
Abstract
The gliding motility of the protozoan parasite Toxoplasma gondii and its invasion of cells are powered by an actin–myosin motor. We have studied the spatial distribution and relationship between these two cytoskeleton proteins and calmodulin (CaM), the Ca2+-dependent protein involved in invasion by T. gondii. A 3D reconstruction using labeling and tomographic studies showed that actin was present as a V-like structure in the conoidal part of the parasite. The myosin distribution overlapped that of actin, and CaM was concentrated at the center of the apical pole. We demonstrated that the actomyosin network, CaM, and myosin light-chain kinases are confined to the apical pole of the T. gondii tachyzoite. MLCK could act as an intermediate molecule between CaM and the cytoskeleton proteins. We have developed a model of the organization of the actomyosin-CaM complex and the steps of a signaling pathway for parasite motility.
T
As we have recently demonstrated (Pezzella et al. 1997b), invasion by T. gondii is calcium-dependent. Calcium channel blockers and calmodulin antagonists significantly reduce the invasion index. The ubiquitous regulatory protein calmodulin plays an important role in processing calcium signals. Ca–CaM complexes bind to and regulate many functions, including cell secretion, cell motility, the organization of the cytoskeleton, and the activation of enzymes. Calmodulin is present in T. gondii (Pezzella et al. 1997a, b) and may be involved in cytoskeletal movement and conoid extrusion. However, unlike actin (Cintra and de Souza 1985; Yasuda et al. 1988; Dobrowolski et al. 1997b), no pattern for CaM distribution has been reported. The functional significance of the distribution of CaM in relation to tachyzoite structures remains to be studied. Our recent work on CaM distribution during tachyzoite invasion of KB cells (human epidermoid carcinoma epithelium cell line 86103004; ECA CC, Salisbury, UK) showed that CaM accumulates at the apical pole of the parasite (Pezzella et al. 1997a, 1998). This indicates that tachyzoite calmodulin plays a part in cytoskeletal movements during invasion.
Materials and Methods
Parasites
Tachyzoites of the T. gondii RH strain were maintained by serial passages at 3–4-day intervals in a culture of THP1 cells (myelomonocytic cell line; American Type Culture Collection, Rockville, MD, non-adherent cells) in RPMI medium supplemented with 10% fetal calf serum, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Tachyzoites were removed when the THP1 cells were lysed, centrifuged at 500 × g for 15 min, and suspended in RPMI medium.
Cells
KB cells (human epidermoid carcinoma epithelium cell line 86103004) were grown in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum, 2 mM glutamine, and streptomycin–penicillin (100 μg/ml-100 U/ml) at 37C in an atmosphere of saturated 5% CO2/95% air. The cells were harvested by trypsination (0.05% trypsin–0.02% EDTA) and seeded at 5.105 cells/25-ml flask.
Infection of KB Cells
KB cells were plated on coverslips (104 cells) for 48 hr to obtain a subconfluent culture. The coverslips were then placed in 24-well microtiter plates (Nunc; Polylabo Block, Strasbourg, France) containing 1 ml RPMI per well. The monolayers of KB cells were then infected with washed tachyzoites with a cell:parasite ratio of 1:10 and were left in contact for 24–96 hr.
Fluorescent Immunolabeling of Actin
Extracellular tachyzoites were placed on glass slides. Intracellular tachyzoites in their KB host cells plated on coverslips were treated by permeabilizing the host cell membrane by immersion for 3 min in 0.1% Triton X-100 and then fixed in 3% paraformaldehyde in PBS, pH 7.2, for 30 min. The cells were first incubated for 30 min with PBS containing 0.2% gelatin, 3% BSA, and then with a rabbit polyclonal anti-actin (chicken back muscle) antibody (Chemicon; Euromedex, Souffelweyersheim, France) diluted 1:30 in the same buffer for 1 hr at room temperature (RT). Tested in Western blotting with tachyzoite homogenate, this antibody recognized particularly a band around 40 kD. The samples were then washed six times with PBS–gelatin-BSA and incubated with biotinylated donkey anti-rabbit IgG antibody (diluted 1:50 in the same buffer) for 1 hr at RT. Cells were washed three times in PBS–gelatin–BSA and three times in PBS and then incubated with streptavidin–Texas Red (diluted 1:50 in PBS) for 15 min in the dark, and washed again with PBS. Their nuclei were stained with 150 mM MgCl2 and 100 μM chromomycin A3 for 15 min in the dark, and finally with MgCl2. The cells were treated with Citifluor and examined by confocal microscopy (microscope Zeiss Axioplan, confocal part: MRC 600).
Fluorescent Immunolabeling of Myosin
Myosin was labeled in the same way as actin, except that the cells were first fixed using 3% paraformaldehyde before per-meabilization with 0.1% Triton X-100. The primary antibody was a rabbit polyclonal anti-(bovine uterus smooth muscle) myosin antibody (diluted 1:50) (Valbiotech; Paris, France). This antibody recognized particularly two bands at 120 kD and 110 kD and a more intense band at 90 kD when tested by Western blotting with a tachyzoite homogenate. These seem to correspond to TgM-C, TgM-B, and TgM-A.
Fluorescent Immunolabeling of Calmodulin
This experiment was performed using the myosin protocol, except that the primary antibody was a mouse monoclonal anti-(Dictyostelium, bovine, rat, and chicken) calmodulin antibody (Tebu; Le Perray en Yvelines, France) diluted 1:80 (Amersham; Les Ulis, France) and the secondary antibody was a biotinylated sheep anti-mouse IgG antibody diluted 1:50 (Amersham).
Fluorescent Immunolabeling of Myosin Light-chain Kinases
MLCK was labeled according to the CaM labeling protocol. The primary antibody was a mouse monoclonal anti-chicken MLCK antibody (Sigma; St Quentin Fallavier, France) diluted 1:50 and the secondary antibody was a biotinylated sheep anti-mouse IgG antibody diluted 1:50 (Amersham.
Controls without the primary antibody were prepared in four experiments.
Double Calmodulin–Actin Immunolabeling in Extracellular Tachyzoites. The main problem with this experiment was to balance the detergent extraction before fixation (for the cytoskeleton protein) against calmodulin removal (calmodulin being a soluble protein). We used extraction for 1 min before fixation to keep the CaM in place. We first labeled the CaM using the protocol used for CaM immunodetection alone (see above). Actin was then labeled by incubating the samples with rabbit polyclonal anti-actin (diluted 1:30), then with a digoxigenin sheep anti-rabbit IgG F(ab′)2 fragment (diluted 1:100) for 1 hr, and finally with a fluorescein sheep anti-digoxigenin Fab fragment (diluted:150) for 30 min.
Double Calmodulin–Myosin Immunolabeling in Extracellular Tachyzoites. Cells were fixed with 3% paraformaldehyde in PBS, permeabilized with 0.1% Triton X-100, and CaM was immunolabeled, followed by myosin. Labeling was performed according to the CaM–actin double-labeling protocol.
Confocal Microscopy, Optical Sections, and 3D Reconstruction
Specimens were examined with an MRC 600 (BioRad; Richmond, CA) confocal laser scanning microscope equipped with two lasers (argon and helium–neon) mounted on a Zeiss Axioplan microscope (Zeiss; Thornwood, NY). The Texas red fluorophore was visualized with the 543-nm excitation wavelength of a helium–neon laser and the chromomycin A3 with the 457-nm excitation wavelength of an argon laser. Images were preprocessed with the Comos software package (BioRad) to increase the contrast and to merge the two labelings. Photomicrographs of the double labeling (protein–nucleus) were obtained by direct visualization, which corresponds to the fluorescence emission from one plane of the object. Serial optical sections (around 20) (Z-series) were processed on specimens at 0.2-μm steps. Three-dimensional reconstruction was performed by converting the 2D images of a Z-series into a volume in which we could make virtual cuts of the reconstructed labeling. The 3D reconstruction was performed with Analyze software (Analyze Mayo Bir; CN Software, Southwater, West Sussex, UK) on a Sun Microsystems workstation (Sun Microsystems; Velizy, France). This software makes it possible to move the 3D-reconstructed objects around and to present the most informative viewing angles.
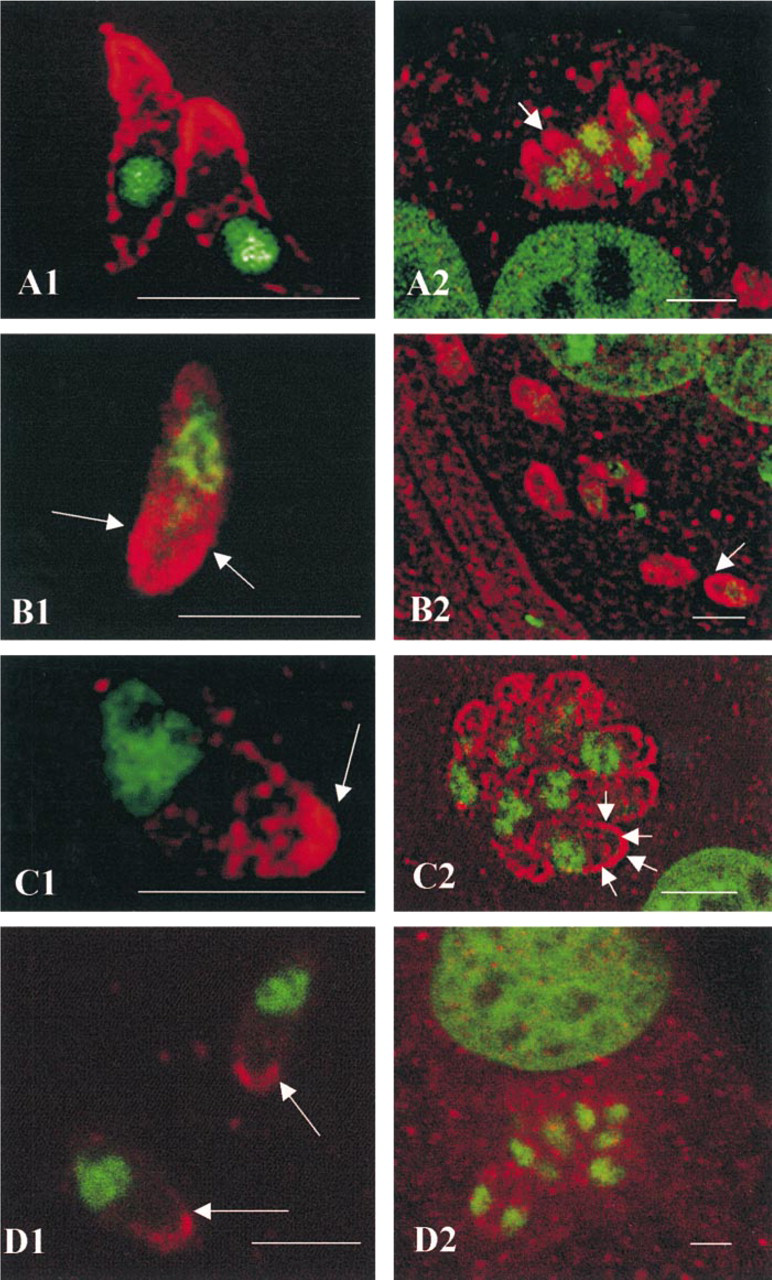
Immunolocalization of actin, myosin, calmodulin, and MLCK by confocal microscopy. (
Results
Localization of Actin, Myosin, Calmodulin, and Myosin Light-chain Kinases of T. gondii
Nuclei stained with chromomycin A3 appeared as green fluorescence. The apical pole of the parasite was at the opposite end from the nucleus.
Actin. Actin was mainly located in the anterior third of the parasite, with a circumferential pattern beneath the parasite membrane complex. The labeling at the apical end appeared to lie under the membrane with a specific V structure (Figure 1A1). The intracellular parasites had similar staining at their apical end (Figure 1A2).
Myosin. Extracellular T. gondii were diffusely stained with the rabbit polyclonal anti-myosin antibody in the broad anterior third of the parasite, with the greatest intensity at the periphery of the conoid (Figure 1B1). Intracellular parasites (Figure 1B2) had a similar myosin distribution.
Calmodulin. Most of the CaM immunostaining was at the pole opposite the green nucleus, the apical end (Figure 1C1). The CaM in intracellular tachyzoites (Figure 1C2) was always prominent at the apical end, but was also distributed beneath the length of the membrane complex of the parasites (arrowed).
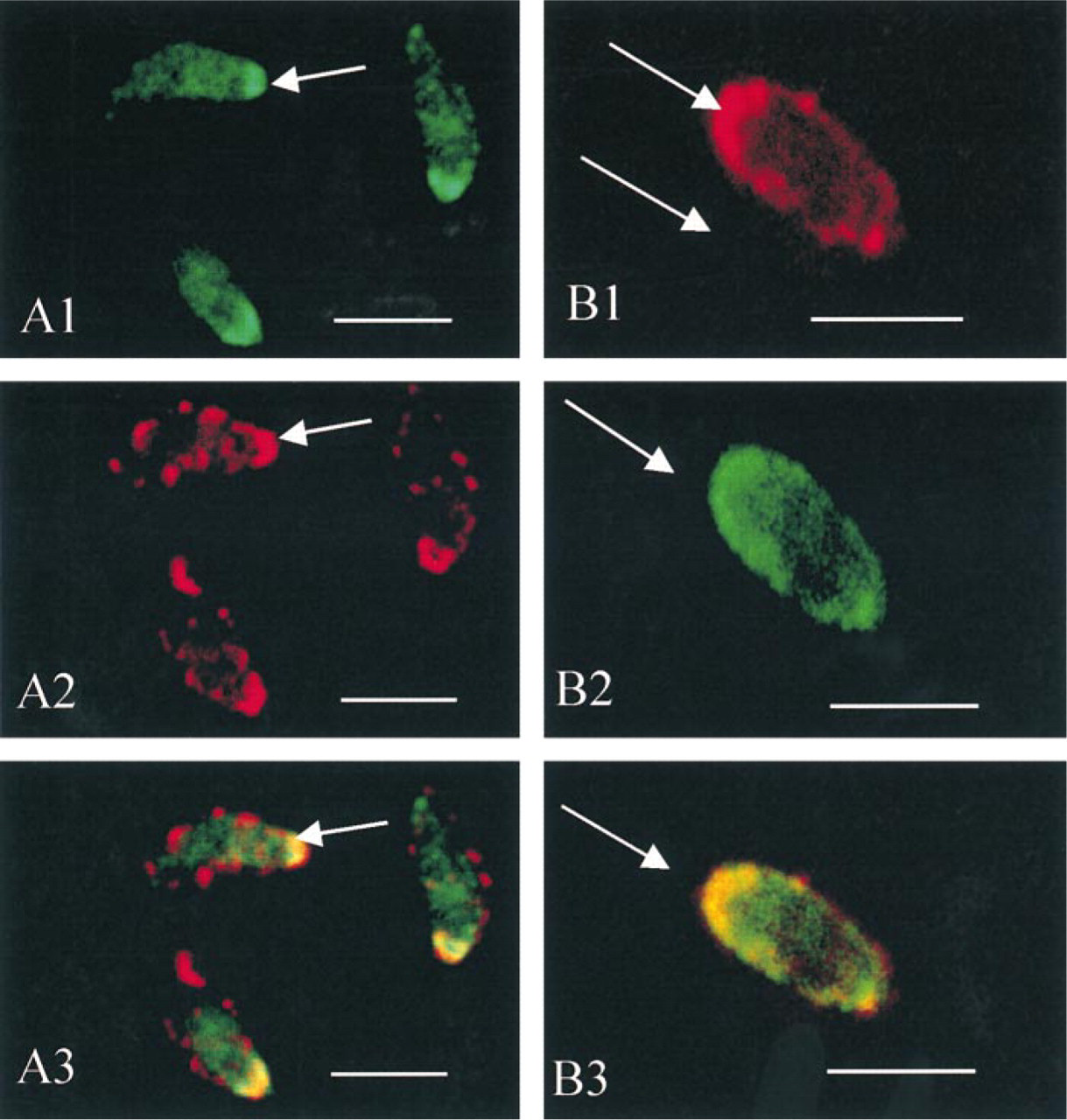
Double immunolabeling of actin (
Myosin Light-chain Kinases. The red fluorescence was less intense but essentially at the apical pole of the extracellular parasite; it was redistributed in the cytosol of the intracellular parasites (Figures 1D1 and 1D2).
Examination of the controls (without primary antibody) showed no labeling, except for the nuclei stained with chromomycin A3.
Extracellular parasites stained with antibodies to actin (Figure 2A1) and CaM (Figure 2A2) showed both CaM and actin at the anterior pole of the tachyzoites. The specific V structure seen in the single labeling was not observed, probably because of the treatment used for the double immunolabeling (the rapid extraction before fixation needed to keep CaM in place made it impossible to detect the V structure).
Fig 2B shows the labeling for myosin (Figure 2B1) and CaM (Figure 2B2), with both proteins at the apical end of the extracellular parasites.
Superimposition of the two images (Figures 2A3 and 2B3) showed a co-localization (actin–calmodulin, myosin–calmodulin) which appeared in yellow fluorescence at the apical extremity of the parasites.
3D Reconstruction of Optical Sections and Visualization of the Labeling
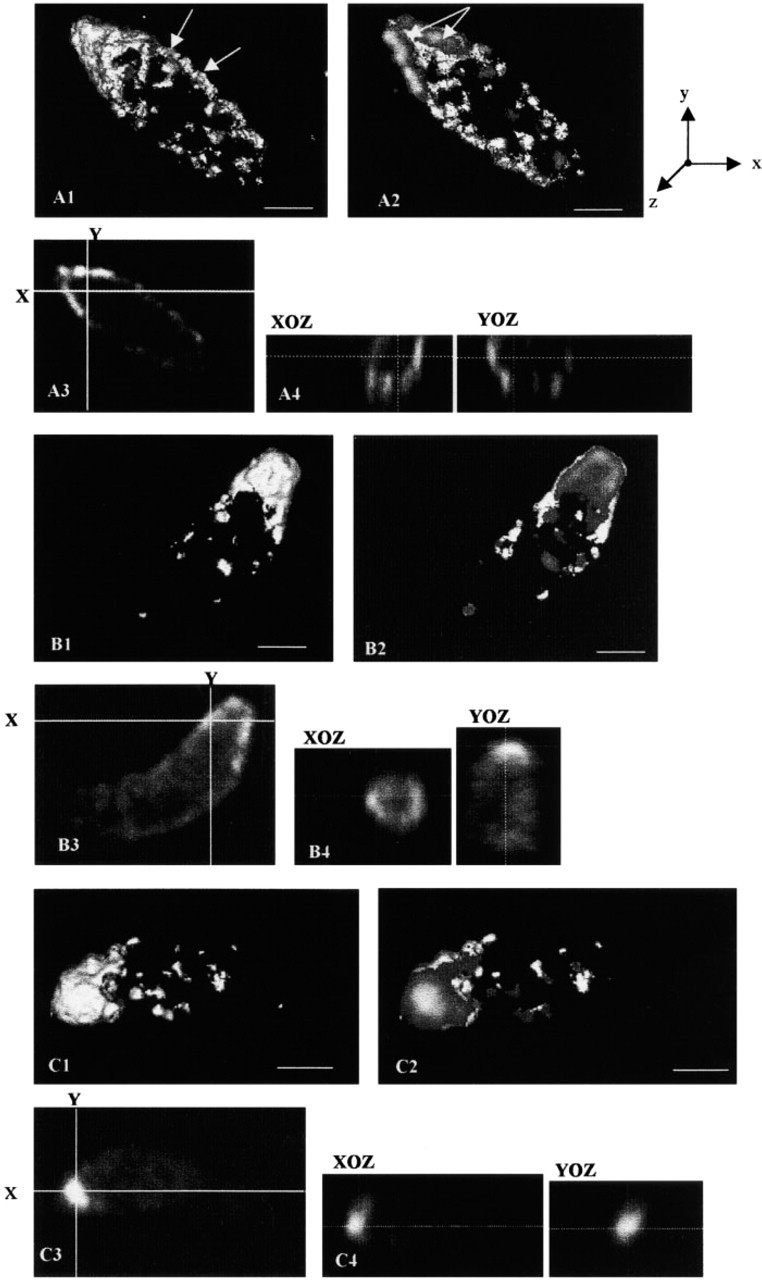
The optical sections of a Z-series were reconstructed to form the volume of the labeled object. The numbers in Figure 3 give an overall view of the reconstructed labeling (1), and a slice (2) in the volume to show the internal part of the object labeling. Orthosections were prepared (4) from a single optical section (3) according to X and Y axes.
The 3D reconstruction of the actin labeling in the extracellular parasites (Figure 3A1) confirmed the apical concentration of this protein, as well as the circumferential actin distribution over the entire tachyzoite with “bow structures” (arrows). The apical end of the parasite appeared to be “hooded” by this cytoskeleton protein. A slice through the object (Figure 3A2) revealed that most of the apical actin labeling was in the periphery, with the specific V structure (arrows), without inner labeling. We conclude that T. gondii actin lies below the membrane. The single optical section (Figure 3A3) and orthosections in the anterior pole along the X and Y axes (Figure 3A4) gave the same circumferential distribution of actin (as a ring) with punctate staining.
The overall 3D visualization of myosin (Figure 3B1) suggested that it was concentrated at the anterior pole of the parasite with a more diffuse pattern. A slice (Figure 3B2) showed differences in the gray scale labeling of intensity in the apical staining, indicating that myosin is distributed along a concentration gradient, with pronounced labeling at the periphery (light gray scale) and weaker labeling in the center (dark gray scale). Orthosections confirmed the diffuse labeling of myosin with a circumferential accumulation (Figures 3B3 and 3B4)
Calmodulin (Figure 3C1) was concentrated in the apical end of the parasite. A slice through the global volume (Figure 3C2) showed that the apical CaM labeling was compact and entire. The resulting CaM concentration gradient showed most CaM in the center, inside the apical end (light gray scale) and less towards the outside. This was supported by the X–Y orthosections showing CaM staining in the shape of an intense full sphere (Figures 3C3 and 3C4).
Discussion
The actomyosin system generates biochemical forces for cell movement and cytokinesis. The motility of T. gondii is critically dependent on actin filaments and is driven by a myosin motor. Previous studies using heterologous antisera or actin-specific antibodies have indicated that actin is essentially confined to the apical end of the T. gondii tachyzoite (Cintra and de Souza 1985; Endo et al. 1988; Yasuda et al. 1988). It also occurs faintly throughout the cytoplasm and is more intense at the perimeter (Dobrowolski et al. 1997b). Three-dimensional reconstruction of labeled tachyzoites showed a discontinuous submembranous distribution of the actin throughout the extracellular parasite, with a pronounced accumulation at the apical end where actin formed a “cap.” There is an intense apical V structure of actin. Some investigations of T. gondii actin have given controversial results. Invasion by T. gondii is inhibited by cytochalasins, which disrupt actin filaments (Dobrowolski and Sibley 1996). However, no microfilaments have been detected in extra- or intracellular parasites (Russel and Sinden 1981; Cintra and de Souza 1985). This suggests that T. gondii actin exists primarily in a globular form (Dobrowolski et al. 1997b). DNase I staining of extracellular tachyzoites revealed G-actin at the apical end, beneath the membrane complex, and in punctate staining scattered throughout the cytoplasm. This is more pronounced in intracellular parasites (Pezzella et al. 1997a, 1998). An actin depolymerization factor (ADF) located beneath the plasma membrane was not found at the apical end of T. gondii (Allen et al. 1997). The depolymerization of actin may be very rapid, and F-actin might be transient. T. gondii may contain actin-binding proteins that sequester monomeric actin and control the assembly of actin filaments, such as toxofilin (Poupel et al. 2000). The organization of actin filaments in cells, as well as their changes in response to [Ca2+]i and other intracellular signals, depends on the interactions of various actin-binding proteins. Such proteins may also determine where in the cell myosin can attach to actin filaments. Five myosins have been identified in T. gondii, three around 90 kD (including TgM-A), one at 114 kD (TgM-B), and one at 125 kD (TgM-C). These three unconventional myosins described by Heintzelman and Schwartzman (1997, 1999), have the head domain responsible for generating mechanochemical forces along actin filaments using energy derived from ATP hydrolysis. These myosins have sequences similar to those of Plasmodium falciparum myosin, and they are both concentrated in the apical part of the parasite (Pinder et al. 1998). TgM-A is concentrated in the apical pole, TgM-C is present in the juxtanuclear region towards this apical pole, and the other unconventional myosins have not been yet localized (Heintzelman and Schwartzman 1999). Our 3D reconstruction of the myosin labeling shows a more diffuse pattern than for actin, with a concentration in the anterior third of the tachyzoite and greater intensity at the periphery of the conoid (confirmed by the orthosections). We also find an actin–myosin network inside the apical pole, with myosin overlapping the distribution of the actin, which is essentially peripheral, with a V form. One of the proteins associated with the cytoskeleton system, MLCK, stimulates the contractile activity of myosin at high [Ca2+] in a CaM-dependent fashion. The primary task of MLCK is to phosphorylate the 20-kD light chain of myosin (shown for a subset of the myosin IIs) and to regulate the actin–myosin crossbridge cycling. According to Heintzelman and Schwartzman (1997), whereas TgM-A has no light chain binding domain, TgM-B and TgM-C have a single well-conserved IQ motif, that indicates a putative light chain-binding site. Because MLCK is present in the conoid and because KT5926 clearly inhibits both the motility and invasion of T. gondii, then MLCK might be the link between the actomyosin motor and CaM, the universal calcium-dependent regulator, in the tachyzoite. The distribution of CaM reflects its function. CaM resides in the locomotion organelles of the ciliates Tetrahymena (Schultz et al. 1983), Paramecium (Momayesi et al. 1986), and the flagellates Trypanosoma rhodesiense and T. congolense (Ruben et al. 1984). As in P. falciparum merozoites (Scheibel et al. 1987), CaM is confined to the apical pole of the T. gondii tachyzoite. This suggests that T. gondii calmodulin is mobilized during parasite invasion.
3D fluorescence reconstruction of actin, myosin and calmodulin. (
(
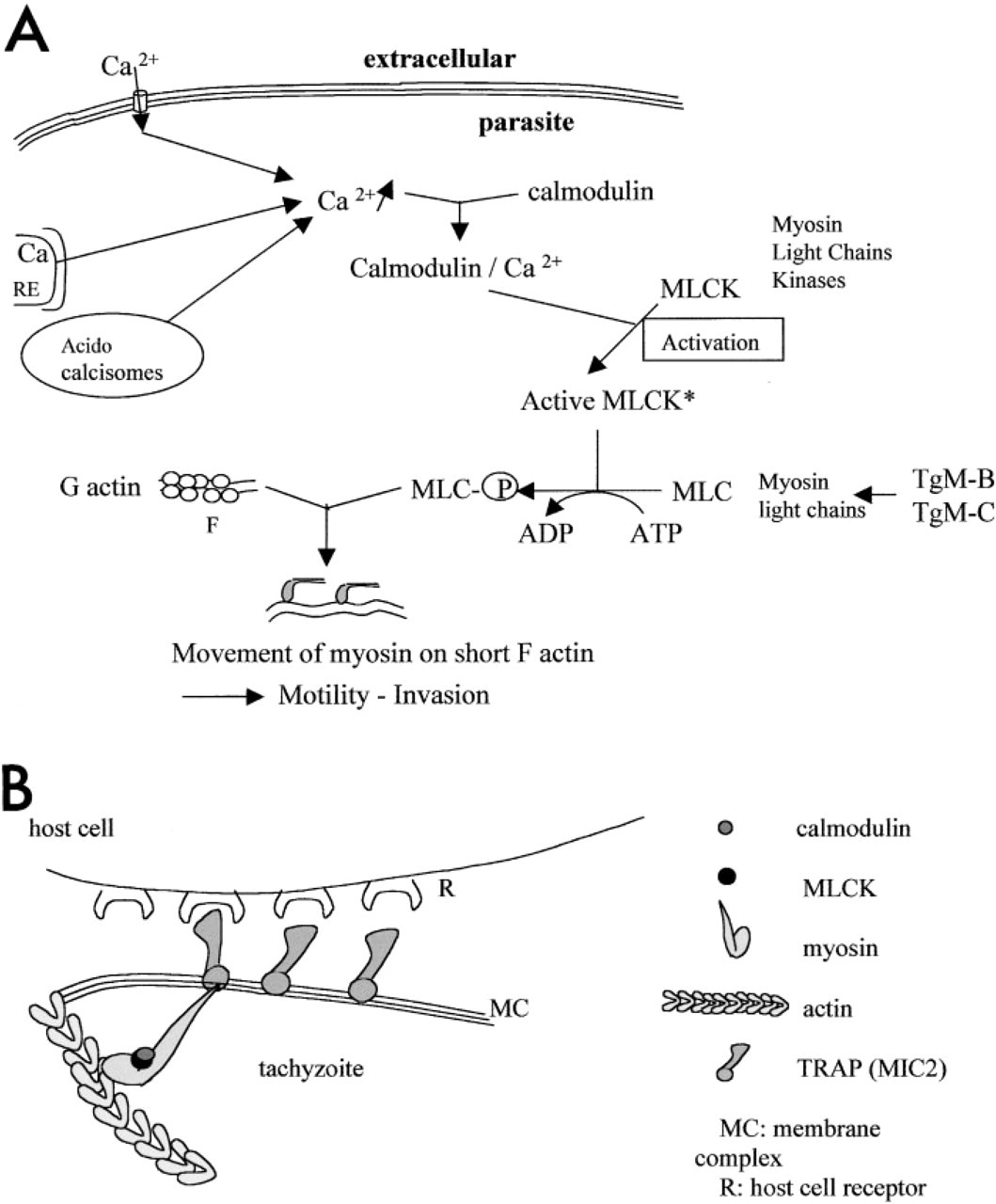
The CaM inhibitors calmidazolium and trifluoperazine significantly reduce the parasite invasion index in vitro (Pezzella et al. 1997b) and prompt tachyzoites to undergo shape changes. However, the distribution of CaM is not significantly modified, suggesting that the production of CaM by T. gondii is not affected. Our results also reveal a redistribution of the T. gondii CaM from extracellular to intracellular, CaM being redistributed from its normal apical location into a circumferential pattern. Changes in CaM distribution in other parasitic protozoans are associated with their metabolite state as dense granular secretions, a Ca–CaM-dependent process. This is found in Entamoeba histolytica during invasion (De Lourdes–Munoz et al. 1992) and during excystation of Giardia lamblia (Bernal et al. 1998). Changes in CaM distribution can be linked to the secretory state in T. gondii. We suggest that CaM regulates the conoid extrusion (a Ca2+-dependent process; Mondragon and Frixione 1996) and also the secretion of lytic enzymes from rhoptries (the exocytosis of which is Ca2+-dependent; Lycke et al. 1975) such as PLA2 (Saffer et al. 1989; Gomez–Marin et al. 1996) to fluidize the host cell plasma membrane when the extracellular tachyzoites attach to their host cell. Second, the redistribution of CaM in a circumferential pattern when T. gondii has entered the host cell could be implicated in the regulation of dense granule secretion (like GRA3, a Ca-dependent protein). Third, when T. gondii attaches to its host cell, particularly via microneme secretion (such as MIC2, a transmembrane protein of the TRAP family adhesins; Sibley et al. 1998), myosins (TgM-B, TgM-C) might become associated with this protein and might be activated by phosphorylation of the light chains by a CaM-dependent MLCK. KT5926 also reduces the attachment to host cells, particularly by blocking MIC secretion (Dobrowolski et al. 1997a). Activated myosin could move along the newly formed actin filament. The actin–myosin motor and the free energy of actin polymerization would provide sufficient driving force for conoid extrusion and then for the penetration of the parasite into the host cell. We suggest that a series of signals (Figure 4A) could lead to the activation of the actomyosin system via Ca2+ and calmodulin, and we have developed a model (Figure 4B) of the organization of cytoskeletal actomyosin and calmodulin in the apical part of the parasite when it is attached to its host cell.
Footnotes
Acknowledgements
We thank Hervé Kaplan and Francis Deligny for technical assistance and Dr Owen Parkes for revising the English text.