
Abstract
The relative insensitivity of nonradioactive mRNA detection in tissue sections compared to the sensitive nonradioactive detection of single-copy DNA sequences in chromosome spreads, or of mRNA sequences in whole-mount samples, has remained a puzzling issue. Because of the biological significance of sensitive in situ mRNA detection in conjunction with high spatial resolution, we developed a nonradioactive in situ hybridization (ISH) protocol for detection of mRNA sequences in sections. The procedure is essentially based on the whole-mount ISH procedure and is at least equally sensitive. Increase of the hybridization temperature to 70C while maintaining stringency of hybridization by adaptation of the salt concentration significantly improved the sensitivity and made the procedure more sensitive than the conventional radioactive procedure. Thicker sections, which were no improvement using conventional radioactive ISH protocols, further enhanced signal. Higher hybridization temperatures apparently permit better tissue penetration of the probe. Application of this highly reliable protocol permitted the identification and localization of the cells in the developing heart that express low-abundance mRNAs of different members of the Iroquois homeobox gene family that are supposedly involved in cardiac patterning. The radioactive ISH procedure scarcely permitted detection of these sequences, underscoring the value of this novel method.
T
Nonradioactive ISH is generally believed to be less sensitive than the radioactive procedure. A number of recent studies challenge this view (van Gijlswijk et al. 1997; Hopman et al. 1998; Speel et al. 1998; Yang et al. 1999). These studies have introduced a novel detection method based on the peroxidase-mediated deposition of haptenized or fluorochromized tyramide, dubbed catalyzed reporter deposition (CARD), or tyramide signal amplification (TSA) (Bobrow et al. 1989, 1992; Speel et al. 1997). It remains nevertheless incomprehensible that a few kilobases of DNA sequences can easily be detected on metaphase chromosomes without tyramide amplification (Wiegant et al. 1991; Korenberg et al. 1995), whereas detection of RNA sequences would require considerable amplification which, in turn, is potentially prone to artifacts. It also appears illogical that rare mRNA sequences are more easily detectable by whole-mount ISH, without the need for advanced amplification systems, than by ISH in sections. Apparently the whole-mount ISH protocol permits a more sensitive detection of mRNA sequences than the section ISH protocol. We therefore set out to adapt the whole-mount ISH protocol to make it suitable for ISH on sections. Application of this sensitive, rapid, and robust protocol on mouse embryo sections has allowed visualization of the expression of a series of Iroquois homeobox genes with high spatial resolution, permitting differentiation of expression in endocardium, cardiac cushions, epicardium, conduction system, and myocardium.
Although the protocol has been specifically described for application on mouse embryos, the procedure worked equally well on sections of chicken embryos, rat liver, and human aorta (data not shown), and is likely to work on other tissues of different species as well.
Materials and Methods
Tissue Processing
Detailed practical protocols for fixation, paraffin embedding, mounting, and sectioning have been described recently (Moorman et al. 2000). In short, embryos from FVB mice or Wistar rats were obtained from timed-pregnant animals. For ISH on sections, embryos were fixed for 4 hr to overnight in freshly prepared 4% formaldehyde in PBS by rocking at 4C. Embryos were dehydrated in a graded ethanol series, paraffin-embedded, cut into sections, and carefully mounted on aminoalkylsilane-coated slides to prevent loss of the tissue sections during the extensive treatments of the ISH procedure. The coating deserves special attention for the nonradioactive ISH on sections, because the sections are easily lost owing to the use of higher temperatures.
For whole-mount ISH, embryos were briefly washed in PBS/0.1% Tween-20 (PBST) and fixed by rocking for 4 hr to overnight in freshly prepared 4% formaldehyde/PBS at 4C. Embryos were dehydrated in a graded methanol series in PBST and stored in 100% methanol until use.
RNA Probes and Probe Specification
35S-labeled probes were made by in vitro transcription, followed by alkaline hydrolysis to yield fragments with a mean size of 100–150 nucleotides as assessed by polyacrylamide gelelectrophoresis (Moorman et al. 2000). Digoxigenin-labeled probes were made according to the manufacturer's specifications (Roche; Mannheim, Germany) and were not subjected to alkaline hydrolysis because this treatment had no or few adverse effects on the sensitivity of hybridization. RNA probes complementary to the mRNAs encoding SERCA2a (Moorman et al. 1995), myosin light chain (MLC) 2v (O'Brien et al. 1993), β-myosin heavy chain (MHC) (Boheler et al. 1992), atrial natriuretic factor (ANF) (Zeller et al. 1987), and Iroquois (Irx) homeobox transcription factors 1–5 (Christoffels et al. 2000), were used.
In Situ Hybridization
Radioactive In Situ Hybridization. The radioactive ISH detection of mRNA sequences has been extensively reviewed elsewhere (Moorman et al. 2000). After removal of paraffin, sections were pretreated for 10 min at 70C in 2 × SSC (1 × SSC = 0.15 M NaCl, 15 mM sodium citrate, pH 7.2), washed for 5 min in double-distilled water, digested for about 10 min, depending on the tissue used, at 37C in 0.1% pepsin dissolved in 0.01 N HCl, rinsed two times for 30 sec in 0.2% glycine/PBS, followed by two washes with double-distilled water for 30 sec and 5 min, respectively, and incubated for 5 min in 10 mM dithiothreitol to reduce nonspecific binding of 35S-labeled probe to the sections.
Sections were allowed to hybridize overnight at 54C in 50% formamide, 10% dextran sulfate, 2 × SSC, 2 × Denhardt's [1 × Denhardt's = 0.02% Ficoll 400, 0.02% polyvinylpyrrolidone, and 0.02% bovine serum albumin (Fraction V)], 0.1% Triton X-100, 50 mM dithiothreitol, and 200 ng/μl salmon sperm DNA. Probe concentration was about 25 pg/μl with a specific activity of 1700 cpm/pg. Approximately 6 μl hybridization mix was applied to the sections. No coverslips were used to improve efficiency of hybridization and to allow easy comparison of serial sections incubated with different probes and controls.
After hybridization, sections were successively washed in 1 × SSC at room temperature (RT), twice in 50% formamide/1 × SSC at 54C for 15 min, and in 1 × SSC for 15 min at RT, followed by an RNase treatment (10 μg RNase/ml) for 30 min at 37C, and additional washes in 1 × SSC and in 0.1 × SSC for 15 min at RT. Sections were dried, dipped in autoradiographic emulsion (Ilford Nuclear Research Emulsion G-5), and exposed for 7–14 days. After development of the photographic emulsion, sections were counterstained with nuclear fast red and mounted in Malinol. In this procedure, the autoradiographic signal is linearly related to the radioactivity present in the sections for a fixed time of exposure and time of development (Jonker et al. 1997).
Nonradioactive In Situ Hybridization. At variance with the radioactive ISH protocol, the pretreatment of the sections consisted only of proteolytic digestion for 5–15 min at 37C with 20 μg/ml proteinase K dissolved in PBS, followed by a 5-min rinse in 0.2%glycine/PBS and two rinses of 5 min in PBS. Sections were then re-fixed for 20 min in 4% formaldehyde/0.2% glutaraldehyde dissolved in PBS to ensure firm attachment of the sections to the microscope slides, and washed twice in PBS for 5 min.
Sections were pre-hybridized in hybridization mix without probe for 1 hr at 70C and then hybridized overnight at 70C. Hybridization mix is composed of 50% formamide, 5 × SSC, 1% block solution (Roche), 5 mM EDTA, 0.1% Tween-20, 0.1% Chaps (Sigma; St. Louis, MO), 0.1 mg/ml heparin (Becton-Dickinson; Mountain View, CA), and 1 mg/ml yeast total RNA (Roche). Probe concentration was about 1 ng/μl. Approximately 6 μl hybridization mix was applied to the sections and no coverslips were used. After hybridization sections were rinsed in 2 × SSC, pH 4.5, washed three times for 30 min at 65C in 50% formamide/2 × SSC, pH 4.5, followed by three 5-min washes in PBST. Probe bound to the section was immunologically detected using sheep anti-digoxigenin Fab fragment covalently coupled to alkaline phosphatase and NBT/BCIP as chromogenic substrate, essentially according to the manufacturer's protocol (Roche). For tyramide signal amplification, the NEN (Boston, MA) Renaissance TSA kit was used. Sections were washed with double-distilled water, dehydrated in a graded ethanol series and xylene, and embedded in Entellan.
Whole-mount In Situ Hybridization. A slightly modified procedure adapted from Wilkinson (1992) and Riddle et al. (1993), was used. Before the procedure, the brains of the embryos were punctured to avoid probe trapping. Embryos were rehydrated in a graded methanol series in PBST and digested for 15–30 min with 10 μg/ml proteinase K dissolved in PBST, followed by a 5-min rinse in 0.2%, glycine/PBST and two rinses of 5 min in PBST. Postfixation was for 20 min in 4% formaldehyde, 0.2% glutaraldehyde dissolved in PBST. Prehybridization and hybridization were performed as described in the nonradioactive ISH protocol (see above). Probe concentration was about 1 ng/μl. After hybridization, embryos were rinsed a few times with 50% formamide, 2 × SSC, 0.1% Tween-20, pH 4.5, followed by three washes with this solution for 30 min at 65C. Embryos were then washed three times for 30 min at 65C in 50% formamide, 2 × SSC, pH 4.5, followed by three 5-min washes in PBST at RT. Probe bound to the embryos was immunologically detected as described above.
Photography
Images were taken using a Photometrix CCD camera or Nikon color camera attached to a Zeiss Axiophot microscope, and digitized files were recorded.
Results
This study was initiated to explore whether adaptation of the whole-mount ISH protocol for use on tissue sections would result in an equally sensitive procedure. This would yield a procedure for the detection of mRNA sequences with superior sensitivity and spatial resolution to the method using radioactively labeled probes.
Sensitivity of Whole-mount ISH Compared to Radioactive ISH on Sections
Recently, we described the expression of five mRNAs encoding Iroquois homeobox-containing transcription factors 1–5 during cardiac development (Christoffels et al. 2000). As measured by quantitative PCR experiments in the mouse embryo, these sequences are about 1000-fold less abundant than the so-called housekeeping genes encoding glyceraldehyde-3-phosphate dehydrogenase, elongation factor 1α, or hypoxanthine phosphoribosyl transferase (data not shown). The spatial distribution was initially assessed by whole-mount ISH only, because hybridization on sections using 35S-labeled probes was not sensitive enough to unambiguously determine the cellular localization. Figure 1 shows an example of how signals obtained by whole-mount ISH relate to those obtained using the radioactive ISH protocol. Expression of the low-abundance Irx4 and Irx3 mRNA and of ANF mRNA, used as positive control, can easily be detected by the whole-mount ISH procedure. The radioactive ISH displayed weak signal of Irx4 mRNA, whereas Irx3 and the other Irx mRNAs (data not shown) could not be detected in the heart, indicating the superior sensitivity of the whole-mount ISH procedure. ANF mRNA in the heart and Irx3 mRNA in the endothelial lining of the lung could easily be detected in the radioactive procedure, displaying the correct pattern of expression, indicating that specificity is being achieved.
Stringency and Temperature
In both the whole-mount and radioactive ISH procedures, hybridization is performed at a similar stringency. However, in the whole-mount procedure this stringency is achieved by using a relatively high temperature, whereas in the radioactive ISH protocol it is achieved by a relatively low salt concentration. In both procedures, 50% formamide is used. We reasoned that temperature might have a separate effect on the tissue as well, by increasing the accessibility of the tissue for probe. We therefore compared two conditions of hybridization on sections, in which a similar stringency of 10C below the Tm is achieved either by hybridization at 58C in 1 × SSC or by hybridization at 70C in 5 × SSC (Figure 2). Sections hybridized at 70C clearly displayed a more specific signal than sections hybridized at 58C.
One might wonder whether a similar improvement can also be obtained by optimizing the proteolytic digestion of the sections. This is not the case. As noted by many authors, optimal digestion conditions must be assessed for each tissue type. The conditions we found are in the usual range but do allow detection of low-abundance mRNAs only if sections are hybridized at high temperatures.
Section Thickness
Assuming that temperature effectively results in a kind of permeabilization or “breathing” of the tissue, allowing better probe penetration, signal should increase with section thickness when hybridized at 70C. This is indeed the case, as shown in Figure 3. The low-abundance Irx4 mRNA that can hardly be detected in sections of 5-μm thickness is easily detectable in 15-μm-thick sections. We have been able to increase section thickness to 35 μm. This yields very strong signals indeed, but one has to accept lower spatial resolution. In a quantitative radioactive ISH procedure (Jonker et al. 1997), we were unable to demonstrate a significant effect of section thickness on signal strength when hybridization was done at 54C (data not shown). Use of high temperatures in this procedure introduces artifacts, most likely owing to difficulties in maintaining reducing conditions.
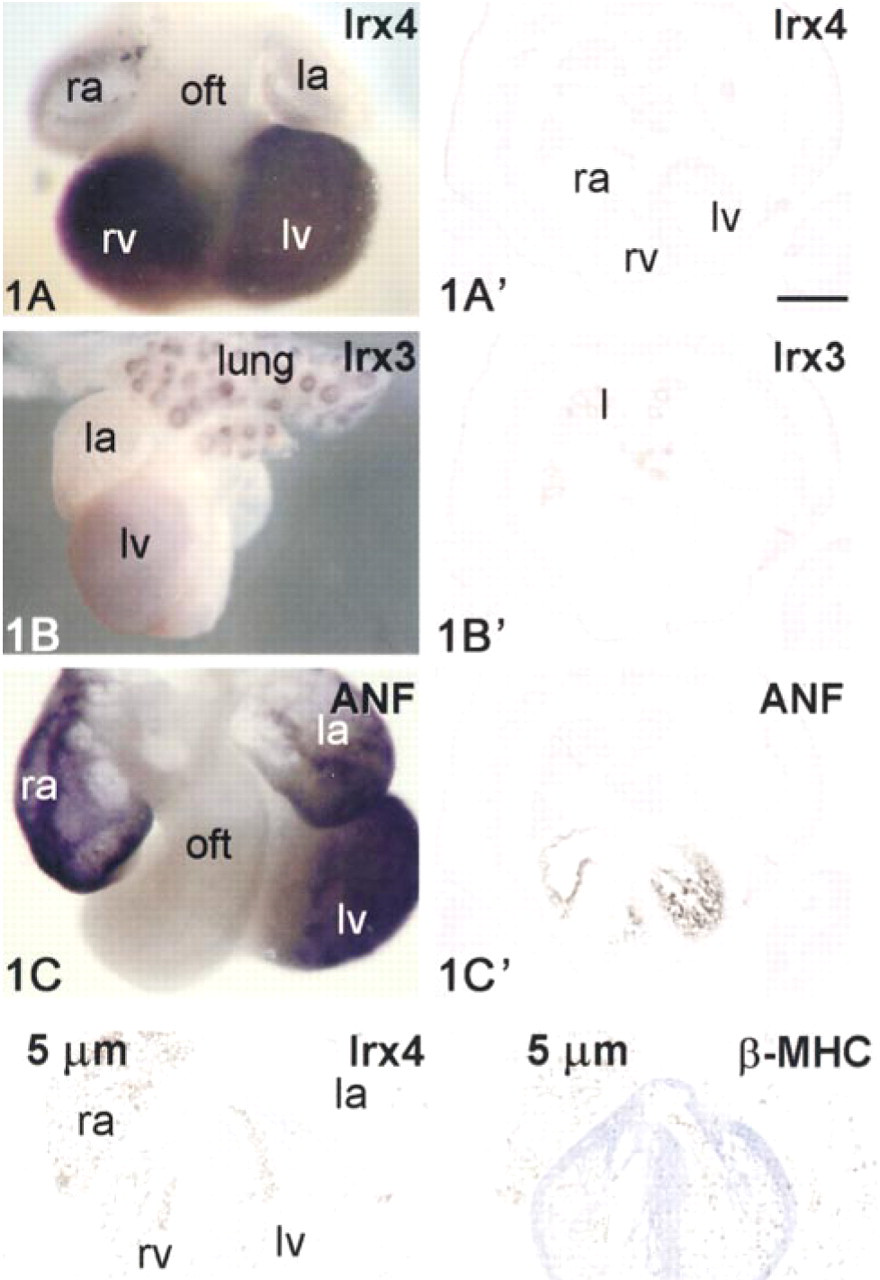
Comparison of whole-mount ISH (
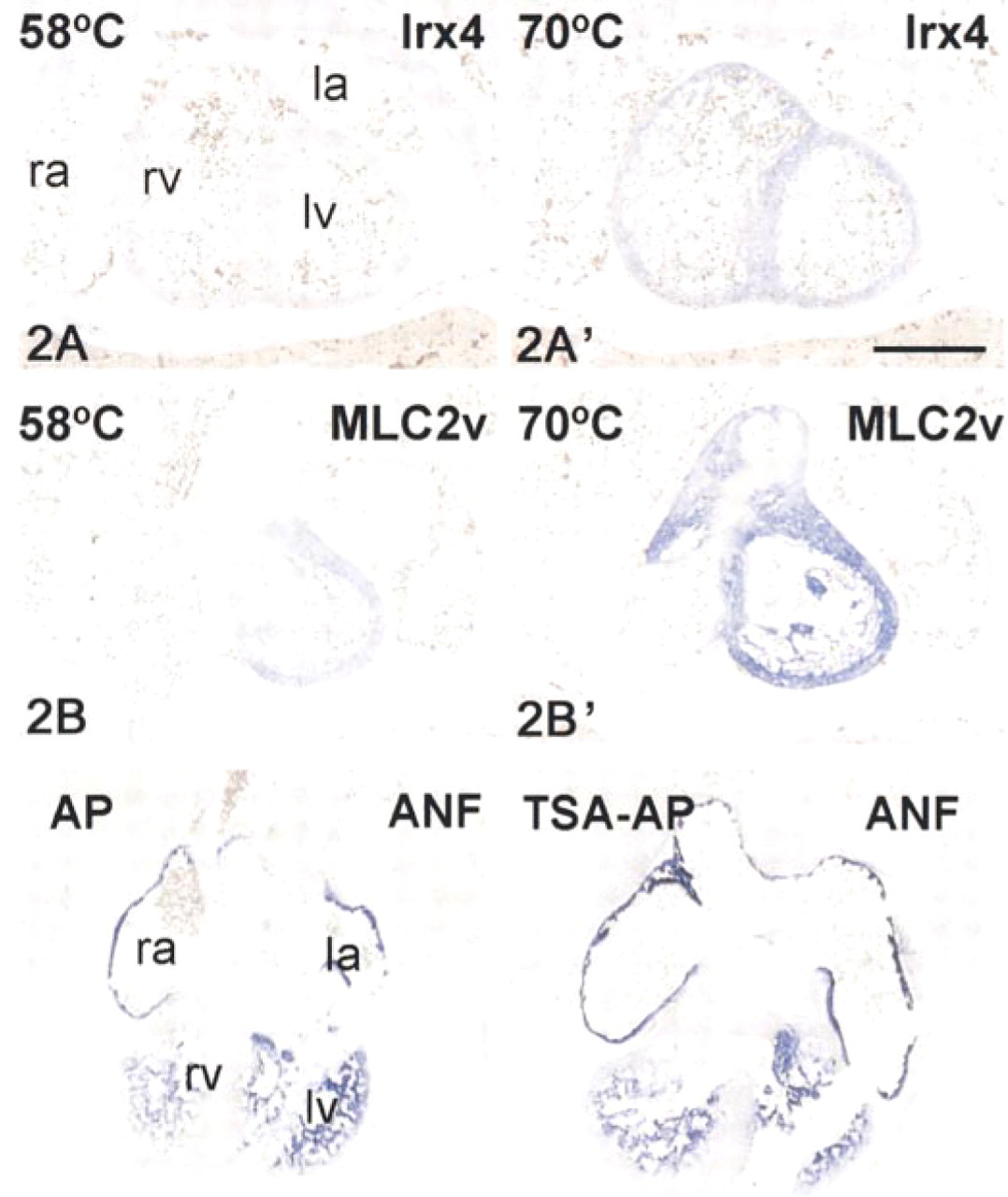
Evaluation of the influence of the hybridization temperature at similar stringency of hybridization. Transverse sections of E13.5 mouse embryos (10 μm) were hybridized at 58C (
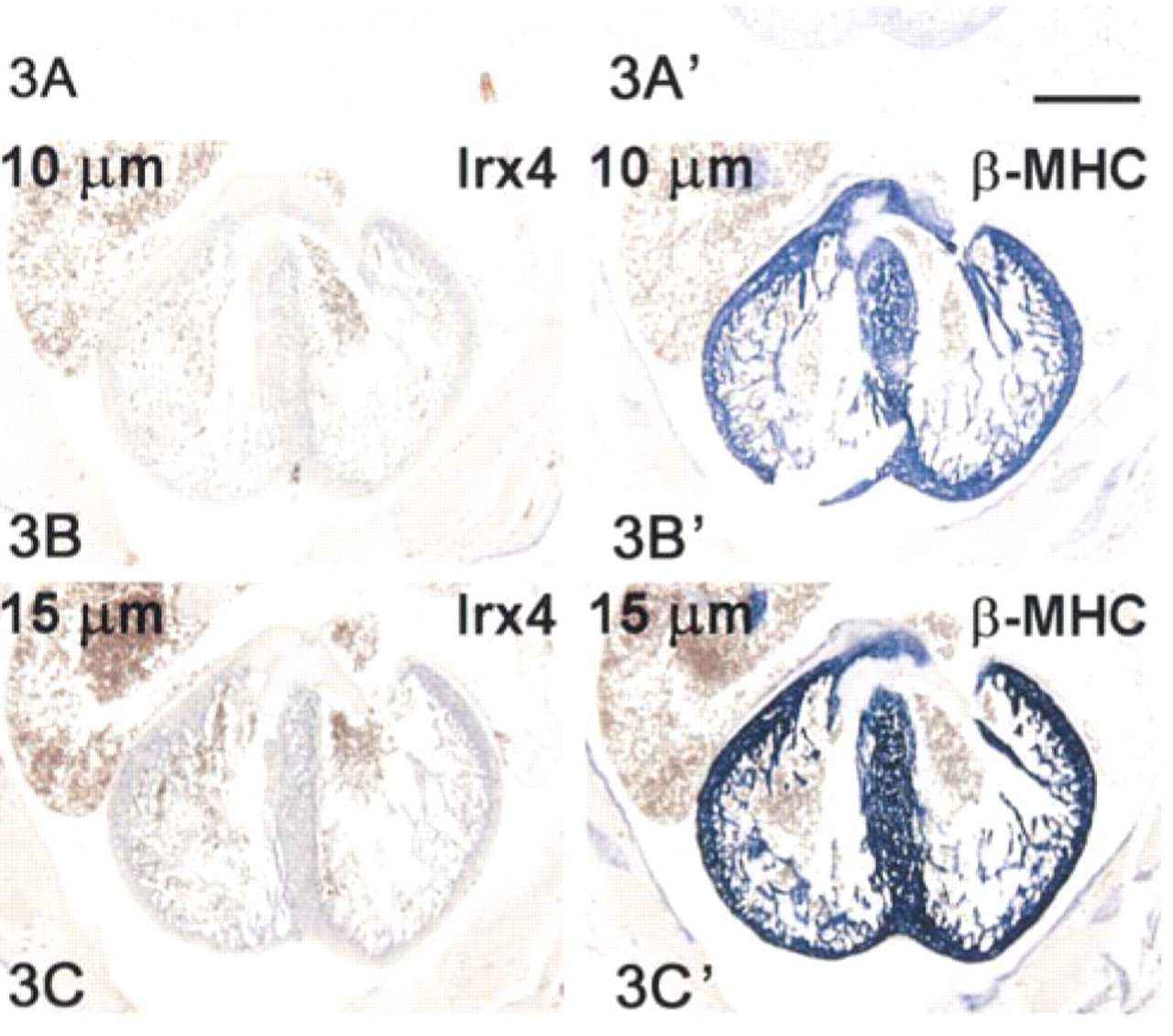
Relationship between section thickness and signal intensity. E15 mouse embryo transverse sections of 5 μm (
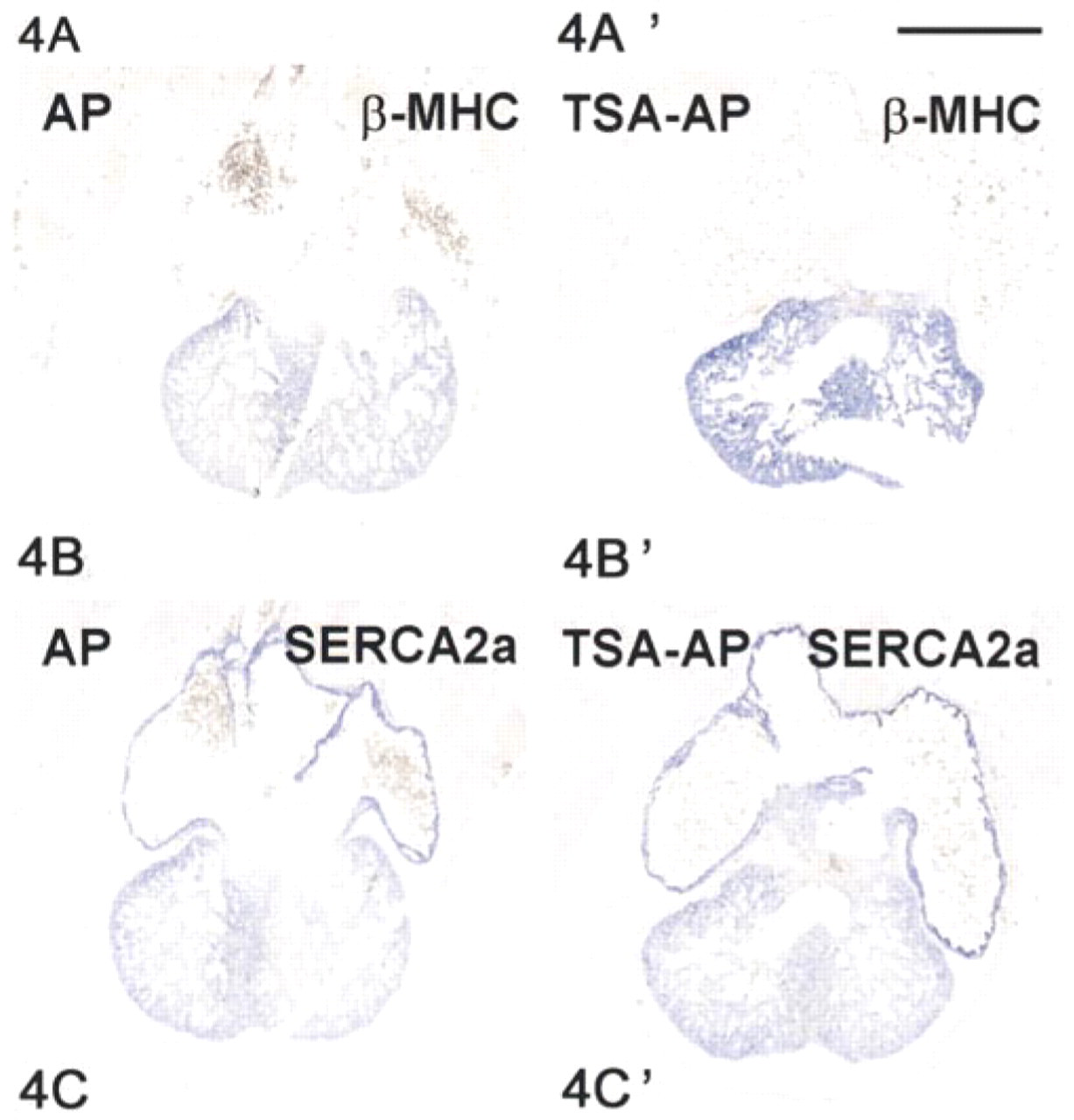
Evaluation of tyramide signal amplification. E14 rat embryo sections (10 μm) were hybridized to antisense digoxigenin-labeled probes to ANF mRNA (
Tyramide Signal Amplification
A next step in our attempts to improve the sensitivity of signal detection was to verify whether an additional enhancement of signal could be achieved by use of the tyramide signal amplification (TSA) system. In this system, horseradish peroxidase is covalently linked to the anti-digoxigenin Fab fragment, which allows deposition of biotinylated tyramide that can subsequently be detected by streptavidin-conjugated alkaline phosphatase. Results are presented in Figure 4. Signal strength is only slightly higher than the conventional signal detection. In agreement with the literature (Yang et al. 1999), probe concentration and color reaction had to be considerably reduced, but the TSA system did not significantly improve the signal-to-noise ratio.
Quality of Spatial Resolution
An important element of nonradioactive ISH on sections is that gene expression can be detected at the cellular level within the topographical context of the tissue, yielding clues about how cells might interact. Figure 5 allows one to assess that the morphological integrity of the tissue has been well preserved after hybridization at higher temperatures. A few examples are dealt with. Based on the whole mount ISH stainings shown in Figures 5A and 5B, a similar cellular localization of Irx4 and Irx5 was anticipated. The nonradioactive ISH on sections clearly demonstrates a myocardial location of Irx4 (Figure 5E) and an endocardial localization of Irx5 (Figure 5D), which lines the developing atrial and ventricular working myocardium. Moreover, the whole-mount ISH did not permit the unambiguous assessment of Irx5 hybridization in the atria, whereas the ISH on sections clearly revealed Irx5 expression in the endocardial cells lining both the atrial and ventricular working myocardium, but not the atrioventricular canal and outflow tract (Figures 5B and 5D). This indicates not only a superior spatial resolution but also a superior sensitivity of the nonradioactive ISH on sections.
Discussion
The power of the in situ detection of mRNA sequences is that the cells that synthesize and accumulate sequences of interest can be identified and placed in a proper morphological context. Consequently, changes in gene expression as measured in homogenates can be corrected for a proper tissue base (Jonker et al. 1997; van den Hoff et al. 1997), and potential cellular interactions can be assessed. We have now described a rapid and sensitive procedure for in situ detection of mRNA sequences in sections, with high spatial resolution. On the basis of quantitative PCR experiments, the level of the Irx mRNA sequences in the embryo is about 1000-fold less than that of housekeeping mRNAs, e.g., glyceraldehyde-3-phosphate dehydrogenase mRNA, indicating the sensitivity of the method. The procedure is largely based on the whole-mount ISH procedure. The significance of the improved procedure is that gene expression can be described at the cellular level with the same or even higher sensitivity than is achieved with the wholemount procedure, thus providing a much better morphological description. Moreover, the procedure turned out to be remarkably robust and highly reproducible.
One could argue that if a few kilobases of DNA sequences can be traced by nonradioactive ISH procedures, even without advanced amplification procedures (Wiegant et al. 1991; Korenberg et al. 1995), a single mRNA molecule of the same length should be detectable as well. If this is not the case, one can argue that either the mRNA sequences are not accessible to the probe or that the detection system fails. Both possibilities deserve additional comment.
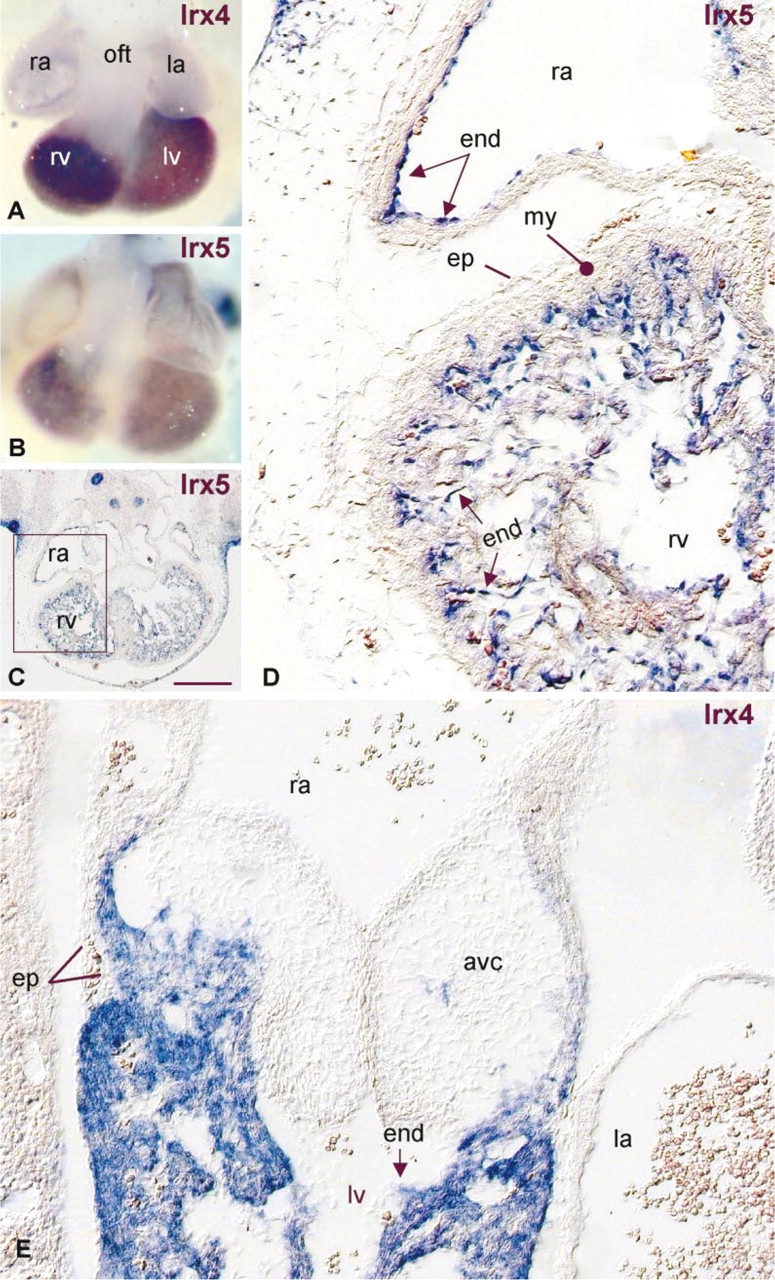
Evaluation of the spatial resolution of the nonradioactive ISH. E13.5 mouse embryos and sections (15 μm) were hybridized to antisense digoxigenin-labeled probes to Irx4 mRNA (
In the protocol described here, we observed only marginal additional beneficial effects of the tyramide signal amplification. This finding is in line with the fluorescent ISH study of van de Rijke et al. (2000), which concludes that loss of target sequence and limited accessibility, rather than the ability to generate sufficient signal, largely determine the sensitivity of ISH. In agreement with Yang et al. (1999), we observed that application of TSA also needs the use of 20–40-fold lower probe concentrations and 20–70-fold shorter times of color reaction to avoid background signal. Although this indicates that the amplification has functioned properly, it also shows that the signal-to-noise ratio has not improved significantly by the amplification. In fact, the gain of signal by the amplification evaporates by the need to lower probe concentration and duration of color reaction. Nevertheless, several studies have reported improvement of sensitivity by the TSA method (van Gijlswijk et al. 1997; Speel et al. 1998; Yang et al. 1999). Apparently our new ISH method already results in a quite specific and sensitive hybridization, the detection of which is only marginally improved by TSA.
Thus far, we have used the novel ISH method to describe the spatial distribution and cellular localization of a number of mRNAs encoding Iroquois-related homeobox transcription factors. We have demonstrated for the first time a heterogeneous pattern of gene expression in the endocardium, Irx5 being confined to the endocardium lining the developing chambers, suggesting an important role in cardiac chamber formation. The formed heart has a complex morphology. A plethora of transcription and growth factors, and interactions among endocardium, cushion tissue, myocardium, and epicardium are involved in its intricate morphogenesis (Rosenthal and Harvey 1999). The patterns of expression of most of these factors are predominantly described at the mRNA level by whole-mount ISH. The ISH method on sections that we have developed is at least equally sensitive as the whole-mount procedure and excellently preserves morphology. Therefore, the method will be pre-eminently suited for analyzing the complex patterns of gene expression during cardiac morphogenesis, yielding clues about the underlying molecular mechanisms.
Footnotes
Acknowledgments
AC Houweling, PAJ de Boer, and VM Christoffels are supported by The Netherlands Heart Foundation grants nos. 96.002 and 98.042.
We express our gratitude to Mr Jaco Hagoort for excellent image acquisition and processing support, Dr Ronald H. Lekanne for performing quantitative PCR experiments, Dr Marc A. Vos for stimulating discussions, and Dr Wouter H. Lamers for carefully reading the manuscript.