
Abstract
We used a proteolytically modified and biotinylated derivative of the cholesterol-binding θ-toxin (perfringolysin O) to localize cholesterol-rich membranes in cryosections of cultured human lymphoblastoid cells (RN) by electron microscopy. We developed a fixation and immunolabeling procedure to improve the preservation of membranes and minimize the extraction and dislocalization of cholesterol on thin sections. We also labeled the surface of living cells and applied high-pressure freezing and subsequent fixation of cryosections during thawing. Cholesterol labeling was found at the plasma membrane, with strongest labeling on filopodium-like processes. Strong labeling was also associated with internal vesicles of multivesicular bodies (MVBs) and similar vesicles at the cell surface after secretion (exosomes). Tubulovesicular elements in close vicinity of endosomes and the Golgi complex were often positive as well, but the surrounding membrane of MVBs and the Golgi cisternae appeared mostly negative. Treatment of cells with methyl-β-cyclodextrin completely abolished the labeling for cholesterol. Our results show that the θ-toxin derivative, when used in combination with improved fixation and high-pressure freezing, represents a useful tool for the localization of membrane cholesterol in ultrathin cryosections.
C
Apart from caveolae, membrane cholesterol appears to be involved in the formation of microdomains at the cell surface by preferential association with glycosphingolipids and sphingomyelin to form the so-called liquid ordered phase (reviewed by Brown and London 1997). These microdomains or rafts are involved in many cellular functions, such as sorting processes, cell-cell interaction, and signal transduction (Simons and Ikonen 1997; Brown and London 1998; Hakomori et al. 1998; Mukherjee and Maxfield 2000). It has been shown that cholesterol-rich microdomains form in the Golgi complex and function in targeting raft-associated proteins to the apical plasma membrane in polarized cells (Keller and Simons 1998; Scheiffele et al. 1998). Microdomains also function as sorting platforms in endocytosis (Mayor et al. 1998; Mukherjee and Maxfield 2000). The subcellular localization of intracellular cholesterol-rich membranes is largely unknown but is of particular interest because many aspects of the intracellular transport of cholesterol are still obscure.
The polyene antibiotic filipin (Kinsky 1970) has been used in many recent studies to localize cholesterol by fluorescence microscopy. This approach does not allow resolution of the various types of intracellular membranes and organelles. Filipin was also used to demonstrate cholesterol at the ultrastructural level (Verkleij et al. 1973; Elias et al. 1979; Robinson and Karnovsky 1980; Orci et al. 1983). However, filipin cytochemistry suffers from important limitations, the main being filipin-induced extraction of cholesterol from membranes and loss of membrane integrity (Behnke et al. 1984a, b). On the other hand, some cholesterol-containing membranes are resistant to filipin and escape detection by this method (Severs and Simons 1983; Pelletier and Vitale 1994).
We have studied the usefulness of a biotinylated and non-cytolytic form of θ-toxin (perfringolysin O) from Clostridium perfringens (termed BCθ hereafter) to localize cholesterol at the subcellular level. The specificity of cholesterol binding by BCθ was demonstrated previously (Iwamoto et al. 1997). BCθ was introduced as a cytochemical probe for detection of plasma membrane cholesterol by Fujimoto and coworkers (1997). In a recent study, BCθ was used to specifically detect cholesterol-rich microdomains in the plasma membrane of cells (Waheed et al. 2001). We have used this probe to label cholesterol for electron microscopic detection by various methods, including labeling of living cells and cryosections of high-pressure frozen or chemically fixed cells. All methods revealed a heterogeneous distribution of cholesterol at the plasma membrane, with a concentration of label on curved plasma membrane processes. Cholesterol was enriched in tubular structures adjacent to the Golgi complex and associated with early endosomes, and in internal vesicles of multivesicular bodies. Our studies show that BCθ is a valuable tool to analyze the distribution of membrane cholesterol on thin cryosections of cells.
Materials and Methods
Reagents
Perfringolysin O (θ-toxin) was prepared and digested with subtilisin Carlsberg as reported previously (Ohno-Iwashita et al. 1986; Sekino-Suzuki et al. 1996). Biotinylated θ-toxin (BCθ) was produced from the nicked θ-toxin as described (Iwamoto et al. 1997). Polyclonal rabbit anti-biotin antibodies were obtained from Sanver Tech (Heerhugowaard, The Netherlands) and FITC-labeled anti-rabbit from Jackson Immuno Research Laboratories (Avondal, PA). Colloidal gold particles were prepared according to Slot and Geuze (1985) and conjugated to protein A (Roth et al. 1978). Methyl-β-cyclodextrin was obtained from Sigma (Zwijndrecht, The Netherlands).
Cells
The EBV-transformed human B-cell line RN (HLA-DR15+) was maintained as described (Raposo et al. 1996).
Cell Surface Labeling
For detection of plasma membrane cholesterol with BCθ in living cells, RN cells were washed several times with ice-cold PBS, pelleted, and kept on ice for 15 min. For surface labeling of fixed cells, RN cells were fixed with 2% formaldehyde, 0.2% glutaraldehyde in 0.1 M PHEM buffer for 30 min on ice and washed several times with ice-cold 0.1% glycine in PBS. Then the cells were resuspended in PBS containing BCθ at a concentration of 15 μg/ml and incubated for 30 min on ice with constant shaking. Next, the cells were washed thoroughly with ice-cold PBS and fixed with 2% formaldehyde, 0.2% glutaraldehyde in 0.1 M PHEM buffer (60 mM PIPES, 25 mM HEPES, 2 mM MgCl2, 10 mM EGTA, pH 6.9) overnight at 4C and infiltrated with 2.3 M sucrose in 0.1 M PHEM buffer for 2 hr on ice. Then droplets of cells in sucrose were mounted on pins and frozen in liquid nitrogen. Cryosections were labeled with anti-biotin antibodies and protein A-gold (see below).
Flow Cytometric Analysis of BCθ Binding to Intact Cells
Living or prefixed RN cells were incubated with BCθ as described above. Living cells incubated with buffer only served as negative control. To confirm specificity of BCθ binding, plasma membrane cholesterol was extracted by treatment of cells with 10 mM cyclodextrin for 30 min at 37C in complete medium before incubation with BCθ. After washing with PBS, the samples were fixed with 2% formaldehyde and 0.2% glutaraldehyde in 0.1 M PHEM buffer for 30 min on ice. After washing with 0.1% glycine in PBS, the samples were incubated with anti-biotin antibodies (4 μg/ml PBS with 1% BSA), followed by an incubation with anti-rabbit-FITC (1:50 in PBS with 1% BSA). Cell-associated fluorescence was analyzed by flow cytometry using FACS Calibur (Becton Dickinson; Franklin Lakes, NJ).
Cryosections After Cryofixation
This method was briefly introduced earlier (Liou et al. 1996). RN cells were pelleted in culture medium. After removal of the supernatant, a concentrated suspension of cells was sucked into copper tubes, placed in a high-pressure freezer (EMpact Leica; Vienna, Austria), frozen, and stored in liquid nitrogen. Cryosectioning was performed at — 170C after trimming away part of the copper tube with a trimming diamond using a Leica Ultracut UCT ultramicrotome. Sections were picked up in a mixture of 2% formaldehyde, 0.2% glutaraldehyde, 1% acrolein, 0.2% uranyl acetate, 1% methylcellulose, and 0.67 M sucrose in 30 mM PHEM buffer, thawed, and transferred to formvar-coated grids. Then the sections were fixed with 2% formaldehyde, 0.2% glutaraldehyde, 1% acrolein, and 0.2% uranyl acetate in 0.1 M PHEM buffer for 5 min at RT before labeling.
After five brief rinses with 0.1 M PHEM buffer, sections were incubated with BCθ at a concentration of 15 μg/ml in 0.1 M PHEM buffer for 30 min at RT, washed, and fixed with 1% glutaraldehyde for 15 min at RT. After several washes with 0.1% glycine in 0.1 M PHEM buffer, sections were incubated for 30 min at RT with anti-biotin antibodies (4 μg/ml in 0.1 M PHEM buffer with 1% BSA), followed by 20-min incubation with protein A-gold in 0.1 M PHEM buffer with 1% BSA. After fixation with 1% glutaraldehyde for 3 min, sections were washed with water, contrasted, and dried as described (Liou et al. 1996). Sections were examined and photographed at 80 kV with a JEOL 1200 EX electron microscope (Tokyo, Japan).
Cryosections After Chemical Fixation
RN cells were washed with 0.1 M PHEM buffer. According to the conventional protocol of Tokuyasu (1973) adapted by Geuze and Slot (1980), cells were fixed with 2% formaldehyde, 0.2% glutaraldehyde in 0.1 M PHEM buffer for 2 hr or overnight at 4C and infiltrated in 2.3 M sucrose for 2 hr or overnight at 4C and frozen in liquid nitrogen. After ultracryomicrotomy, cryosections were picked up and thawed in 2.3 M sucrose. To improve the preservation of membrane cholesterol, cells were fixed with 2% formaldehyde, 0.2% glutaraldehyde, and 1% acrolein in 0.1 M PHEM buffer for 30 min on ice. Fixation was followed by infiltration with 1.8 M sucrose in 80 mM PHEM buffer containing 2% formaldehyde, 0.2% glutaraldehyde, 1% acrolein, or 1.8 M sucrose in 80 mM PHEM buffer containing 0.5% OsO4 and 0.2% uranyl acetate for 2 hr on ice. In case of a weak fixation, cells were fixed with 4% formaldehyde in 0.1 M PHEM buffer and infiltrated with 1.7 M sucrose in 75 mM PHEM buffer containing 4% formaldehyde. Then droplets with cells were put on cutting pins and frozen in liquid nitrogen. After cryosectioning, sections were picked up and thawed according to Liou and co-workers (1996) in a 1:1 mixture of 2.3 M sucrose and 2% methylcellulose. Sections were labeled immediately after pick-up and thawing as described above. For samples fixed with formaldehyde only, sections were incubated with BCθ for 3 hr at 4C. To confirm the specificity of cholesterol labeling, sections were incubated with 10 mM methyl-β-cyclodextrin for 20 min and washed with 0.1 M PHEM buffer before labeling to also remove intracellular membrane cholesterol.
Quantification
To determine the labeling density of the plasma membrane cholesterol, living RN cells were incubated with BCθ as described above. After washing the cells were fixed with 2% formaldehyde and 0.2% glutaraldehyde in 0.1 M PHEM buffer and processed for cryosectioning. To make sure that three independent samples were used for quantification, 15 μm of the block was trimmed away between cutting sections. After labeling of sections with anti-biotin and protein A-gold, 10 random pictures were taken from a single section of each of the three grids and printed at a final magnification of ×28,000. We determined the labeling density (gold/μm membrane) by using point and intersection counting with a line lattice (10-mm distance) overlay as described by Weibel (1979) and Griffith (1993) in three different domains of the plasma membrane: flat membrane, large extensions and invaginations, and filopodia. Filopodia were defined as round or elongated profiles with a diameter of 100 nm.
To determine the density of cholesterol labeling on endosomes and the Golgi complex, sections were cut as described above and incubated with BCθ for 30 min at RT, followed by labeling with anti-biotin antibodies and 10-nm protein A-gold. For quantification of labeling on the Golgi complex, five pictures of different Golgi areas with clearly discernible stacks were taken from a single section of each of the three grids at a magnification of ×20,000. The labeling densities on Golgi cisternae and TGN associated vesicles/tubules were determined on prints with a final magnification of ×47,000 using a curvilinear lattice with 5-mm distance between lines (isotopic test system according to Merz 1967) for point and intersection counting. Gold particles were assigned to a structure when they lay within a distance of twice the particle size from its membrane.
For quantification of labeling on endosomes, pictures of seven to nine endosomes (Type 3 in Kleijmeer et al. 1997) with not more than five internal vesicles were taken from a single section of each of the three grids. These endosomes represent tubulovesicular early endosomes. Labeling densities were determined for the limiting membrane, the internal vesicles, and the surrounding vesicular and tubular profiles at a distance of not more than 800 nm from the vacuolar part of the endosome. For point and intersection counting, we used a square line lattice with 5-mm distance between lines on prints at a final magnification of ×47,000.
Results
Search for Optimal Cholesterol Labeling
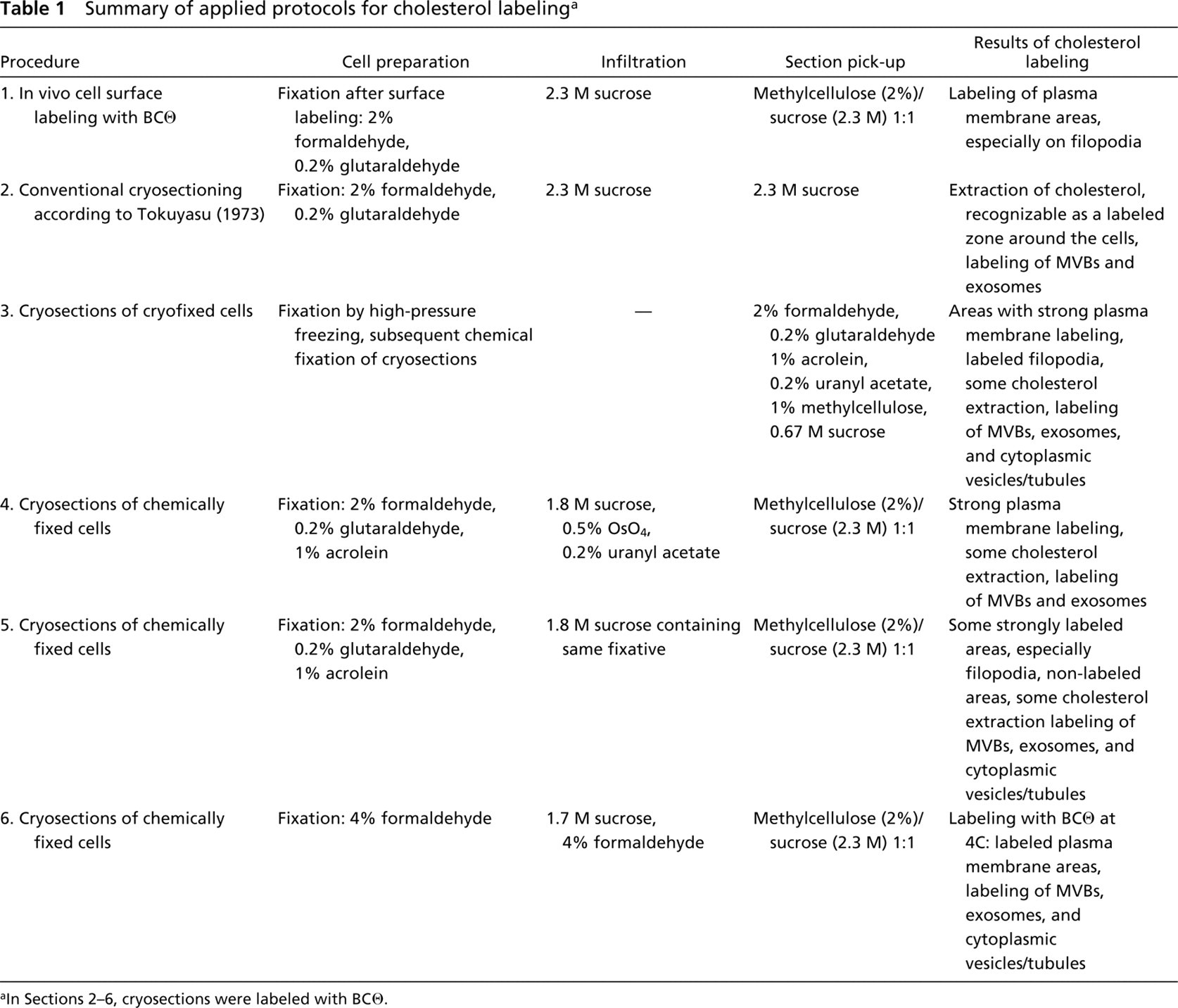
Labeling of Cholesterol on the Cell Surface. It is well known that the major fraction of the cell's membrane cholesterol resides in the plasma membrane. Accordingly, we found significant labeling of the cell surface when we incubated living RN cells with BCθ at 4C (Table 1), fixed the cells, and processed them for cryosectioning and immunolabeling with anti-biotin and protein A-gold (Figure 1A). Removal of cholesterol by pretreatment with 10 mM methyl-β-cyclodextrin completely abolished plasma membrane labeling. If we changed the sequence of steps in the cell surface labeling procedure so that cells were first fixed and then incubated with BCθ, the plasma membrane labeling was significantly reduced (Figure 1B). This was not due to cholesterol extraction during the fixation procedure because we could still achieve BCθ binding to cryosections of these preparations (see below). A possible explanation for the failure to label intact cells after fixation is that a coat of crosslinked surface proteins makes the membrane cholesterol inaccessible to the probe. To confirm these morphological data, we performed a quantitative analysis using flow cytometry and compared the extent of surface labeling in living cells to that of fixed cells. As shown in Figure 2, indeed less BCθ bound to fixed cells (Curve 3) than to living cells (Curve 4). The binding was even more reduced after cholesterol extraction with 10 mM cyclodextrin (Curve 2).
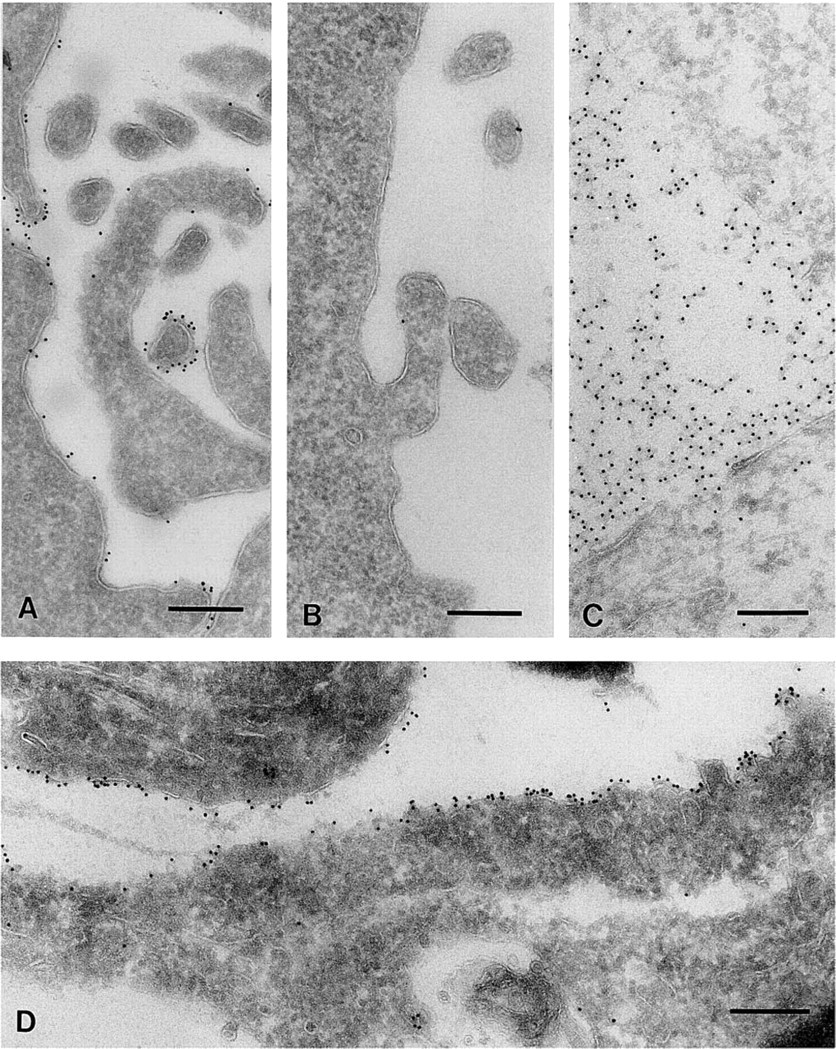
Summary of applied protocols for cholesterol labeling a
aIn Sections 2–6, cryosections were labeled with BCθ.
Labeling of Plasma Membrane Cholesterol in Conventional Cryosections. Because we wanted to study the intracellular cholesterol distribution, we explored the possibility of using cryosections for BCθ binding and subsequent immunogold labeling. First, cryosections were prepared in the conventional way as described in Materials and Methods, incubated with BCθ and immunolabeled with anti-biotin and protein A-gold (Table 1). The pattern of labeling on these sections showed that the distribution of cholesterol was seriously disturbed (Figure 1C). The plasma membrane itself was usually not labeled, but a wide zone of gold particles was present at the extracellular side, indicating extraction of cholesterol.
Labeling of Plasma Membrane Cholesterol in Cryosections of Cryofixed Cells. Next, we circumvented chemical prefixation and used high-pressure frozen cells (Table 1). Cryosections were cut at — 170C on a dry diamond knife. To achieve maximal preservation of ultrastructure, it was important that the cryosections were cut flat without compression, picked up, thawed without stretching, and transferred flat to a grid. The pick-up solution contained a mixture of sucrose, methylcellulose, formaldehyde, glutaraldehyde, acrolein, and uranyl acetate. Including methylcellulose and uranyl acetate in the pick-up solution was crucial for good preservation of the ultrastructure. As shown previously (Liou et al. 1996), such sections can be successfully used for immunolabeling after the fixatives have been washed away. When these sections were labeled with BCθ, prominent plasma membrane labeling was observed (Figure 1D). Although extraction of cholesterol was still apparent in some plasma membrane areas, this result clearly demonstrated that under these conditions localization of plasma membrane cholesterol is feasible.
Living RN cells were incubated with BCθ at 4C. Cryosections were labeled with anti-biotin and protein A-gold (10 nm). Labeling of cholesterol was frequently detected on the tips of filopodium-like plasma membrane protrusions (
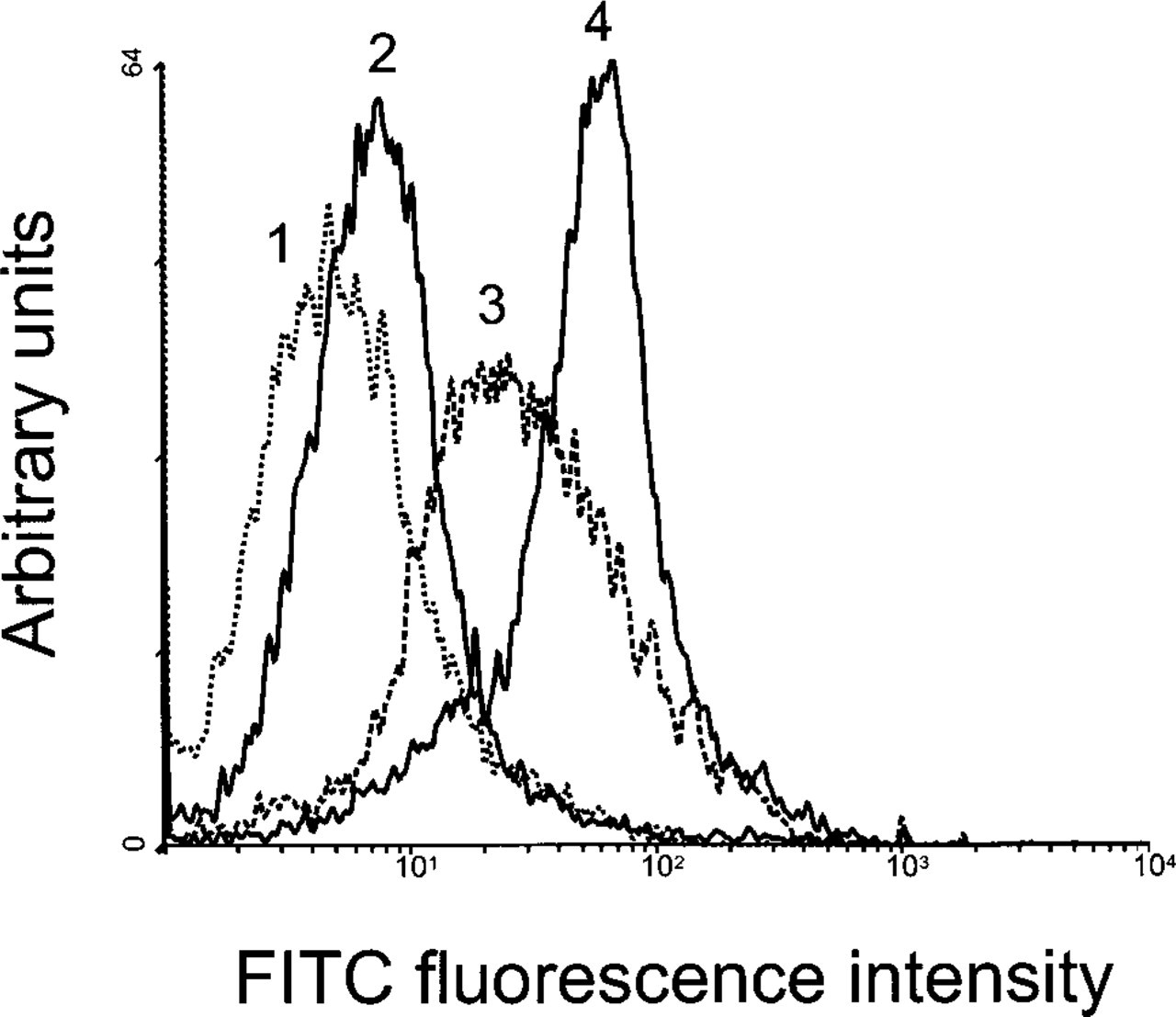
Flow cytometric analysis of BCθ binding to intact cells. Living RN cells were incubated with BCθ (Curve 4). Prefixation of cells reduced BCθ binding (Curve 3). Extraction of cholesterol with 10 mM methyl-β-cyclodextrin reduced BCθ binding significantly (Curve 2). Living RN cells were incubated with buffer alone as a negative control (Curve 1).
Effect of Fixation Procedures on Labeling of Plasma Membrane Cholesterol. The results with the high-pressure frozen samples showed that the tissue processing of chemically fixed material and the way of section retrieval are critical for the maintenance of cholesterol within the membrane. In the initial experiments with fixed cells, we used fixatives containing formaldehyde and glutaraldehyde. These fixatives bind to proteins without any known direct interaction with membrane lipids. Therefore, we next investigated whether fixatives such as acrolein, uranyl acetate, and OsO4 which act on membrane lipids (Hayat 1981), retain cholesterol (Table 1). The strongest and most consistent labeling of the plasma membrane was obtained with a mixture of formaldehyde, glutaraldehyde, and acrolein in the initial fixation and OsO4 and uranyl acetate in the infusion sucrose (Figure 3A). Because acrolein and OsO4 cannot be combined directly, we choose to use them in this sequence. In general, major improvements were achieved by the introduction of acrolein and OsO4 in the procedure, while the influence of uranyl acetate was less definite. However, OsO4 and uranyl acetate are known to severely affect protein antigenicity. Therefore, OsO4 and uranyl acetate were omitted from the sucrose infusion and replaced by formaldehyde, glutaraldehyde, and acrolein (Table 1). This resulted in strong plasma membrane labeling in some areas but still non-labeled areas occurred as well (Figure 3B).
Influence of Low Temperature During Cholesterol Labeling. Cholesterol-rich microdomains, or rafts, are insoluble in Triton X-100 at low temperature (Simons and Ikonen 1997). Therefore, we tested whether cholesterol extraction from sections could be reduced by labeling sections with BCθ in the cold (Table 1). Surprisingly, labeling at 4C did not enhance the labeling of cholesterol or prevent cholesterol extraction from sections of samples fixed with formaldehyde, glutaraldehyde, and acrolein followed by infusion with sucrose containing the same fixative or OsO4 and uranyl acetate. However, on sections of samples fixed with formaldehyde only and infiltrated with sucrose in the presence of formaldehyde, plasma membrane cholesterol was detectable when labeling was performed in the cold (Figure 3C) but not after labeling at RT.
Subcellular Distribution of Cholesterol
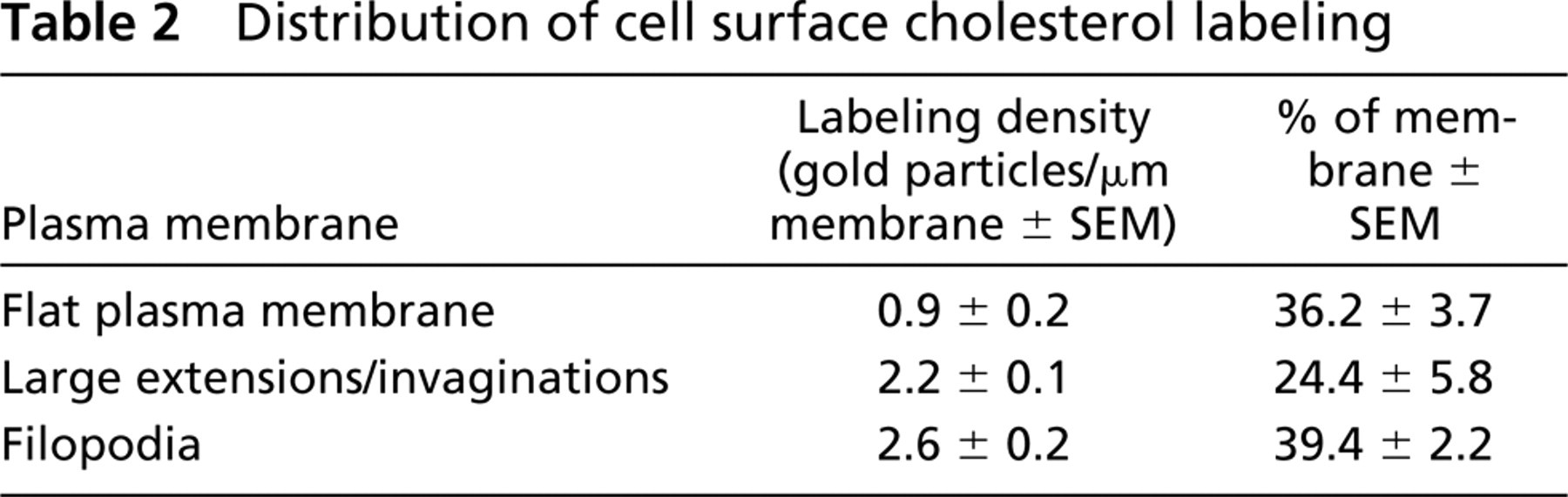
The Plasma Membrane. Cholesterol-enriched membrane areas were heterogeneously distributed and often found on filopodium-like processes of the cell surface. To analyze this finding quantitatively, we determined the labeling density in three different domains of the plasma membrane: flat plasma membrane, large extensions/invaginations, and filopodia after labeling the surface of living cells with BCθ at 4C (Table 2; Figure 1A). Filopodia showed 2.9 times more labeling than the flat areas of the plasma membrane. Large plasma membrane extensions and invaginations were 2.4 times more labeled than flat membrane. On cryosections of cryofixed cells (Figure 1D) or chemically fixed cells (Figures 3A and 3B), this pattern was also frequently observed but was less reproducible due to cholesterol extraction from the sections.
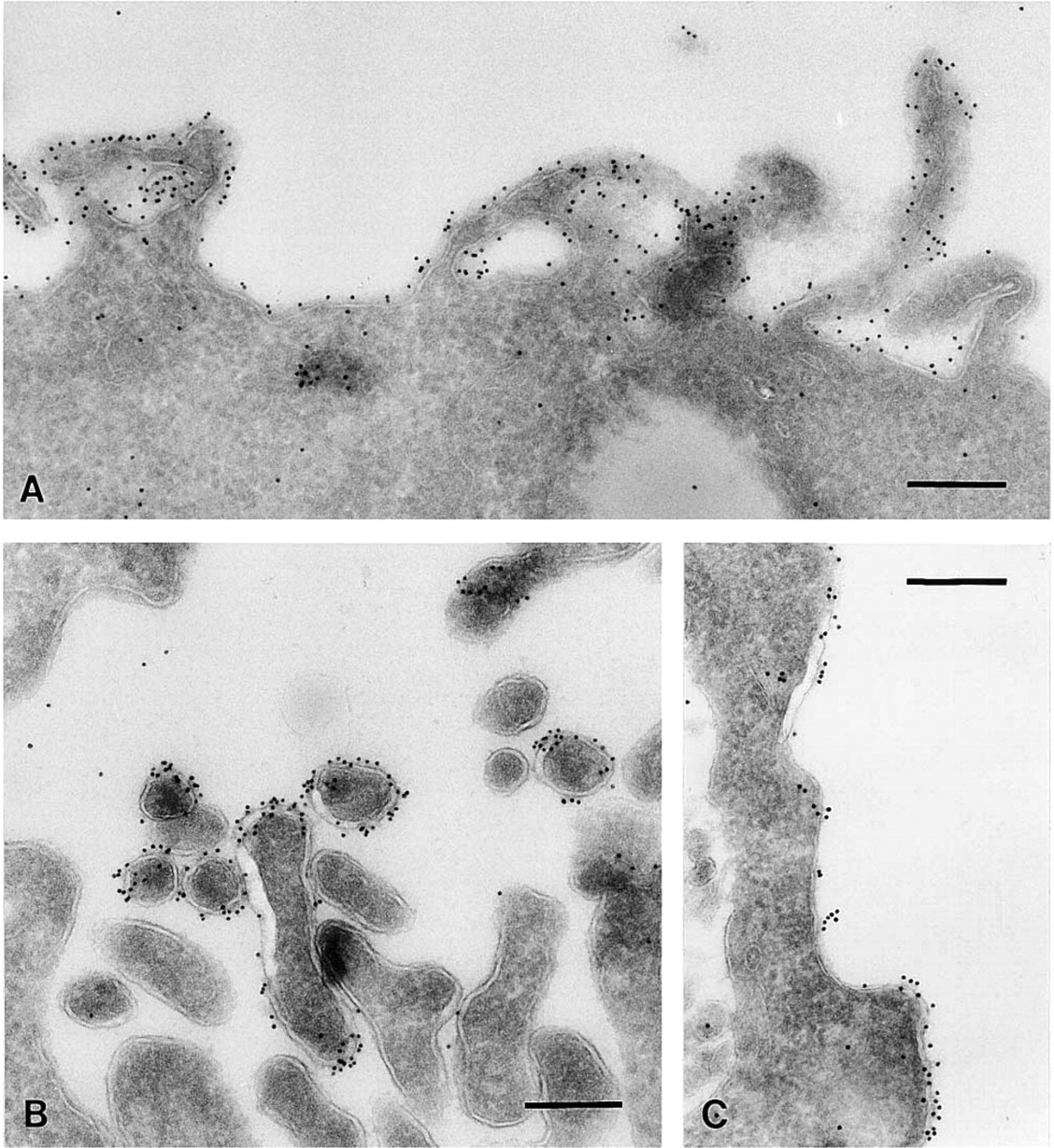
Including acrolein in the fixative and OsO4 and uranyl acetate in the infusion sucrose resulted in strong plasma membrane labeling of RN cells. Cholesterol labeling was often confined to the plasma membrane and sometimes enriched in filopodium-like protrusions (
Removal of plasma membrane cholesterol by incubating cells with 10 mM cyclodextrin before fixation abolished plasma membrane labeling completely, confirming the specificity of labeling. The consistency of the heterogeneous distribution of cholesterol obtained by different approaches indicates that this pattern of plasma membrane labeling is not due to relocation artifacts during tissue processing.
Intracellular Membranes. The modified fixation and infiltration procedure was also suitable to localize intracellular cholesterol. Cholesterol in intracellular membranes appeared less susceptible to extraction than plasma membrane cholesterol. With all different approaches we observed essential similar cholesterol labeling. The results could also be reproduced with cryosections of high-pressure frozen cells that were fixed after sectioning (data not shown). We used sections prepared according to Table 1, section 5 for detection of intracellular membrane cholesterol because this fixation resulted in a very well-preserved ultra-structure. Cholesterol was not detectable in the ER. Labeling of cholesterol was observed on vesicles and tubules associated with the TGN, whereas the Golgi cisternae were mostly unlabeled (Figure 4A). As shown in Table 3, the labeling density on the vesicles and tubules was 25 times higher than the labeling associated with the Golgi cisternae. Along the endocytic pathway, cholesterol was regularly localized to small cytoplasmic vesicles associated with MVBs (Figure 4B). MVBs often showed strong cholesterol labeling of their vesicular content, indicating cholesterol-rich membranes, whereas the perimeter membrane remained mostly unlabeled (Figure 4C). For a quantitative analysis of this observation, we selected early endosomes according to Kleijmeer and co-workers (1997). These typically contained not more than five internal vesicles. We determined the relative membrane surface areas and labeling densities of the limiting membrane, the internal vesicles, and the surrounding vesicles/tubules (Table 4). The internal vesicles displayed an 11-fold higher labeling density than the limiting membrane. For the tubular portion of the endosome the factor was 7.
Sometimes labeled particles in the lumen of MVBs were observed that were smaller than the internal vesicles and probably represented LDL particles. Interestingly, lysosomes with a multilaminar content next to the MVBs were almost devoid of labeling (Figure 4C). RN cells are able to secrete the vesicular content of MVBs by exocytosis (Figure 4D). This process leads to the production of small extracellular vesicles, so-called exosomes (Johnstone et al. 1987; Raposo et al. 1996; Escola et al. 1998). Such released exosomes were frequently observed at the plasma membrane and labeled strongly for cholesterol (Figures 4D and 4E). The cholesterol in exosomes was resistant to extraction both before and after sectioning, independent of the fixation procedure used. Extraction of sections with 10 mM methyl-β-cyclodextrin before incubation with BCθ abolished labeling for cholesterol.
Discussion
BCθ, a non-cytolytic and biotinylated derivative of perfringolysin O (Iwamoto et al. 1997), was introduced as a cytochemical probe for plasma membrane cholesterol by Fujimoto and co-workers (1997) and used in a study on lymphocytes (Hagiwara et al. 1999). In this study we evaluated the use of BCθ for subcellular localization of cholesterol by labeling thin cryosections with the probe and then visualizing probe-binding sites with anti-biotin antibodies and protein A-gold. In principle, this approach allows the localization of intracellular cholesterol in situ with high-resolution electron microscopy.
Distribution of cell surface cholesterol labeling
As an important prerequisite, novel procedures and techniques had to be worked out to preserve the cholesterol distribution in cell membranes. Apart from the preservation of cholesterol, the properties of BCθ itself may influence the results. An ideal cytochemical probe should detect cholesterol in any membrane in which it is present and the amount of labeling should represent the concentration of cholesterol in the labeled area. The specificity of cholesterol binding by θ-toxin and its derivatives was confirmed previously by detection of cholesterol on TLC plates and by using liposomal assays and cells (Ohno-Iwashita et al. 1988, 1991; Iwamoto et al. 1997; Waheed et al. 2001). However, it has been shown that the nicked form of θ-toxin (Cθ) binds membrane cholesterol with two different affinities depending on the amount of cholesterol and the lipid composition in a given membrane (Ohno-Iwashita et al. 1988,1991,1992). It is therefore very likely that the actual arrangement of cholesterol and other membrane components, such as phospholipids or proteins, in a biological membrane influence the efficiency of cholesterol detection by BCθ. By using cholesterol depletion, Waheed and co-workers (2001) demonstrated that binding of BCθ to intact cells requires a high concentration of membrane cholesterol. Therefore, BCθ is a useful tool for selective detection of cholesterol-rich membranes rather than for quantitative in situ determination of membrane cholesterol.
The access of a relatively large protein such as BCθ to a small molecule such as cholesterol in a membrane can also be restricted by steric hindrance. As depicted in the model (Figure 5), our observations on intact living cells show that BCθ could bind plasma membrane cholesterol (Figure 5A) but that prefixation prevented this (Figure 5B). This was probably due to a penetration barrier formed by crosslinking of membrane proteins.
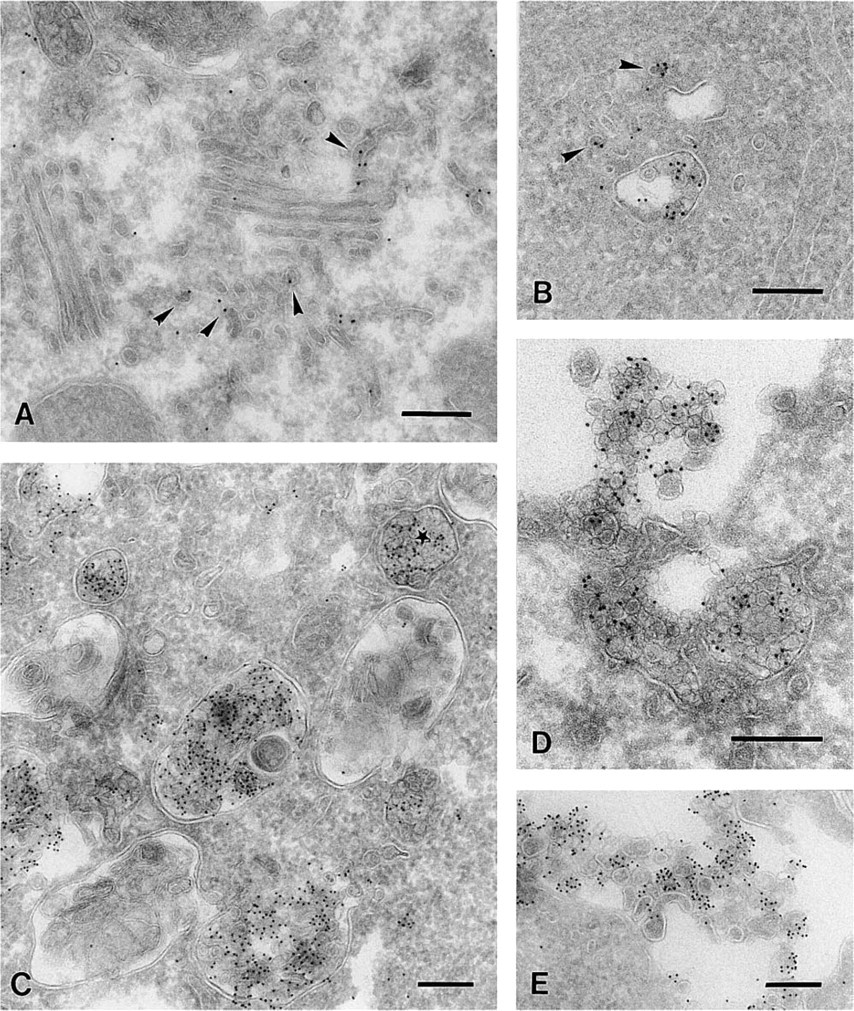
Intracellular cholesterol was frequently detected on highly curved membranes such as tubulovesicular elements associated with the Golgi apparatus (
Distribution of cholesterol labeling on the Golgi complex

In particular, cholesterol-binding proteins could mask membrane cholesterol. The accessibility of cholesterol to binding of the probe in sectioned membranes (Figure 5C) differs from that in intact cells, which may influence labeling efficiency.
In cryosections, a large proportion of the plasma membrane cholesterol is extracted and dislocated, most likely during section pick-up and thawing. It is conceivable that, during the process of thawing, the membrane lipids diffuse and re-seal the hydrophobic interior of the cross-sectioned membrane (Figure 5C). This would be especially relevant for the localization of lipids because these are mostly unfixed and easily displaced. Therefore, it is crucial to adjust the composition of the pick-up solution in such a way that damage and lipid extraction are minimized. It was shown previously that introduction of methylcellulose into the sucrose pick-up solution improves the integrity of cryosections (Liou et al. 1996). For cryosections of high-pressure frozen material we used, in addition to methylcellulose, membrane-stabilizing agents such as uranyl acetate (Liou et al. 1996) and acrolein in combination with the more common fixatives formaldehyde and glutaraldehyde in the pick-up solution. This procedure resulted in a well-preserved ultrastructure of the cryofixed cells and reduced cholesterol redistribution. Chemical fixation of cryofixed cells after sectioning has several advantages. Fixatives penetrate easily and quickly into thin sections, fixation starts during thawing of the sections and, as a result of short diffusion distances, fixation of cryosections takes less time than fixation of whole cells. This new method may also be of value to localize small molecules that are easily redistributed during conventional fixation and cryosection handling. A disadvantage is that the sectioning of high-pressure frozen cells is technically demanding. Further studies will explore the full capacity of high-pressure freezing technique and its application in immunocytochemistry.
Distribution of cholesterol labeling in early endosomes
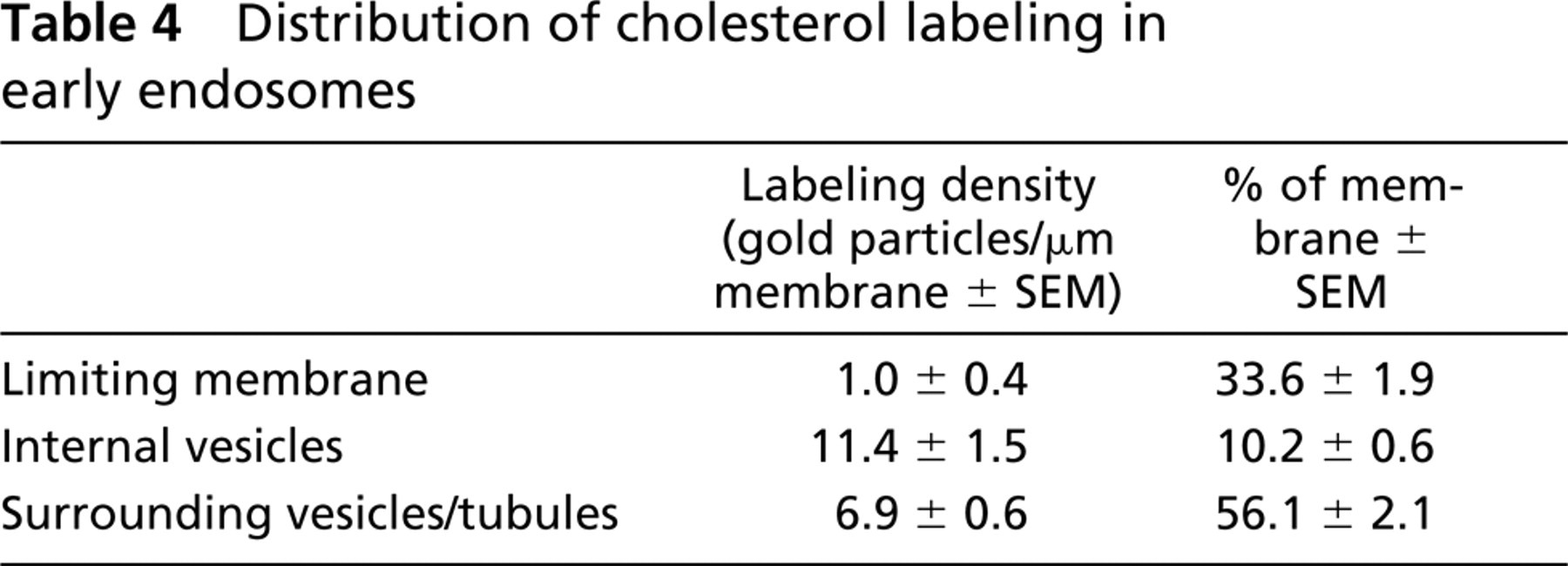
In this study we developed an improved protocol for the preparation of chemically fixed samples for intracellular cholesterol localization. Fixation and temperature during sample preparation and incubation with BCθ were crucial for the detection of membrane cholesterol on cryosections. As an important modification of the conventional protocol, fixatives were included in the infusion sucrose. The continued fixation during sucrose infiltration, in particular using fixatives reacting with membrane lipids such as acrolein, OsO4, and uranyl acetate, minimized cholesterol extraction. When a weak fixation with formaldehyde alone was used, low temperature during incubation of cryosections with BCθ reduced cholesterol extraction. Compared to the plasma membrane, cholesterol in intracellular membranes of chemically fixed cells appeared more resistant to extraction, and therefore the detection was less dependent on the processing conditions. In particular, MVBs and exosomes showed strong labeling with BCθ independent of the fixation protocol. Because exosomes from RN cells have a special protein composition that differs from that of other cell membranes (Escola et al. 1998), we conclude that the susceptibility of cholesterol to extraction depends also on the protein environment in a membrane.
In agreement with Hagiwara and co-workers (1999), we demonstrated in RN cells a preferential localization of cholesterol on filopodia. This cholesterol labeling pattern appeared consistent with three different approaches: cell surface labeling of living cells and on cryosections of chemically fixed and high-pressure frozen cells. Therefore, it is very likely that we indeed detected cholesterol-rich areas at these plasma membrane processes, even though it can not be completely ruled out that differences in the degree of accessibility of cholesterol on a curved membrane compared to a flat membrane contributed to the observed differences in labeling density. Our interpretation is supported by studies showing that the newly identified membrane protein prominin is specifically enriched in such plasma membrane protrusions (Weigmann et al. 1997) and that this localization is dependent on cholesterol-rich microdomains in these areas (Röper et al. 2000).
Intracellular cholesterol was specifically detectable in small cytoplasmic vesicles and tubules associated with the TGN and endosomes. These results support the hypothesis that cholesterol-enriched microdomains or rafts function in sorting processes in the bio-synthetic route in polarized cells (Simons and Ikonen 1997; Keller and Simons 1998; Scheiffele et al. 1998) and along the endocytic pathway (Mayor et al. 1998; Mukherjee and Maxfield 2000). Whether the cholesterol-rich membranes detected by our method represent rafts remains to be determined. An important pool of cholesterol was found in the internal vesicles of MVBs and the exosomes that are released from MVBs via exocytosis. This observation raises the question of whether high levels of cholesterol may be a structural requirement for the formation of these highly curved vesicles.
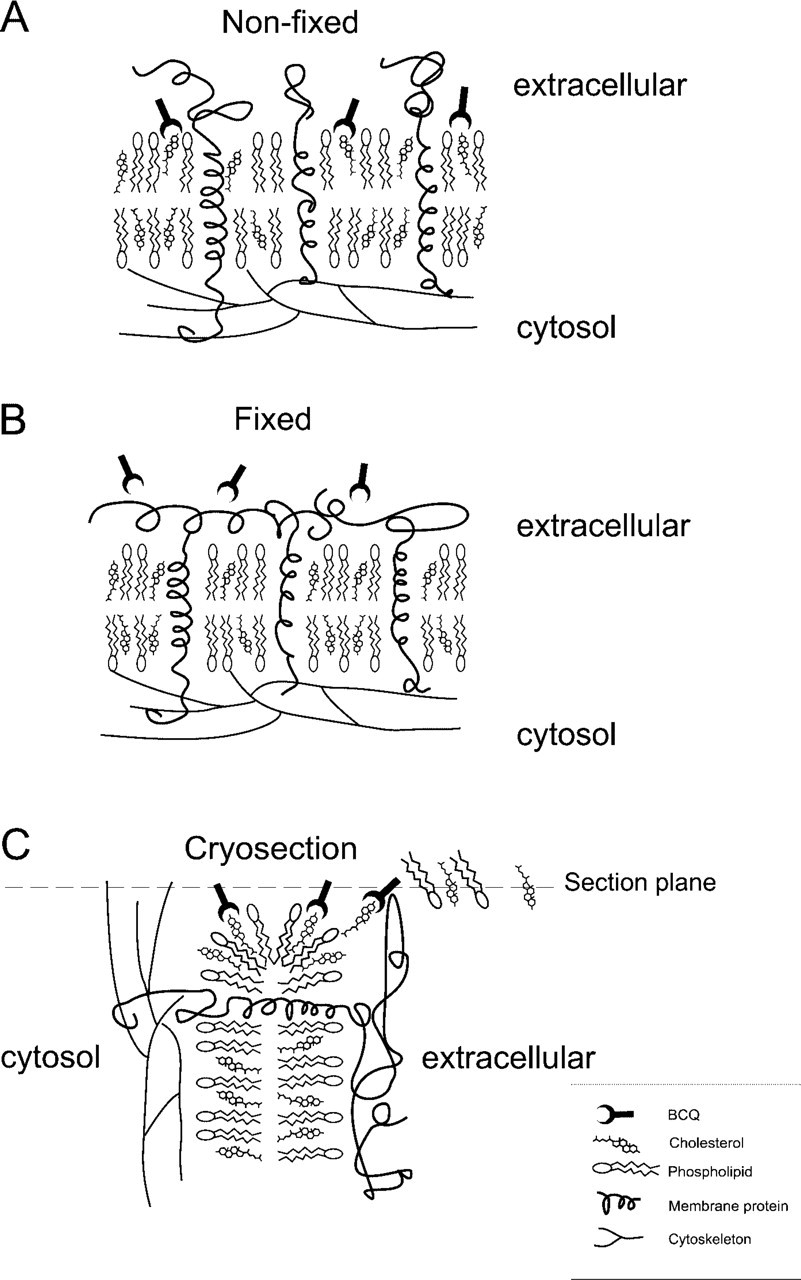
Model of binding of BCθ to membrane cholesterol. (
In conclusion, we have shown that, under well-defined conditions, membrane cholesterol can be demonstrated at the ultrastructural level. Our results indicate relative high concentrations of cholesterol in highly curved membrane structures. Further studies are required to elucidate the functional aspects of such a restricted localization.
Footnotes
Acknowledgements
WM is a CBG (Centre of Biomedical Genetics) postdoctoral fellow at the UMC Utrecht, The Netherlands. This work was supported by a short-term fellowship of the Human Frontier Science Program Organization (Strasbourg, France).
We thank R.M.C. Scriwanek and M. van Peski for excellent photographic work.