
Abstract
Herpes simplex virus type 1 (HSV-1) is a common human pathogen of skin and mucous membranes and is potentially dangerous when the infection is disseminated. Viral morphogenesis, especially the mechanism of viral envelopment and the exact pathway for processing and transport of HSV-1 glycoproteins, is still unclear. We report the results of optimized immunogold-labeled cryosection electron microscopy of HSV-1-infected cultured human fibroblasts (MRC-5). The simplified method presented has proved necessary to obtain reproducible results on cellular distribution of viral glycoproteins. It is now possible to demonstrate the viral glycoprotein gD-1, but not gC-1, in the nuclear membranes and to demonstrate gD-1- and gC-1-labeled viral particles in the perinuclear space, and to show the fate of the viral particles in the endoplasmic reticulum and Golgi area in infected cells.
T
With the light microscopic method, one can state only that the HSV-1 glycoproteins are located near the nucleus, in the cytoplasm, or in the plasma membrane (Jensen et al. 1995a). Confocal microscopic scanning images present the light microscopic findings in a spatial way (Jensen et al. 1995b). Electron microscopy is necessary to identify the fine structure of the cell, but the HSV-1 glycoproteins gC and gD do not remain immuno-demonstrable after the process of Epon embedding (unpublished data). In this study we present a detailed, refined method and extend the investigation to examine the ultrastructural localization of the viral glycoproteins by electron microscopy of immunogold-labeled ultrathin cryosections. In comparative studies, especially if the level of antigenicity is low, it is important to make use of methods that are as simple as possible. Otherwise, there is a great risk for losing information, obtaining unreproducible results, or even false-negative and false-positive results.
Materials and Methods
Reagents, Antibodies, and Conjugates
All chemicals were acquired from Merck (Darmstadt, Germany) unless otherwise indicated.
The monoclonal antibodies (MAbs) against HSV-1 gD (
General Remarks
All experiments were repeated at least twice and duplicates of both grids and slides were made. In each experiment, 200–300 cells were examined in the electron microscope and the results were scored semiquantitatively.
The optimal working dilution and incubation time in a humid chamber at room temperature (RT) were determined for all batches of antisera and conjugates. Gold conjugates were titrated by anti-β-tubulin immunostaining of non-virus-infected cells (MOCK). Antisera were stored at -80C, avoiding repeated thawing and freezing. Dilutions of antisera and conjugates were made just before use. PBS (10 mM Na2HPO4/KH2PO4, 150 mM NaCl, pH 7.4) and PBS-buffered solutions were renewed every second week. Glycine and bovine serum albumine (BSA) were stored at -20C for 6 months. Sucrose, methyl cellulose, and aqueous uranyl acetate were renewed every fourth week and stored at 4C. Great care was taken in preparing solutions, and attention was directed to controlled temperature conditions through all procedures.
Quality control of the immunogold-labeled ultrathin sections for electron microscopy included preservation of ultra-structural morphology, no precipitation or clumping of the gold probe, and minimal but just visible background staining with few gold particles over the nuclei to ensure saturation of the antigen-antibody reaction. Further, it was required that cross-sectioned tubulin in MOCK cells was labeled with about 10 G5 particles and that longitudinal sectioned tubulin showed gold particles with a linear disposition.
Light Microscopy
Conditions for the low-fading immunofluorescence indirect labeling with propidium iodide contrast in whole cells and semithin cryosections have been described earlier (Jensen et al. 1995a).
Silver Enhancement
Silver amplification with IntenSE M (no. RPN 491; Amersham) was performed as set out in the pack leaflet at 18C in a darkroom under red safelight and was compared to physical development (Danscher 1981), which was applied under the same conditions.
Serum
The goat serum (State Serum Institute; Copenhagen, Denmark) and FBS (fetal bovine serum sterile, no. 29-101-54; Flow Laboratories, McLean, VA) were decomplemented by heat inactivation for 30 min at 56C (Ejercito et al. 1968).
Viral Stock and Titration Assay
Viral stocks of HSV-1 strain F (Ejercito et al. 1968) were replaced every 6 months. They were made phenotypically concordant by two passages in human embryonic lung cells, passage below 35 (MRC-5, no. 02–021; Flow Laboratories) at an MOI (multiplicity of infection) of 0.01 pfu (plaque forming units) per cell in EBME (essential basal medium Earle (modified) with Earle's salts, no. 14–000; Flow Laboratories) supplemented with 1% FBS. The HSV-1-infected MRC-5 cells were carefully scraped off the culture flasks with a plastic policeman, centrifuged at 1500 rpm for 10 min, and the pellet from 107 infected cells was resuspended in 1.5 ml EBME medium with 1% FBS. This cell suspension was sonicated at 14 μm four times for 5 sec and in between kept on ice, then centrifuged at 1500 rpm for 5 min to remove cell debris, and afterwards the supernatant was stored in aliquots of 500 μl at -80C.
The viral titers of the viral stocks were determined by plaque assay (Ejercito et al. 1968) after 1-h adsorption of 1 ml diluted viral suspension in log10 steps on confluent Vero cell (African green monkey kidney cells, no. 03-230; Flow Laboratories) monolayers (passage 137–144).
Cell Preparations
MRC-5 delivered at passage 26–28 were grown and proved mycoplasma-free as described previously (Jensen et al. 1995a). The media were renewed every second week and glutamine (no. 16–801; Flow Laboratories) added just before use. Attention was given to careful handling of the cells. Their condition was light microscopically examined every day and the cells were not allowed to grow too heavy. The cells were passaged twice a week just at confluence corresponding to 1.5 X 106 cells in a 25-cm2 cell culture flask (no. 163371; Nunc, Roskilde, Denmark). Three days before HSV-1 infection, the cells were plated on sterile glass cover-slips (Smethwick; Warley, UK) for investigation of whole cells or in 25-cm2 culture flasks in preparation for cryosections. At passages below 35, the just confluent cells were infected with HSV-1; after 1 hr of adsorption at 37C, the inoculum was removed and EBME containing 1% FBS was added. At 12 hr post infection at 37C, the medium was removed and the cells were washed briefly in sterile 37C PBS. The cells were then fixed for 10 min in PBS-buffered freshly prepared paraformaldehyde (PFA, no. P 026; TAAB Laboratories Equipment, Aldermaston, UK), to which was added glutaraldehyde (GA, no. G 002; TAAB Laboratories Equipment). The fixations with 8% PFA, 3% PFA + 0.2% GA, 3% PFA + 1% GA, and 3% PFA + 2% GA were compared. MOI of 10, 20 and 30 pfu per cell in EBME supplemented with 1% FBS were investigated.
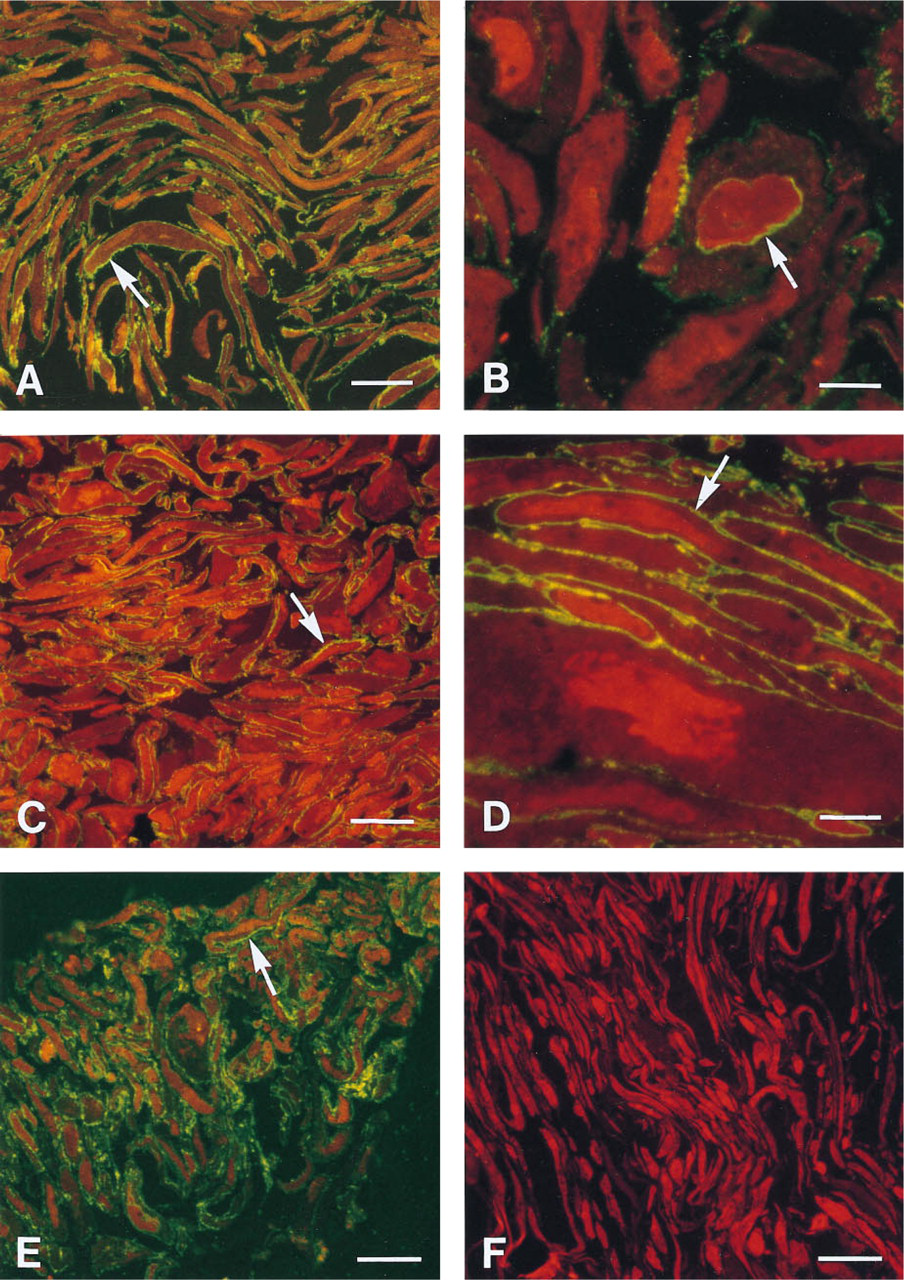
Immunofluorescence with propidium iodide contrast in semithin cryosections of HSV-1 (
To make frozen cell cultures, the cells were scraped off the culture flasks with a plastic policeman, then embedded in gelatin without air bubbles, and after 30 min at 37C centrifuged at 12,000 rpm for 5 min. After 1 hr at 4C, the pellet was cut with razor blades into cubes about 1 mm3 and cryoprotected in 2.3 M PBS-buffered sucrose (no.7651; Merck) with 1% PFA overnight for roughly 18 hr at 4C, frozen, and stored in liquid nitrogen (Van Bergen En Henegouwen 1989). Embedment in 10% and 20% gelatin from Merck (no. 4078, grain size 800 μm) and from Sigma Chemical (St Louis, MO; G 6144, 60 Bloom), and 20% IGSS quality gelatin (RPN 416; Amersham) was compared.
Semithin (about 500 nm thick) cryosections for light microscopy and ultrathin (about 100 nm thick) cryosections for electron microscopy were cut with a glass knife in an RMC MT 6000 XL cryoultramicrotome. The sections were removed from the knife with aid of a sucrose droplet in a wire loop (Van Bergen En Henegouwen 1989). The ultrathin sections were put on formvar (1% solution, no. F 005; TAAB Laboratories Equipment)-covered and carbon-coated 200-mesh nickel grids (HR 24, Ni-3-0 mm; Graticules, Tun-bridge, UK). The sections were stored at 4C in PBS-buffered 1% PFA and immunolabeled within 24 hr.
Optimized Immunogold Labeling
The ultrathin cryosections on the grids were transferred by aid of the smallest possible 0.25-mm platinum wire loop through great drops on a sheet of Parafilm, as follows. In addition to washing (twice for 5 min) with PBS, pH 7.4, glycine (no. 4201; Merck) 0.02 M in PBS was put on for 10 min to inactivate any remaining aldehyde. After washing with PBS (twice for 5 min), PBS containing 0.8% BSA (A 4503; Sigma Chemical) and 0.1% IGSS quality gelatin followed for 5 min. Thereafter, incubation was carried out for 45 min with the primary antibody (Fd 138–80 1:250, DL6 1:1500, 1D3 1:125, HD1 1:1000, HCl 1:800 or anti-β-tubulin 1:600-1:1000 in different batches) diluted in PBS + BSA + gelatin solution with 1% goat serum. The primary antibody was washed off with PBS + BSA + gelatin solution (six times for 4 min). In the indirect three-layer (GAR-G5 or GAR-G1) but not in the two-layer (GAM-G5) labeling method, this was followed by incubation for 30 min with affinity-purified RAM 2.5 μg/ml in PBS + BSA + gelatin solution containing 1% goat serum and subsequent washing with PBS + BSA+ gelatin solution (six times for 4 min). Incubation followed for 20 min with GAR-G1, GAR-G5 or GAM-G5 in suitable concentrations of 1:25–1:75 diluted in PBS + BSA + gelatin buffer with 1% goat serum. Washing followed with PBS + BSA+ gelatin buffer (six times for 4 min), PBS, pH 7.4 (six times for 4 min), and postfixation with 2% GA in PBS, pH 7.4 (10 min). An intervening wash was performed with PBS, pH 7.4 (four times for 4 min), and glass-distilled water (four times for 4 min). If desired, the silver enhancement of the gold particles was done for 8.5 min, followed by washing in glass-distilled water three times for 5 min. The sections were then stained and protected against air-drying artifacts (Van Bergen En Henegouwen 1989) by a solution of ice-cold 1.35% methyl cellulose (64610 Mithocel MC 25 mPa.s USP, no. 18.804–2; Aldrich, Steinheim, Germany) containing 0.4% aqueous uranyl acetate (no. 8473; Merck). Any excess of methyl cellulose was removed after 10 min by a piece of filter paper and the grids were dried at room temperature. The ultrathin sections were examined in a Zeiss electron microscope 900, operating at 80 kV, and photomicrographs were taken on Agfa Scientia EM film (3.25 X 4 inches, HJQ7B). Results of this optimized method were compared with the earlier published recommendations (Nielsen et al. 1989) and with increased time of incubation with primary, secondary, and tertiary antibodies.
Controls
Immunocytochemical controls were carried out by (a) immunostaining of MOCK cells, (b) omission of the primary or secondary antibody and replacement with buffer or type-matched unrelated MAb UCHL1 1:25, or (c) with anti-β-tubulin serving as a positive control. The specificity of the MAbs and the reliability of the immunogold labeling were controlled by immunoblotting and immunofluorescence light microscopy.
Results
Increasing the MOI from 10 (50% infected cells) to 30 (90% infected cells) caused diffuse labeling rather than infection visible in spots of the monolayer cell culture. Electron microscopic studies investigate only a minor part of the sample, which is why the most convenient MOI of 30 was chosen for future studies. Furthermore, the labeling intensity appeared to increase with rising MOI. Exceeding MOI 30 was risky because the cells tended to fall off the culture flask before the time for harvesting.
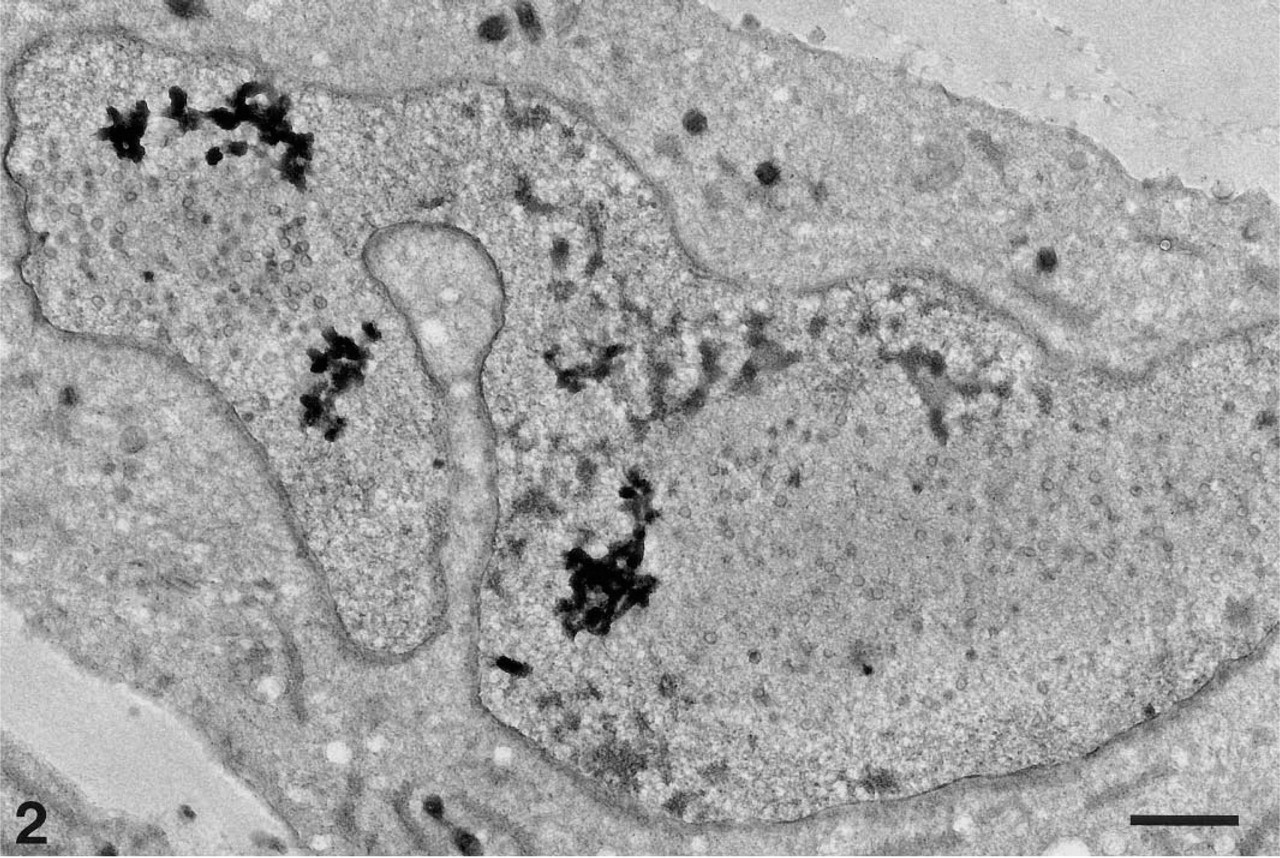
In overview, ultrathin cryosection of HSV-1-infected fibroblast immunolabeled with HC1 and GAR-G5. Bar = 1 μm.
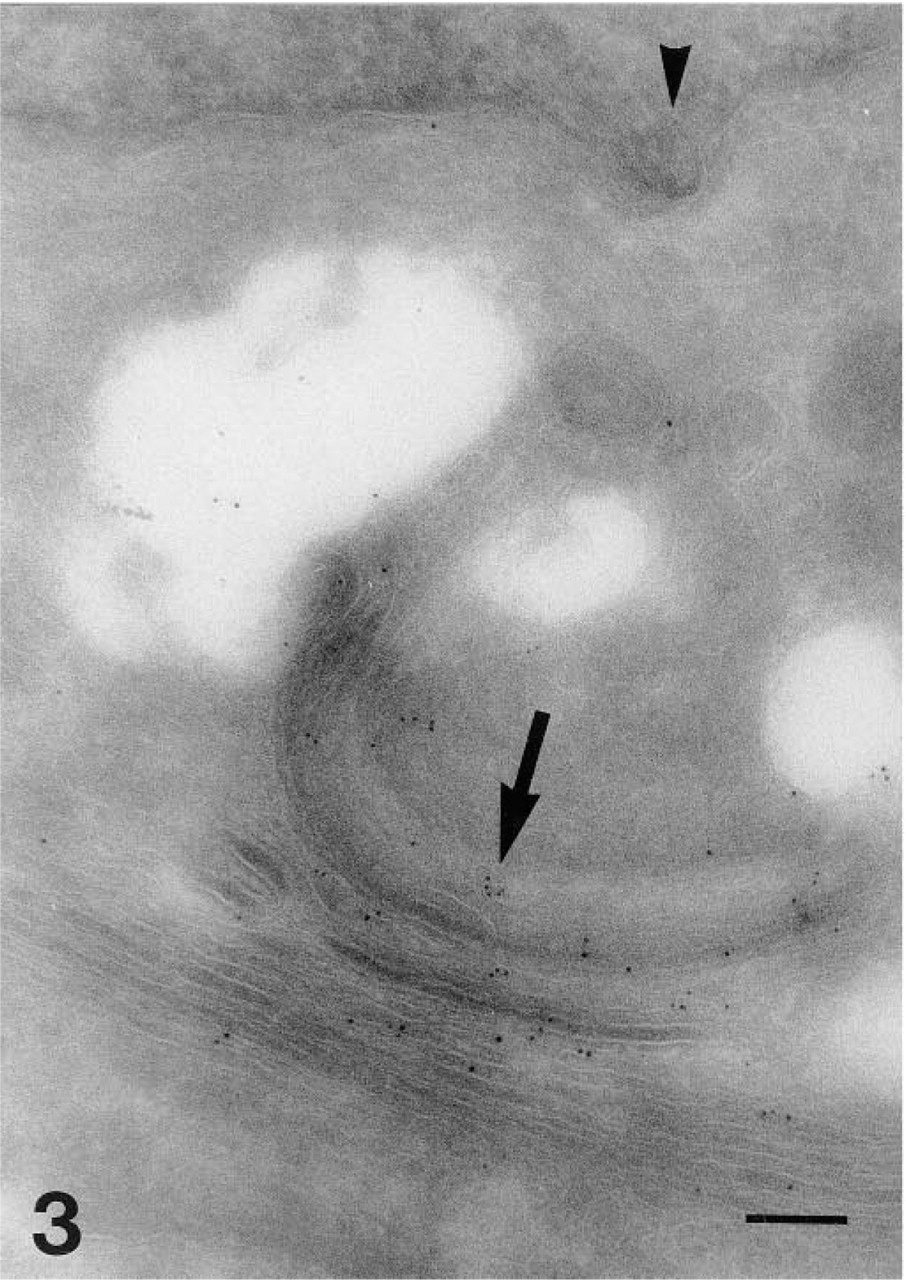
Two-layer indirect 5-nm immunogold identification of gD-1 (antibody DL6) in well-represented membranes of the Golgi complex (arrow). A nucleocapsid deforms the outline of the nuclear envelope (arrowhead). Bar = 0.125 μm.
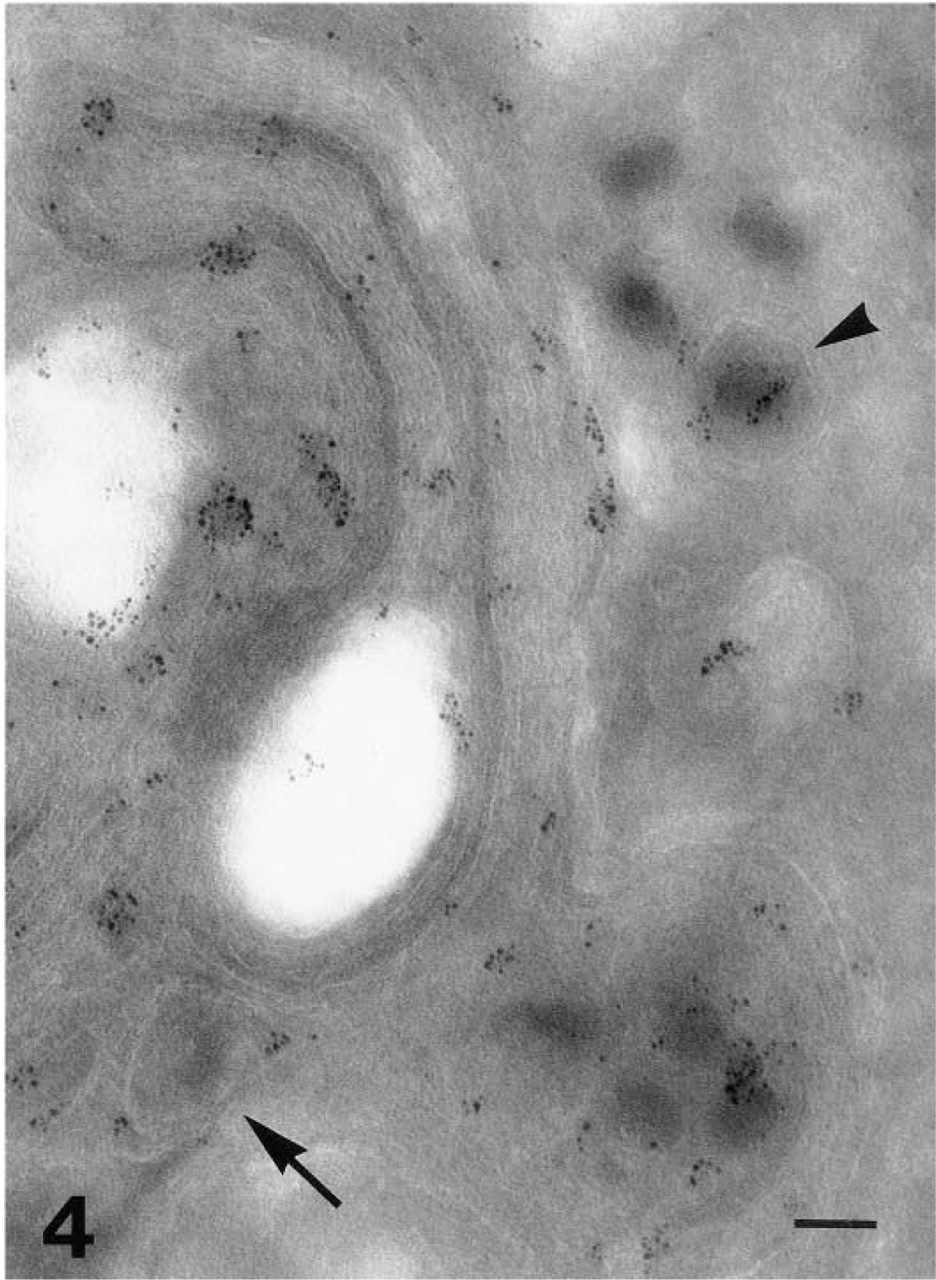
Intense gD-1 (Fd 138–80 antibody) labeling is observed in the Golgi region with the physically enhanced 1-nm gold probe. Enveloped viral particles are labeled both inside (arrow) and outside (arrowhead) the Golgi complex. Bar = 0.125 μm.
Morphology
The MOCK cells were long and slender, with regular nuclei and insignificant vacuolization of the cytoplasm (Figure 1F). Infection with HSV-1 rendered the cell larger, with variable rounding (Figures 1A–1E), the nuclei became irregular with margination of the nuclear chromatin, and the cytoplasm was vacuolated in part, caused by dilatation of the endoplasmic reticulum and the Golgi sacs (Figure 4). The membranous ultrastructure was well preserved (Figures 2 and 3).
Fixation
Compared to 8% PFA, the impact of adding GA to the fixative consisted of improved ultrastructure and, to some extent, reduced intensity of the specific labeling, especially at the plasma membrane. Basically, the labeling was independent of the applied concentrations of GA. Therefore, for further investigations, the fixative of 3% PFA added 2% GA was preferred.
Embedment
The 20% Merck gelatin was preferable for embedment because it yielded vivid labeling, because the ultrastructure was uniform in the compared gelatins, and because it was difficult to cut samples embedded in the other gelatins. In fact, chatter was now the only troublesome technical problem.
Immunolabeling
The simplified cell preparation and immunoincubation instructions using only PBS-buffered solutions described were reproducible, and cryopreparations have been stored in liquid nitrogen for 5 years without losing immunoreactivity. Specific labeling of HSV-1 glycoproteins was observed, although to a lesser degree, by the two-layer indirect immunogold method (Figure 3) compared to the three-layer technique if the concentration of the primary antibody was doubled and the incubation time with GAM increased to 60 min. No further significant labeling was obtained by higher concentrations, longer incubation times at room temperature, incubation overnight at 4C, or incubation according to the scheme but at 37C. Compared to the 5-nm gold, the 1-nm gold particles (Figure 4) intensified the labeling but did not provide new information. The enhanced particles vary in size and shape, and there is a risk for producing artifacts.
Glycoproteins
The 1D3 antibody worked in a three-layer indirect immunofluorescence labeling of 8% PFA-fixed cells (Figure 1E), but convincing ultrastructural specific labeling was never observed. Thus far, we have not seen different localization of the labeling of the discontinuous epitope visualized by HD1 and the continuous gD-1 epitope labeled by the antibody DL6. Optimized immunolabeling of cryosections allowed the identification of both gC-1 (Figure 6) and gD-1 on the surface of extracellular HSV-1 viral particles, on the plasma membrane (Figures 1A, 1C–1E, 6, and 8), on the surface of intracellular enveloped viral particles (Figure 4), in the Golgi apparatus (Figure 4), and in the endoplasmic reticulum (Figure 7). It was possible to demonstrate both gD-1 and gC-1 in the cell adhesion areas and sometimes on the surface of enveloped viral particles localized in the perinuclear space (Figures 5 and 7). However, distinct labeling of the nuclear membranes was demonstrated only with the gD antibodies (Figures 1B and 5). Capsids were seen close to and in the nuclear membranes (Figure 3), but complete budding and evidential acquirement of envelope from the nuclear membranes were never detected. The fate of enveloped and unenveloped viral particles in the endoplasmic reticulum (Figure 8) and in the Golgi area (Figure 4) could be followed.
Controls
Immunostaining of MOCK cells (Figure 1F) and omission of the primary or secondary antibody in HSV-1-infected cells were all gD-1- and gC-1-negative.
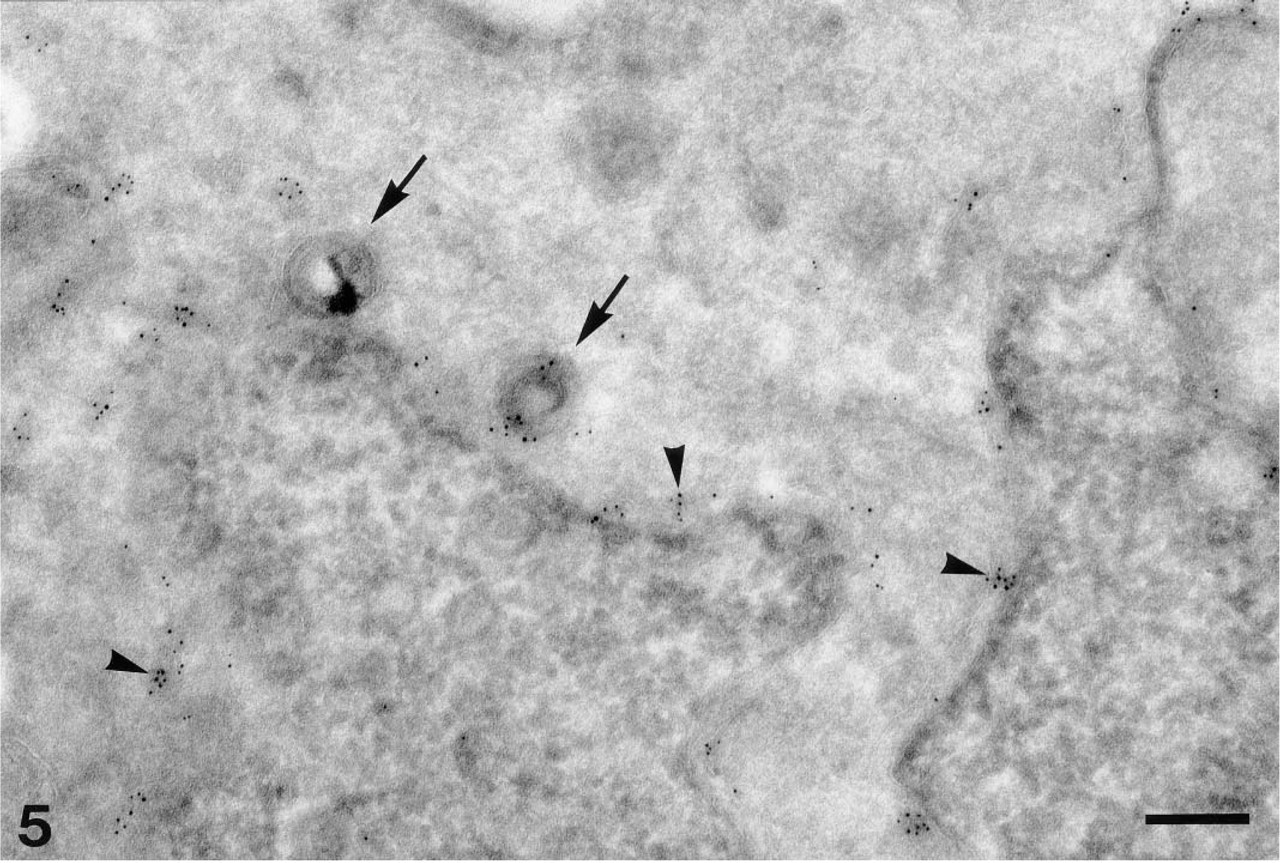
By optimized immunogold cryosection electron microscopy, it is possible with certainty to detect gD-1 (here Fd 138–80 and GAR-G5) in the nuclear envelope (arrowheads) and on the surface of enveloped viral particles situated in the perinuclear space (arrows). Bar = 0.25 μm.
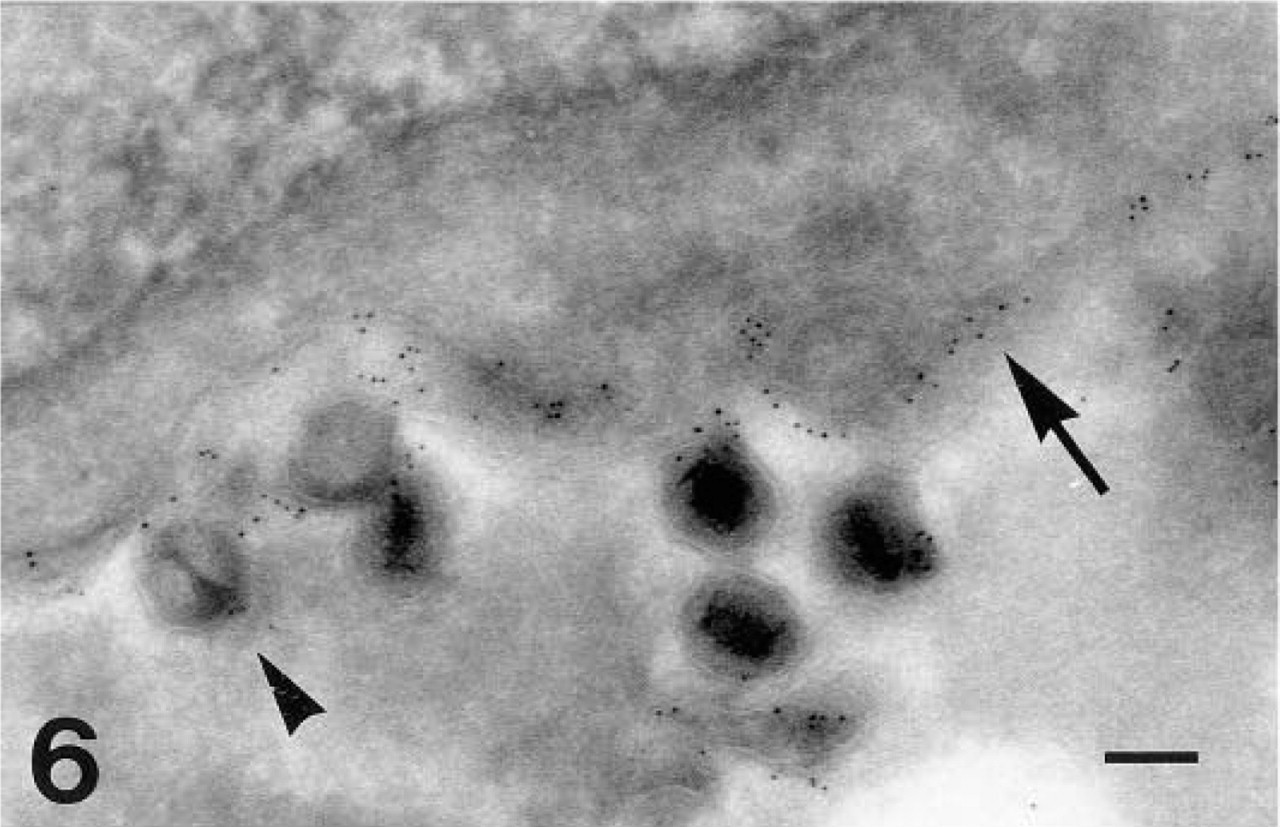
gC-1 labeling (GAR-G5) in HSV-1-infected fibroblasts. Arrow, plasma membrane; arrowhead, extracellular viral particle. Bar = 0.125 μm.
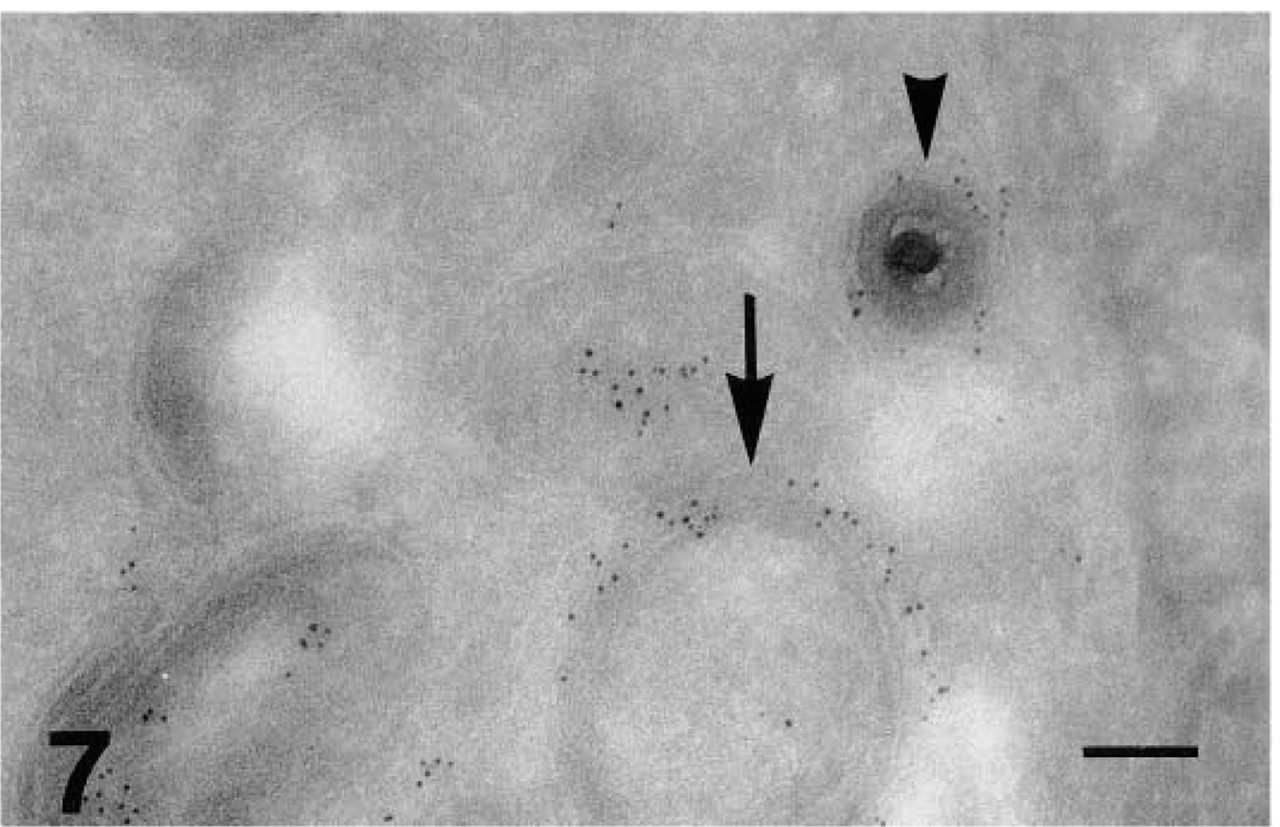
Glycoprotein C labeling (GAR-G5) in ultrathin cryosection of HSV-1-infected fibroblasts. Arrow, endoplasmic reticulum; arrowhead, labeled enveloped viral particles in the perinuclear space. Bar = 0.125 μm.
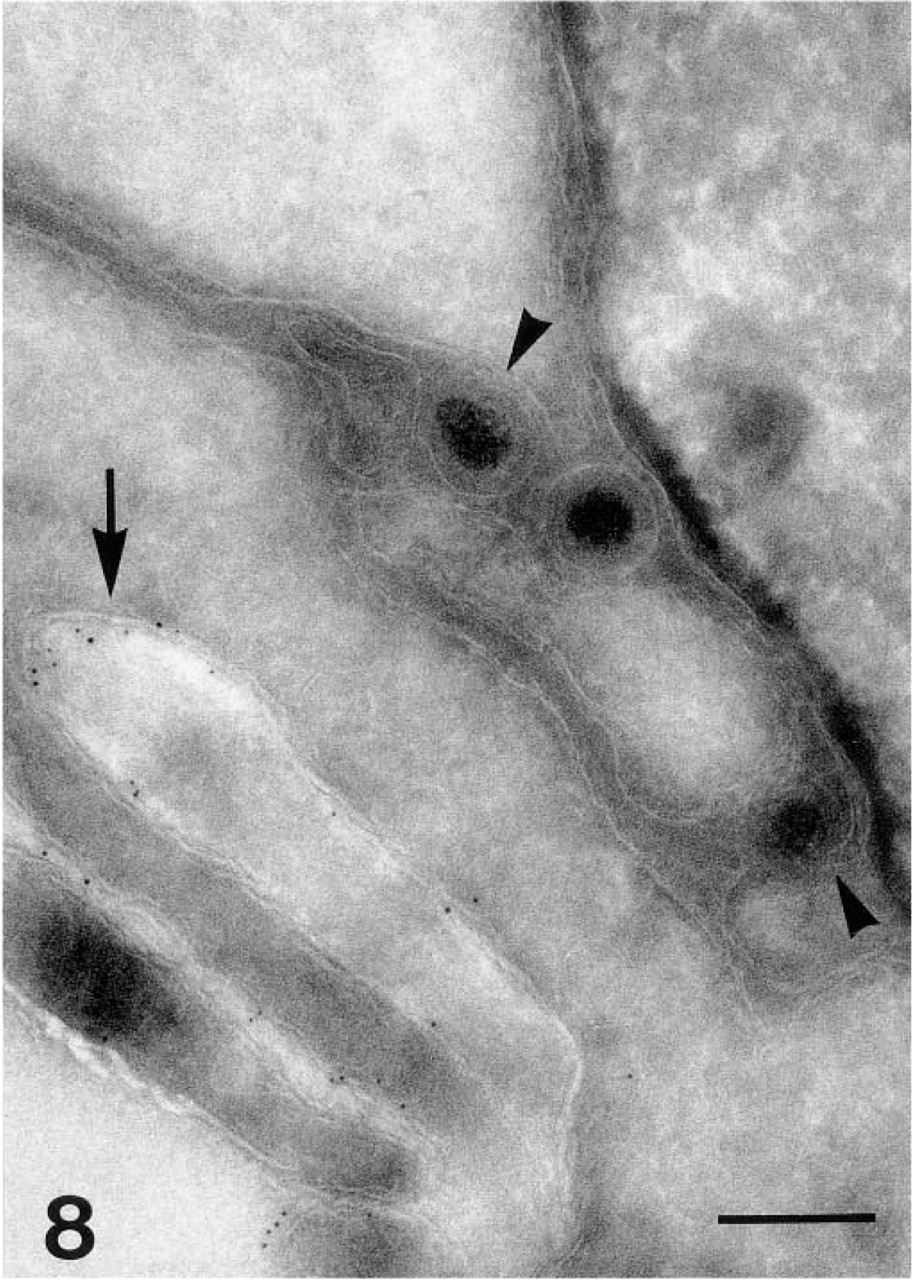
Viral particles in the endoplasmic reticulum (arrowheads). gC-1 is located on the cell surface (arrow). Bar = 0.25 μm.
Discussion
There are three essential problems in immunoelectron microscopy of Herpes simplex-virus-infected cells: (a) the number of labeled cells, because only minor areas are investigated; (b) the intensity of immunostaining; and (c) reproducibility of immunolabeling. An ultrathin section of 100 nm represents approximately 1% of the whole-cell antigenicity (calculated from Figure 2), which calls for special considerations. Moreover, the results can be influenced by the thickness of the sections, the infection of different cells is not synchronized, and even the most optimized procedure allows antigens to be destroyed to various degrees. Therefore, we do not find quantitative analysis of the labeling possible.
Previous immunogold labeling studies of HSV-1-infected human embryonic lung cells processed for ultrathin cryosections (Nielsen et al. 1989; Norrild et al. 1991) demonstrated the glycoproteins gC and gD in Golgi sacs, in membranes of cytoplasmic vesicles and nonmembranous parts of the cytoplasm, on the plasma membrane, and on the surface of intracellular and extracellular viral particles. However, these studies failed to identify viral glycoproteins on the nuclear membranes or in the perinuclear space; the viral particles in the perinuclear space or in cytoplasmic vesicles in close proximity to the outer nuclear membrane remained unlabeled. Complicated techniques with several buffers and many replacements of solutions run the risk for errors, and we find such methods unsuitable for obtaining the reproducible results needed in comparative studies with weak antigen presentation in ultrathin sections.
Ultrastructural studies, in particular, need fixation to preserve the cell structure and to immobilize soluble antigens. Unfortunately, fixatives tend to modify or abolish antigenicity by alteration of the primary, secondary, or tertiary structure of antigens. In accordance with Larsson (1993), we found 3% PFA with an added 2% GA preferable in terms of both morphology and antigenicity. The GA is an irreversible and the PFA a reversible crosslinking fixative (Hop-wood 1985; Larsson 1993). To avoid contaminants such as formic acid and methanol (Larsson 1993), the buffered formaldehyde solution was freshly prepared from paraformaldehyde powder. At fixation around pH 7.4, many primary amino groups are charged and are probably unreactive to formaldehyde (Larsson 1988). Because the speed of the fixation is a limiting factor, it is possible that diffusion of substances during the fixation occurs. The observed intense labeling of the plasma membrane and the lesser, intracellular staining of samples fixed with 8% PFA could be caused by insufficient penetration by the fixative, resulting in washout of the antigens. Moreover, glutaraldehyde fixation has been claimed to induce vesicles in the cytoplasms of fibroblasts (Hopwood 1985), which may have contributed to the observed morphological modifications of HSV-1-infected MRC-5 cells.
The advantages of colloidal gold markers at the electron microscopic level include the electron density, the stability, and the facts that it is easy to prepare in different and even small diameters and is easy to recognize (Larsson 1988; Van Bergen En Henegouwen 1989). The low steric hindrance of 1-nm gold particles and multiple gold particles linked to the immunoglobulin molecule results in increased labeling intensity (Leunissen et al. 1989). However, the immunoglobulin molecule is still large compared to both the 1-nm and the 5-nm gold particles (Larsson 1988; Leunissen et al. 1989), and the outcome of our studies by using these probes was the same. Although carefully performed, the size of silver-enhanced gold particles varied significantly and was impossible to standardize. Quantitation of silver-enhanced gold (Gilbert et al. 1994) and introduction of artifacts by intensification techniques should be avoided. We intensified the labeling signal by using a three-layer technique, being aware of and attentive to the risk for introducing unspecific staining by each additional layer. However, the precision (Larsson 1988) suffers as a consequence of both silver enhancement and three-layer intensification. The pH of the immunoreaction is important to the sensitivity and specificity (Larsson 1988). The statement by Larsson (1988) that it is absolutely necessary to pretreat sections with inert protein or serum before applying the primary antiserum is in direct contradiction to our experience. Unspecific binding was reduced by avoiding a blocking step, by decomplementation of the goat serum, by affinity-purification of RAM, and by careful washing steps.
One of the limitations of electron microscopy is sampling, wherein only a small proportion of the cells can be studied. The electron microscopic examinations revealed areas of cells with extraordinarily high levels of capsids near the nuclear membrane, viral particles in the perinuclear space, gD-1 labeling of the nuclear membranes, or well-preserved endoplasmic reticulum. In addition, there is cell-to-cell variation of the infection and in different levels of the sections, which makes it necessary to study many cells. By examining 200–300 cells at passages below 35 in each experiment, we obtained uniform results from the method used.
To the best of our knowledge, our observation by electron microscopy is the first demonstration of gD-1 in the nuclear membranes and on enveloped viral particles located in the perinuclear space of HSV-1-infected human fibroblasts by immunogold labeling of cryosections. Several conventional and immunoelectron microscopic studies on the morphogenesis of Herpes simplex-infected cells have been carried out over the past three decades. Surface epitopes have been examined by immunolabeling and negative staining of unembedded viral particles (Vernon et al. 1981; La Thangue et al. 1984; Stannard et al. 1987). Periodic acid–thiocar-bohydrazide–SO2 was used to identify unspecific glycoproteins in Epon-embedded samples (Lopez–Iglesias and Puvion–Dutilleul 1988a,b), resulting in occasional and very faint reaction of the nuclear membrane and the nearby enveloped nucleocapsids. Pre- and postembedding immunolabeling with ferritin or gold identified HSV polyclonal unspecified antisera or anti-gB polyclonal antibody in high (Epon)- or low (LR White or Lowicryl K4M)-temperature resin-embedded material stained the nuclear membranes (Nii et al. 1968b; Gilbert et al. 1994) but failed to react with anti-gD MAb (Komuro et al. 1989). The UL11 gene product, a protein presumed to facilitate capsid envelopment and translocation to the extracellular space, has also been shown to be associated with the inner nuclear membrane (Baines et al. 1995). Freeze-fractured, pre- embedding immunogold labeling of cells infected with either HSV-1 (F) or a recombinant lacking the UL20 gene revealed gB (polyclonal antibody) and gD (monoclonal antibody) in the outer and inner nuclear membranes and on the enveloped virions in the perinuclear space (Torrisi et al. 1992; Avitabile et al. 1994). Attempts to demonstrate the HSV-1 glycoproteins in the nuclear membranes of the host cells by immunolabeling of ultrathin cryosections have, up to the present study, been unsuccessful (Nielsen et al. 1989; Norrild et al. 1991), although weak reaction with an MAb against bovine Herpesvirus Type 2 was noted in HSV-1-infected Vero cells (Pietschmann et al. 1989).
In theory, the ability to identify discontinuous epitopes in ultrathin cryosections should be reduced compared to continuous epitopes. Thus far, we have observed only a quantitative but not a qualitative disparity between labeling with the antibody HDl11 to a discontinuous gD-1 epitope and the antibody DL6 to a continuous gD-1 epitope. The presence of HCl antigen in the nuclear membranes might be at a density below the level of detection with the reagents and the technique used, either because of destruction or because of minimal production or excessive release. It has previously been reported that envelopment occurs at the thickened or reduplicated nuclear membrane (Nii et al. 1968a). We studied thousands of infected cells, but envelopment by certain budding with ligation from the inner nuclear membrane was never detected. This suggests that the process of possible envelopment at the nuclear membrane is very rapid or, more likely, that too slow fixation allows the envelopment process to be completed before the cells are harvested. The exact pathway by which the virus enters and leaves the cell is still unclear but appears to exploit the normal metabolic reactions of the host cell. The fact that the majority of glycoproteins are identified in the plasma membrane, the enveloped HSV-1 viral particles, the endoplasmic reticulum, and especially the Golgi-associated membranes might be an indication that the envelopment, either wholly or in part, takes place in the extranuclear compartment.
Footnotes
Acknowledgements
Supported by grants from the Danish Cancer Society, The Foundation of Health Insurance “Danmark,” Erik Hørslev and wife Birgit Hørslev's Fund, the Else and Mogens Wedell–Wedellsborgs Fund, the DMA Research Fund, the Novo Nordisk Foundation, the Danish Foundation for Advancement of Medical Science, and the Aage Bang's Foundation.
We gratefully acknowledge the support of Prof O. Norén, Prof H. Sjöström, and Assoc Prof G.H. Hansen (University of Copenhagen, Denmark) in putting cryosection and electron microscopy facilities at our disposal. Generous donations of monoclonal antibodies DL6 and 1D3 were given by Dr R.J. Eisenberg and Dr G.H. Cohen (University of Pennsylvania, Philadelphia, PA), Fd 138–80 by Dr S. Chatterjee (University of Alabama, Birmingham, AL), and HCl and HD1 by Dr L. Pereira (University of California, San Francisco, CA). We would like to thank J. Grøndahl–Hansen, PhD, for affinity-purification of rabbit anti-mouse immunoglobulin. L.M. Leerhøj, N. Broholm, and L. Bæhr rendered excellent technical assistance.