificial immunospecimen) a
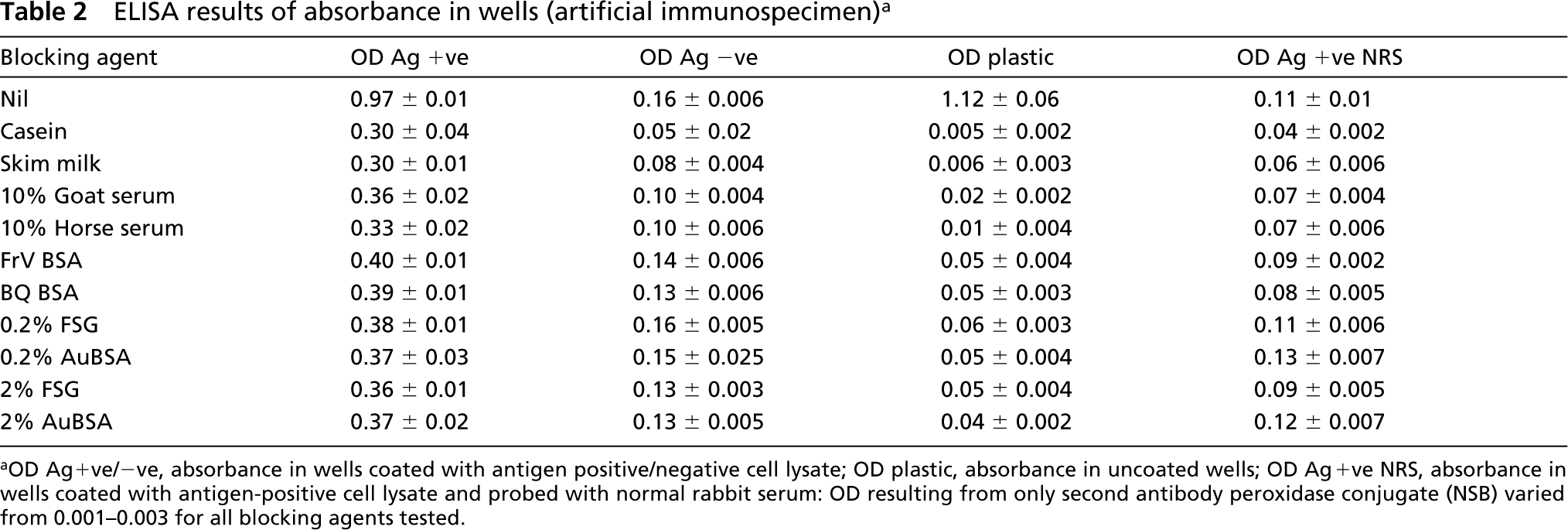
OD Ag+ve/-ve, absorbance in wells coated with antigen positive/negative cell lysate; OD plastic, absorbance in uncoated wells; OD Ag+ve NRS, absorbance in wells coated with antigen-positive cell lysate and probed with normal rabbit serum: OD resulting from only second antibody peroxidase conjugate (NSB) varied from 0.001–0.003 for all blocking agents tested.
TEM results of number of gold particles/μm2 on cells and clear plastic resin a
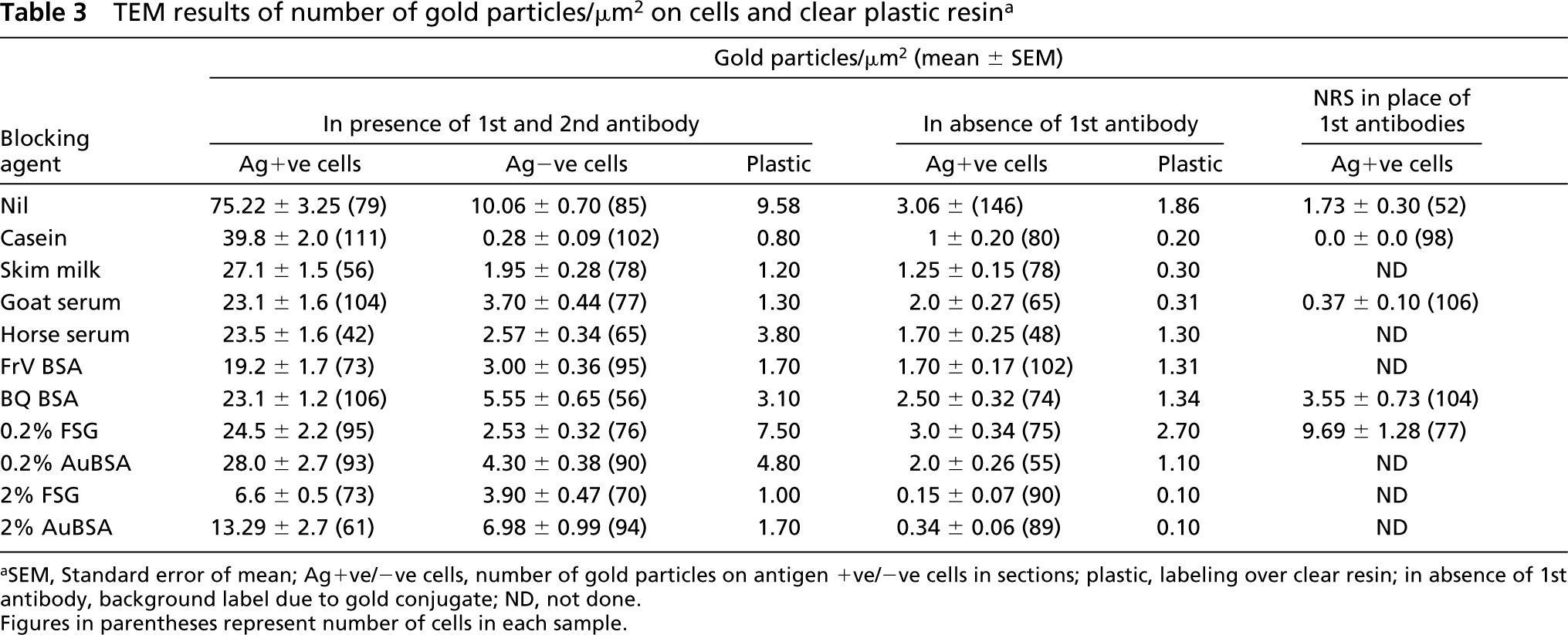
SEM, Standard error of mean; Ag+ve/-ve cells, number of gold particles on antigen +ve/-ve cells in sections; plastic, labeling over clear resin; in absence of 1st antibody, background label due to gold conjugate; ND, not done. Figures in parentheses represent number of cells in each sample.
Probe Density
The numbers of gold particles/μm2 of total cell or clear plastic area are shown in Table 3. The representative areas are shown in Figures 3–6. On cell sections of antigen +ve E. coli cells labeled with anti-VHb antibody (Figures 3A, 3C, 4A, 4C, 5A, 5C, 6A, and 6C), the probe density was always higher than the level of nonspecific binding in adjacent plastic. It was also significantly higher (p<0.001) than the probe density on cell sections of antigen -ve E. coli cells (Figures 3B, 3D, 4B, 4D, 5B, 5D, 6B, and 6D) or sections of antigen +ve E. coli cells probed with; normal rabbit serum (in place of the first antibody) or only with the gold conjugate (not shown). Probe density results confirmed the ELISA finding that CAE and SM gave lowest levels of background labeling. As observed in ELISA, use of CAE as a blocking agent gave the lowest levels (p<0.001) of nonspecific binding on antigen -ve E. coli cells (0.28 ± 0.09) followed by SM (1.95 ± 0.28). Other blocking agents resulted in higher levels of nonspecific binding. Use of CAE for blocking also resulted in the lowest density of nonspecific labeling on clear plastic resin (0.80) and also when NRS was used in place of first antibody to label antigen +ve E. coli cell sections.
Discussion
The current study demonstrates the advantage of ELISA as a convenient method for selection of optimal blocking reagents to carry out immunogold labeling electron microscopy. Blocking to suppress nonspecific labeling is a crucial aspect for the sensitive and specific localization and quantitation of antigen in cells and tissue (Petrusz et al. 1976, 1977; Seymour et al. 1976; Swaab et al. 1977; Hyatt 1989; Bendayan 2000; Burry 2000). We have evaluated the utility of dot-blotting, Western blotting, and ELISA methods in an attempt to predict the best blocking reagent that could be used for immunolabeling of TEM sections. Blocking efficiency in TEM was evaluated in terms of nonspecific labeling by (a) primary antibody onto clear plastic resin, (b) primary antibody to cell components other than the antigen by utilizing antigen-negative cells, (c) gold-conjugated secondary antibody to cells, (d) gold-conjugated secondary antibody to plastic, and (e) binding of control serum (diluted to a similar protein concentration as the primary antibody) on cells.
The dot-blotting method totally failed in accurately predicting the different levels of nonspecific binding, either on clear plastic or on cell components that would occur when samples were evaluated by TEM. The use of this method would lead to the erroneous conclusion that all of the blocking reagents, with the exception of BQBSA, would give similar low levels of NSB because only BQBSA-blocked samples showed some significant background labeling on the nitrocellulose matrix. As in the case of dot-blotting, the use of Western blotting also did not provide any clear indication of the relative merits of various background-suppressing reagents. Moreover, these two methods were also not sensitive enough to detect any NSB of primary antibody to cell components other than the antigen (by use of antigen-negative cell lysates) and were also unable to detect the background labeling that occurs due to control serum (NRS). Inability to differentiate background labeling due to nonspecific binding reflects the limitation in sensitivity of the Western blotting method (even densitometric scans of the blots failed to differentiate among various blocking agents; data not shown). In addition, this lack of ability to differentiate among different blocking agents in terms of NSB may also be due to the biophysical nature of the interaction involving nitrocellulose support, the blocking agent, and the antibody molecules.
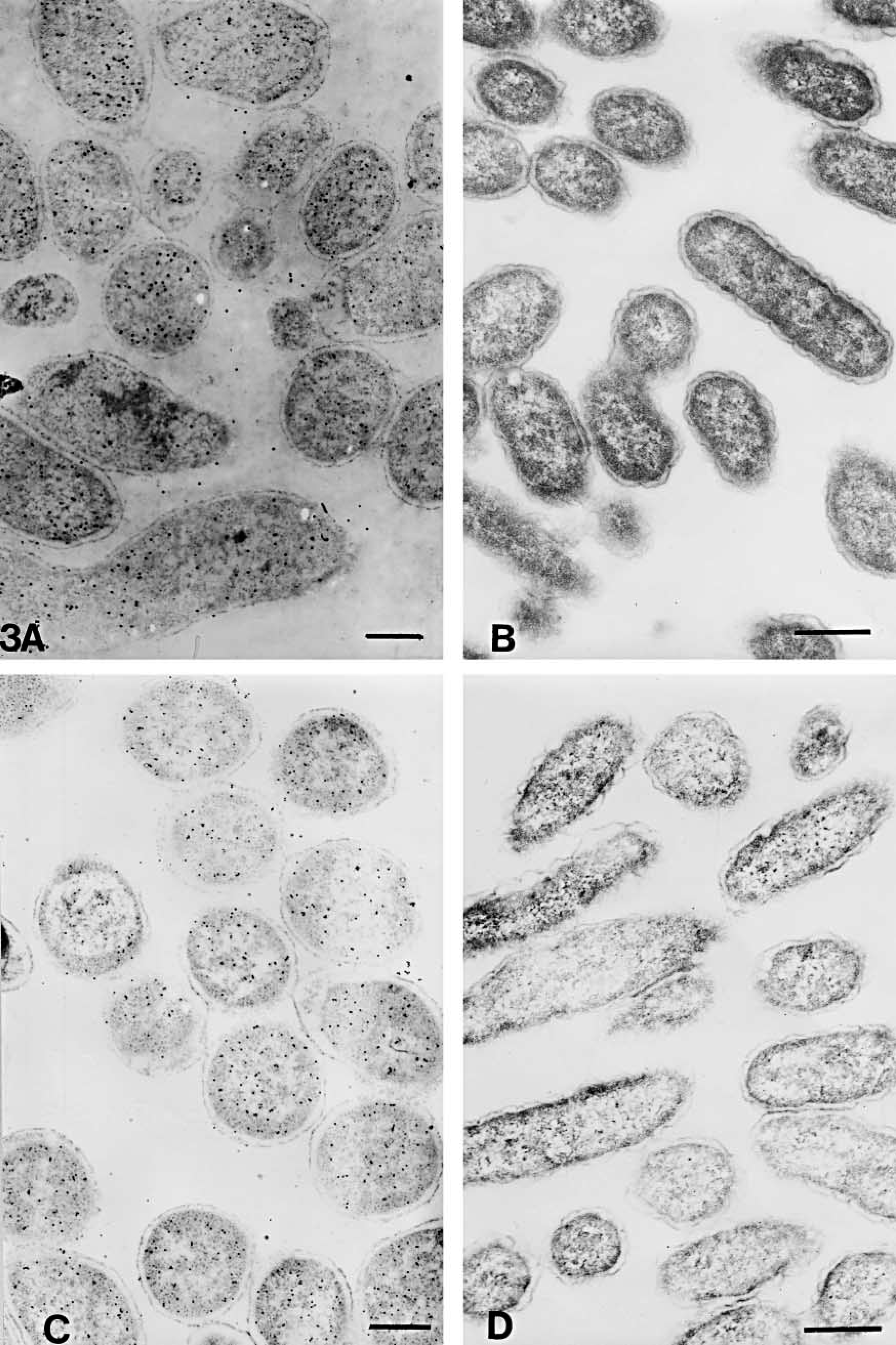
Electron micrographs of antigen-positive (
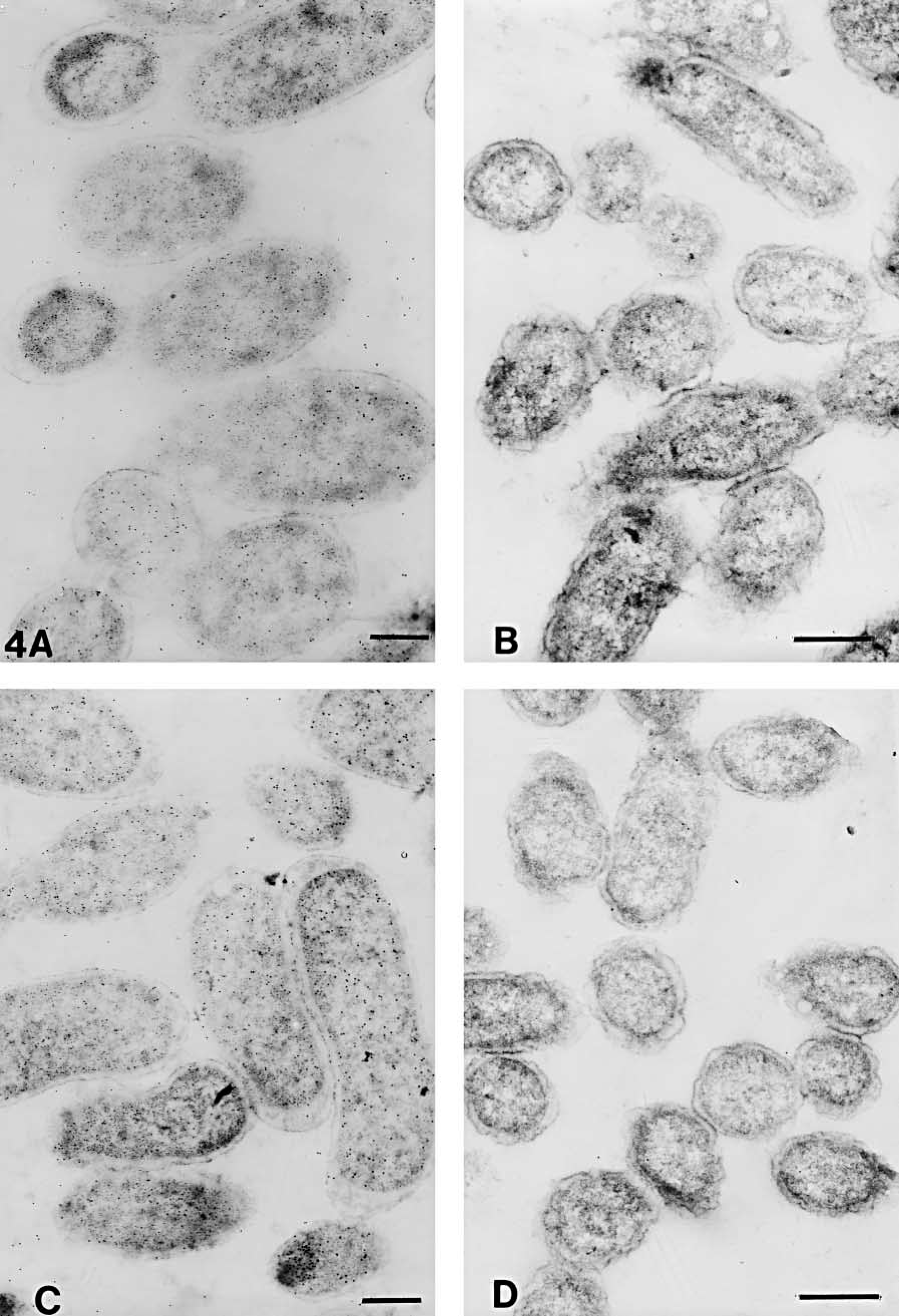
Electron micrographs of antigen-positive (
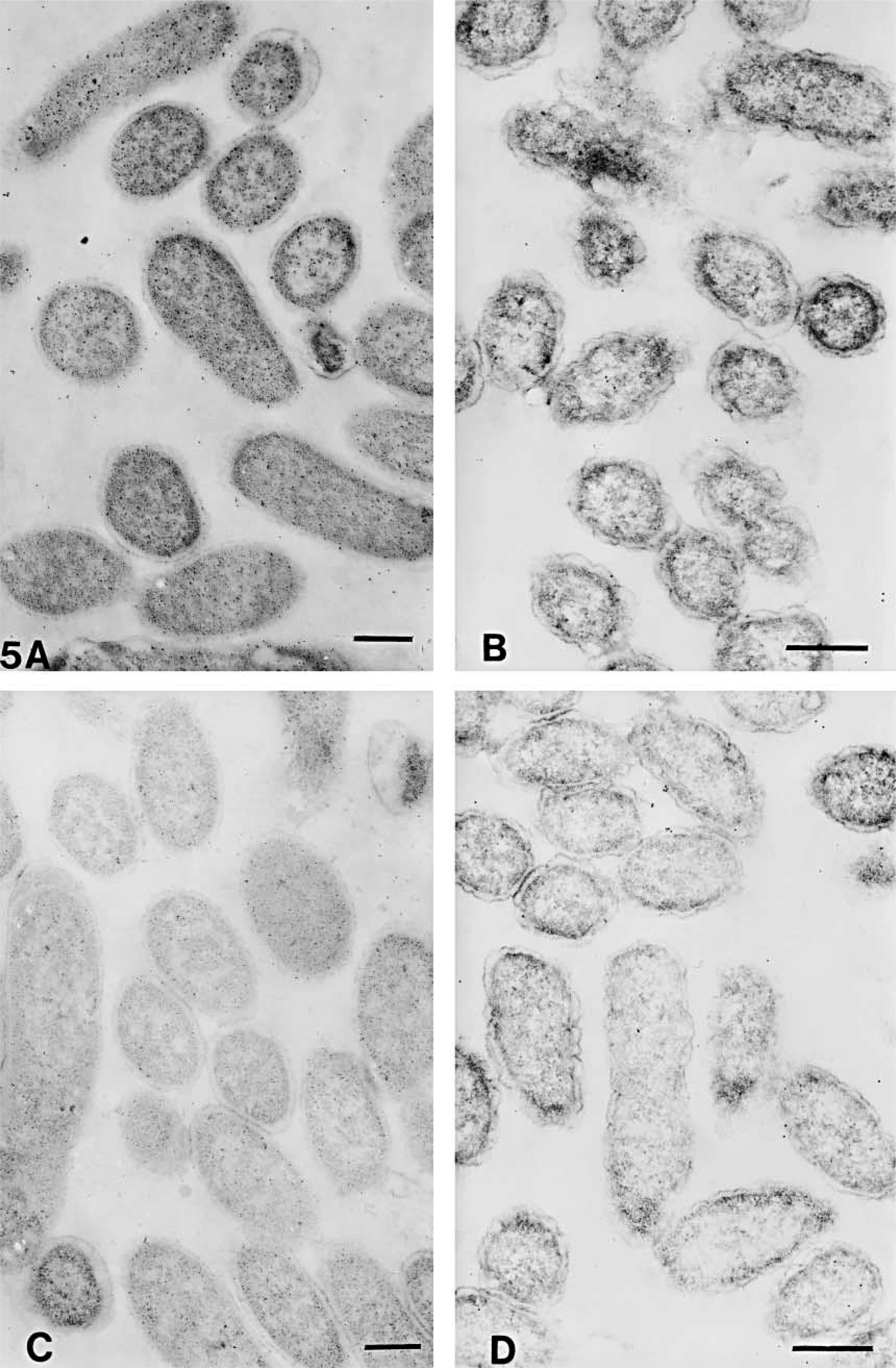
Electron micrographs of antigen-positive (
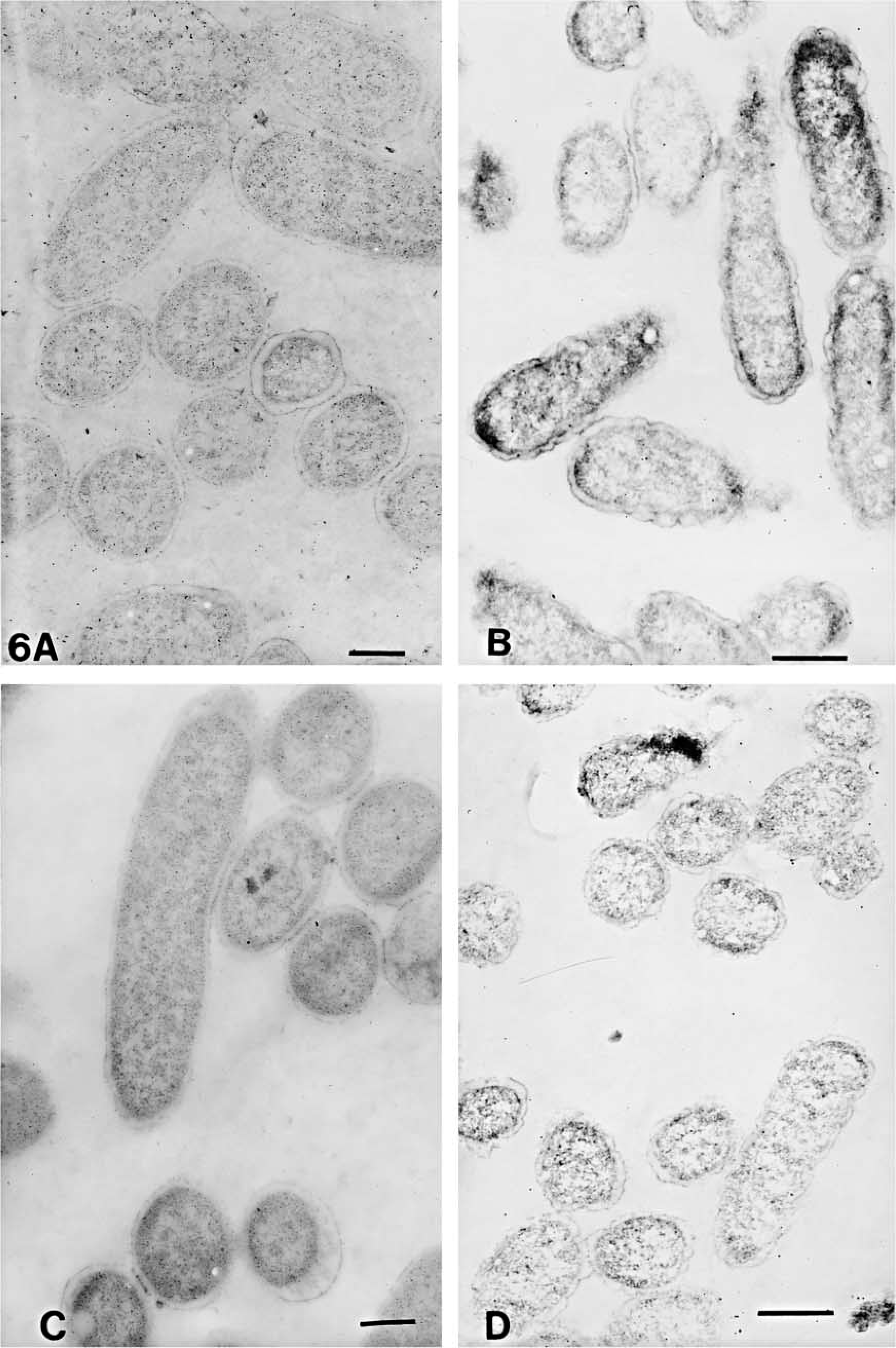
Electron micrographs of antigen-positive (
In contrast to the above two approaches, ELISA provided a clear-cut distinction in background level of signals when different blocking reagents were used. This method accurately predicted the significantly lower (p<0.001) levels of NSB observed in all cases when immunogold labeling TEM was carried out utilizing casein (and, to a lesser extent, skim milk) for blocking. The close similarity between the effects of CAE and SM could be due to the fact that the major component of skim milk is casein. In accordance with ELISA results for nonspecific signal arising from the use of control serum, immunogold labeling showed the lowest level of binding for CAE, 0.0 ± 0.0 gold particles/μm2. In the case of 2% FSG and 2% AuBSA, although the level of NSB by primary antibody to antigen negative cells was significantly higher than CAE and SM, the background labeling by gold conjugate alone, to antigen-positive cells as well as to clear plastic, was much lower than all the other blocking agents. In these two cases the positive signal, due to primary antibody binding to antigen-positive cells was also significantly less (p<0.001) than the labeling on cells when any of the other blocking agents were utilized. This may be caused by obscuring of exposed antigenic sites in cut cell sections at these higher concentrations of FSG and AuBSA, which may be due to the differences in the physical and chemical nature of these two proteins. Casein and BSA are globular proteins, and the major components of horse and goat sera, albumin and globulin, are also globular in nature. On the other hand, gelatin (a preparation from collagen) is a fibrous protein with a preponderance of both charged and hydrophobic amino acids. AuBSA is a partially linearized (to facilitate exposure of hydrophobic groups) form of BSA developed by Aurion and has an increased net negative charge due to acetylation of amino groups. The manufacturers recommend the use of these proteins only at 0.1 to 0.2% rather than at concentrations as high as 2% (Leunissen Jan 1997). A variation in the relative efficiency of different blocking agents has been reported for various immunoassays (Spinola and Cannon 1985; Vogt et al. 1987; Sheng and Schuster 1992; Bendayan 1995a).
Earlier we have demonstrated the utility of ELISA-based methods for rapid selection of sample preparation conditions for quantitative detection of antigen in cell sections by immunogold labeling TEM (Ramandeep et al. 2000). Our present study demonstrates that evaluation of different blocking agents, utilizing cell lysates adsorbed in ELISA wells (artificial immunospecimen), provides information that is useful for selection of optimal blocking reagents to be used in immunogold labeling electron microscopy. This method offers significant advantages over the existing methods that use nitrocellulose membranes. Because this method also utilizes antigen in a cell lysate that has been adsorbed into ELISA wells, the procedure can be combined with that for determining optimal sample processing conditions (Ramandeep et al. 2000). Apart from requiring limited quantities of reagents, the method is also fairly rapid. Multiple blocking conditions and different kinds of controls can easily be carried out simultaneously (the exact number depending on the well format).
In the present work we have presented data using a soluble intracellular antigen. However, in principle the ELISA technique can also be applicable to other proteins including membrane bound antigens (Noteboom et al. 1984; Schmutz et al. 1994). With membrane antigens, special care needs to be taken in interpretation of results because certain antigens and extraction procedures are known to affect the sensitivity of ELISA (Livingston et al. 1997). Isolated cell membranes and some organelles can also be used directly for ELISA (Howard et al. 1980). Alternatively, for membrane antigens exposed on the cell surface, whole-cell ELISA can be considered (Straus et al. 1993). Methods that deal with antigen present in extracted cell lysate may not truly mimic the microenvironment in vivo. This may be especially true for antigens in whole-tissue homogenates or those that are sequestered in certain cell compartments, such as membranes. Nevertheless, it should be emphasized that there exists a strong correlation between the extent of background signal seen using cell lysates for ELISA and direct immunogold labeling TEM. Although the sensitivity of antigen detection and the biophysical interactions between molecules vary tremendously from one protein to another, the technique used and the protocol proposed herein using cell lysates can certainly be applied in many cases to select the most optimal blocking reagent(s).
Footnotes
Acknowledgments
We wish to thank Drs A. Mondal and G.C. Varshney for critical reading and correction of manuscript. The skillful technical assistance of Mr Anil Theophilus is gratefully acknowledged. This is IMTech Communication No. 030/2001.