
Abstract
Because tissue freeze-drying is an excellent way to preserve antigenic conformation, we have tested the feasibility of this technique to reveal nonradioactive in situ hybridization (ISH) of tissue mRNA. We have compared mRNA detection after different methods of tissue preservation, freeze-drying, cryosectioning, and formaldehyde or methanol fixation. Our results show that nonradioactive ISH is more sensitive for tissues preserved by freeze-drying than for other tissue preparations. We have demonstrated that freeze-drying allows combination of ISH and immunohistochemistry for simultaneous detection of mRNA and antigen because with this technique of tissue preservation ISH does not affect the sensitivity or the amount of the detected antigens. This work underscores the fact that tissue freeze-drying is an easy, convenient, and reliable technique for both ISH and immunohistochemistry and achieves excellent structural conditions for nonradioactive detection.
The introduction in the late 1960s of in situ hybridization (ISH) techniques (Gall and Pardue 1969; John et al. 1969; Buongiorno-Nardelli and Amaldi 1970) opened a new era in histology and cell biology. Whereas immunocytochemical methods can demonstrate the presence of synthesized protein molecule in a define conformational state and can aid in characterization of cell types, ISH allows detection of the exact cell location of protein synthesis in a tissue. Coupled with immunodetection, ISH allows screening of different cell populations for specific function or precise location of a cell's differentiation state.
The sensitivity and efficiency of ISH depend on several variables, for which optimal conditions must be determined: (a) probe construction and hybridization conditions; (b) tissue conservation and fixation; and (c) the method used for signal revelation. Radioactively labeled DNA or RNA probes were initially used and are still widely applied for ISH because of their high sensitivity and the amplification effect of autoradiography. Nevertheless, safety problems, reduced stability of radioactively labeled probes, speed, and quality of visualization, as well as the extensive development of immunochemistry, which allows much more precise localization of antigen sites in a tissue section compared to the autoradiographic technique, have stimulated interest in the development of nonradioactive probes (Dix and Eisenberg 1988; Chevalier et al. 1997). Development of digoxigenin-labeled riboprobes provides ease of ISH nonradioactive detection. However, nonradioactive ISH usually requires a 10- to 50-fold higher concentration of probes than radioactive ISH (Bloch 1993; Wilcox 1993), and to decrease this difference processing of tissue conservation must be optimized. The other critical step is the storage and fixation of tissue. Storage methods must ensure the long-term stability of tissue morphology and mRNA, and fixation techniques must be compatible with mRNA retention in situ. Ideally, the storage and fixation methods chosen should also permit the study of tissue by other techniques, such as immunochemistry either alone or in combination with ISH. Compared to cryosectioned or pre-fixed tissue, freeze-drying and subsequent paraffin embedding are well established to be more efficient and easier for antigen conservation (Stein et al. 1984, 1985; Coulter and Elde 1986). More recently Kingsbury et al. (1996) have demonstrated the better sensitivity of cryosections compared to formaldehyde-fixed, paraffin-embedded tissue. However, tissue freeze-drying was not tested for its sensitivity in ISH. In this study we compared sensitivity of tissue labeling by nonradioactive ISH of conventional tissue preparation: (a) pre-fixation and paraffin embedding; (b) cryosectioning; and (c) freeze-drying and paraffin embedding. We combined ISH staining with immunochemical detection of two very sensitive antigens, smooth muscle myosin heavy chain (sm-MHC) and vascular cell adhesion molecule-1 (VCAM-1).
Materials and Methods
Animals
In this study we used two markers of smooth muscle cells: smooth muscle myosin heavy chain (sm-MHC) because in normal aorta the mRNA and protein levels are high and are expressed specifically by SMCs of the medial layer so that they provide two internal negative controls (fibroblasts in the adventitial layer and endothelial cells in the intimal layer); and vascular cell adhesion molecule-1 (VCAM-1) as a marker of inflammatory response because the mRNA and protein are absent in normal aorta and therefore unstimulated animals provide controls. To induce the expression of VCAM-1, three Fauve de Bourgogne rabbits (2.5 kg) were injected (per kg bw) with 250 μg Escherichia coli lipopolysaccharide (Ec LPS) (Fries et al. 1993), sacrificed after 6 hr, and used for VCAM-1 mRNA detection. Three normal Fauve de Bourgogne rabbits (2.5 kg) were used for sm-MHC mRNA and antigen detection and as controls for VCAM-1 expression. All rabbits were sacrificed with an IV overdose of anesthetic agent and the aortas were collected.
Preparation of Tissues
Fresh unfixed aortas were washed with PBS and then divided into several parts. One third of each aorta was fixed with paraformaldehyde. The remaining two thirds were deep-frozen for further study in cryosections or for freeze-drying.
PFA or Methanol Fixation and Paraffin Embedding
Tissue samples were fixed in methanol overnight at 4C or in 4% PFA for 3 hr at 4C, and were dehydrated conventionally in graded alcohol (30–100%) and finally in xylene for 10 min. Tissues were then embedded in paraffin. Before processing for immunostaining or ISH, slides were dewaxed by incubation in xylene twice for 10 min and were rehydrated through four alcohol steps from 100C to 50C, then kept wet in PBS.
Cryosections
Tissue portions were deep-frozen in liquid nitrogen without any prior tissue fixation. For cryosections, tissues were embedded in OCT compound and stored at −70C until sectioning. Freshly cut cryostat sections were air-dried for 30 min, fixed for 10 min in cold acetone, and kept wet in PBS until processing.
Freeze-drying of Tissue Preparation
Tissues were removed from liquid nitrogen and tissue desiccation was performed according to the technique previously described by Stein et al. (1985). To prevent the tissues from thawing, tissues were placed on the pre-cooled plate of an Edwards-Pearce tissue dryer (Edwards High Vacuum; Manor Royal, Crawley, West Sussex, UK) and dried at −40C under 10−2 Torr pressure (in the presence of phosphorous pentoxide) for 8–16 hr. Tissues were then allowed to warm up under vacuum at room temperature (RT) to prevent any water condensation on the dried piece, which was then immersed in paraffin at 56C under vacuum (to ensure perfect tissue infiltration) for overnight incubation. Then the vacuum was broken and the arteries were embedded in paraffin and stored at RT until sectioning. For freeze-dried tissues, sections were dewaxed by two incubations in a xylene bath for 10 min and were fixed by immersion in cold acetone for 10–15 min as described (Stein et al. 1985).
Probes and cRNA Preparation for ISH
Total cellular RNA was prepared from aortic media and was reversed-transcribed. For human sm-MHC probe, human sm-MHC cDNA was amplified by PCR for 30 cycles at 60C for annealing, using primers as already described (Lavie et al. 1999). Briefly, the obtained PCR product was purified, digested in a 392-bp fragment, and subcloned into BamHI-HincII sites of pBluescript vector according to standard procedures. The PCR product specificity was verified by the DNA sequence of the insert determined by using the dideoxy chain determination method by modified T7 DNA polymerase. The specificity of the probe for rabbit sm-MHC was confirmed by Northern blot analysis (not shown). For rabbit VCAM-1, the probe of 700 bp was a generous gift from Dr. Myron Cybulsky (Li et al. 1993).
To construct the riboprobe, pBluescript containing the human sm-MHC cDNA insert or rabbit VCAM-1 cDNA insert was linearized by action of XbaI for antisense VCAM-1 and sm-MHC probes and PsTI for sense VCAM-1 probe, and by KpnI for sense sm-MHC probe. Furthermore, the cDNA probe was in vitro-transcribed using T7 and T3 RNA polymerase for amplification. RNA probe labeling by digoxigenin-UTP was performed by in vitro transcription according to the manufacturer's instructions (Boehringer; Mannheim, Germany). Riboprobe lengths were 700 bp for VCAM-1 and 400 bp for sm-MHC.
Riboprobe Concentration
Evaluation of synthesized riboprobe was done by dot-blot with DIG-labeled RNA controls (Boehringer). As the principal riboprobe amount we used 100 ng per slide. Various concentrations of VCAM-1 riboprobe (100 ng, 10 ng, 1 ng) were tested to determine the limiting dilution for detection of significant signal.
In Situ Hybridization
Each tissue sample was cut at an 8-μm section and transferred to 3-aminopropyl triethoxysilane-coated slides (Sigma; St Louis, MO) to increase adhesion. Sections were treated with proteinase K (1 μg/ml) in 0.1 M Tris-HCl and 0.05 M EDTA for 15 min for frozen sections. For PFA fixation, sections were treated with proteinase K from 1 μg/ml to 20 μg/ml for 30 min. The sections were then rinsed in PBS, treated with glycine (2 mg/ml) for 2 min, and fixed in 4% PFA for 5 min for cryodesiccated sections and 20 min for cryosections and fixed tissues. After several washes in PBS, the sections were incubated in triethanolamine buffer, pH 8, for 5 min. After several washes in PBS, prehybridization buffer (4 × SSC, 5 × Denhart's, 0.1% N-laurylsarcosine) was applied for 30 min at 37C. For hybridization, U-digoxigenin-labeled riboprobes were added and hybridization was allowed to proceed overnight at 50C. For PFA-fixed tissue, we added a step at 90C for 20 min before incubation at 50C overnight. After hybridization, sections were incubated twice with 50% formamide, 2 × SSC at 55C, treated with RNase A (20 mg/ml) for 30 min, and washed twice in 2 × SSC, followed by 1 × SSC washes. To detect the specific hybrids, the sections were incubated for 90 min at 37C with an anti-digoxigenin antibody (Boehringer) conjugated with alkaline phosphatase. Staining was allowed to develop overnight in the dark with nitroblue tetrazolium salt and 5-bromo-4-chloro-3-indol phosphate (Boehringer). Slides were dehydrated by alcohol immersion, mounted in DPX, or kept wet in PBS for immunochemical labeling. For the purpose of this study we performed for each probe all ISH detection for all the tissue fixations at the same time.
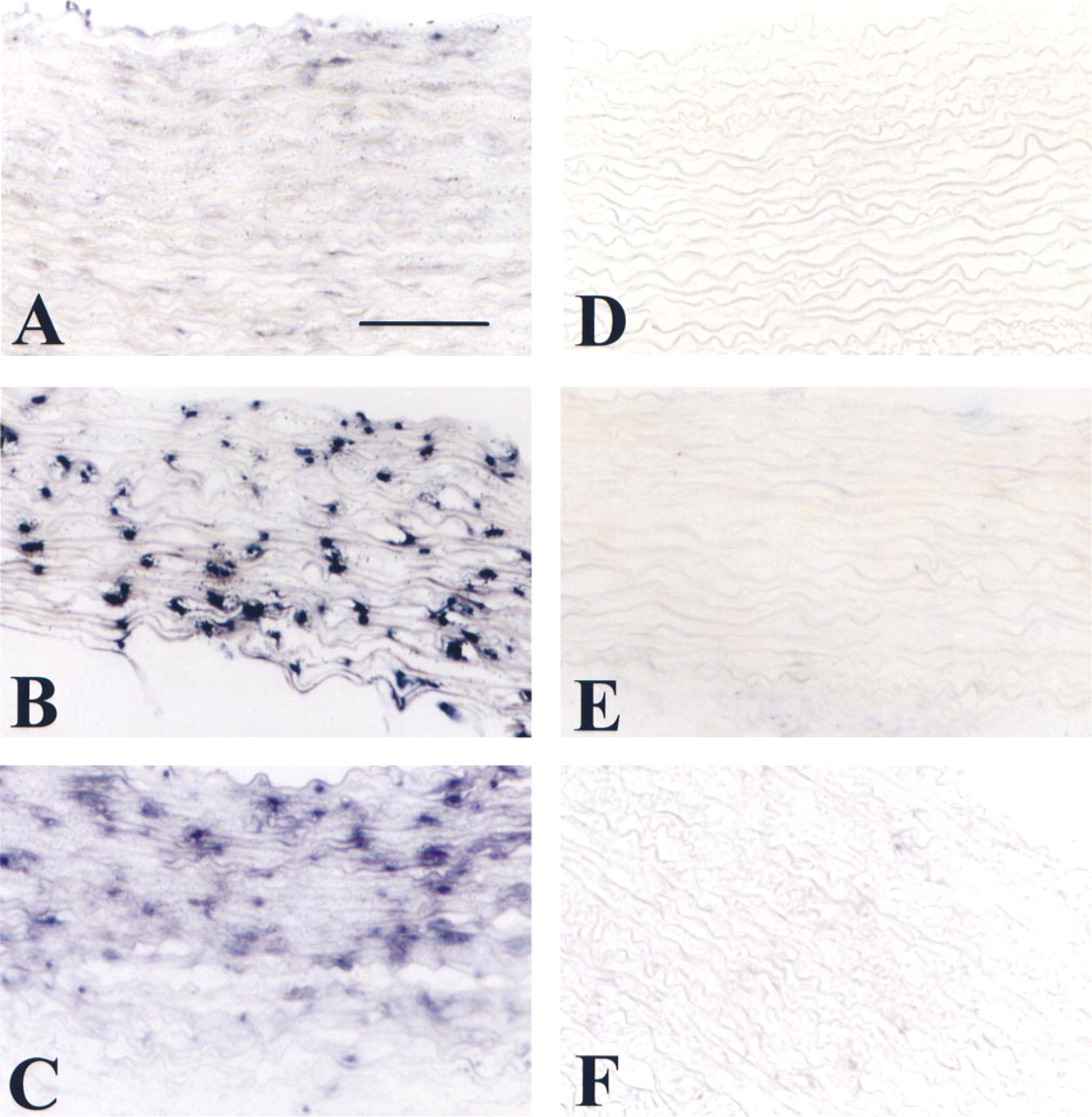
sm-MHC mRNA is detected by nonradioactive ISH in normal rabbit aorta as described in Materials and Methods. (
Antigenic and mRNA semiquantitative determination a
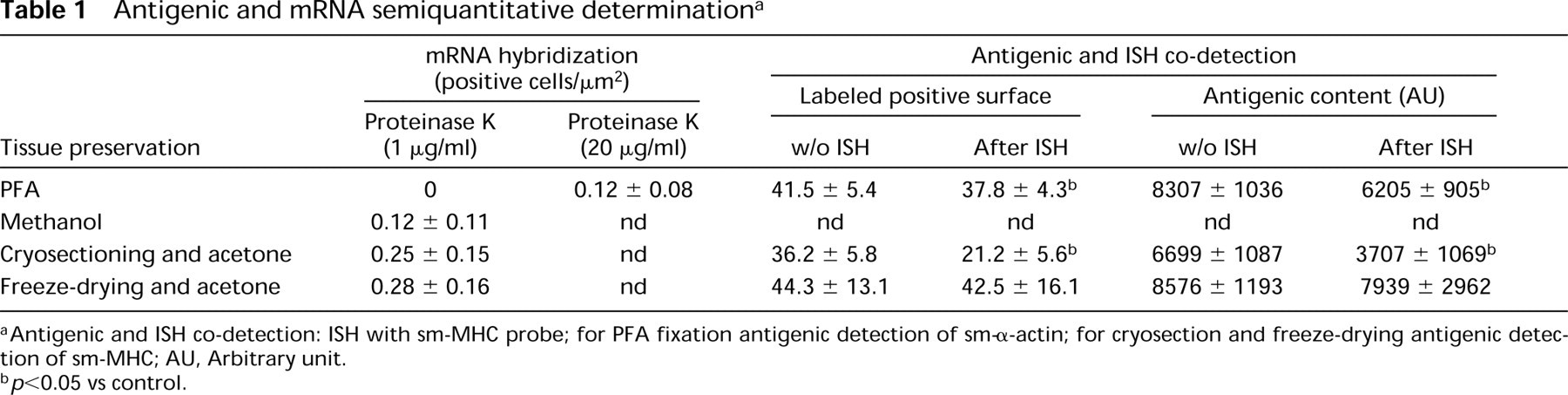
Antigenic and ISH co-detection: ISH with sm-MHC probe; for PFA fixation antigenic detection of sm-α-actin; for cryosection and freeze-drying antigenic detection of sm-MHC; AU, Arbitrary unit.
p<0.05 vs control.
Immunohistochemistry
After ISH, conventional immunohistochemistry was performed. All antigens were detected with very well-characterized monoclonal antibodies (MAbs). For sm-MHC detection we used MAb hSMV (Sigma); Rb 1/9, which recognizes rabbit VCAM-1, was a generous gift from Dr. Cybulsky (Li et al. 1993); and 1A4 for detection of smooth muscle α-actin (sm-α-actin). All dilutions used were done according to the manufacturers instructions. Antigen presence was revealed by the indirect immunoperoxidase method as previously described (Hsu et al. 1981; Daniel Lamazière et al. 1993). Endogenous peroxidase activity was blocked by incubating the sections in of 3% hydrogen peroxide. Before staining, sections were preincubated for 20 min in a blocking solution containing 5% bovine serum albumin (BSA) in Tris-buffered saline (TBS). Sections were first incubated with primary antibody at appropriate dilutions for 1 hr at RT, rinsed three times with an excess of TBS, incubated with biotinylated sheep anti-mouse Ig (Amersham; Poole, UK) for 1 hr at RT, rinsed three times with an excess of TBS, and covered with streptavidin-biotin-horseradish peroxidase complex. After 30 min at RT, the sections were rinsed three times with an excess of TBS. The bound antibody was revealed by incubation with 10% (v/v) 3,3′-diaminobenzidine tetrahydrochloride dihydrate in stable peroxide substrate buffer (Pierce; Rockford, IL). Omission of primary antibodies or use of a nonspecific antibody served as negative controls. Slides were dehydrated by alcohol immersion, then mounted in DPX. Each antigenic detection method was performed for all tissue fixation conditions in the same run.
Semiquantitative Analysis of Distribution of Antigens and mRNA
Immunostaining was quantified at high-power (X 400) magnification by color video image analysis using an IBM PC (Daniel Lamazière et al. 1993). The video camera was connected to an inverted microscope. The software Quancoul (Quant'Image; INSERM U441, Talence, France) defined true colors on the basis of three independent parameters (hue, intensity, and saturation). The parameters were calibrated against a background of control antigen, which enabled extraction of the brown or blue color corresponding, respectively, to the measured antigens or mRNA. The thresholds were adjusted so that all but the faintest staining could be detected. For immunostaining quantitation, to account for the color intensity as an index of the amount of antigenic detection, we expressed the antigenic content in arbitrary units as the product of the mean percentage of positively labeled surface and the hue intensity of the same positive pixels (Daniel Lamazière et al. 1993; Bezie et al. 1998). The degree of antigen expression was expressed by two parameters: percent of positively labeled surface and antigenic contents (AU). At least six sections from each aorta were examined and the values from these sections were averaged to calculate a value for each artery. For ISH quantitation, the number of stained cells labeled by riboprobe were counted by counting the blue-stained area stained by alkaline phosphatase, and numbers of stained cells were expressed per μm2.
Statistical analyses were done by Student's t-test.
Results
ISH Comparative Analyses According to Method of Tissue Preparation
The aim of our study was to describe an easy way to detect antigenic and nucleotide probes in the same tissue preparation. Tissue preparation is of great importance to ensure good and reliable detection of proteins as well as ribonucleotides. Before staining, tissue must be prepared for preservation and to remain antigenic in a stable conformation. In this study we tested three types of tissue preparation: (a) conventional histological fixation with the use of PFA; (b) cryosectioning; and (c) freeze-drying followed by paraffin embedding. For freeze-dried tissues and cryosections, both antigen and ribonucleotide detection was possible. However, for prefixed tissues embedded in paraffin, the sensitivity of mRNA detection was quite low, which necessitated optimized conditions for hybridization by enzymatic treatment, which can further alter antigenic detection.
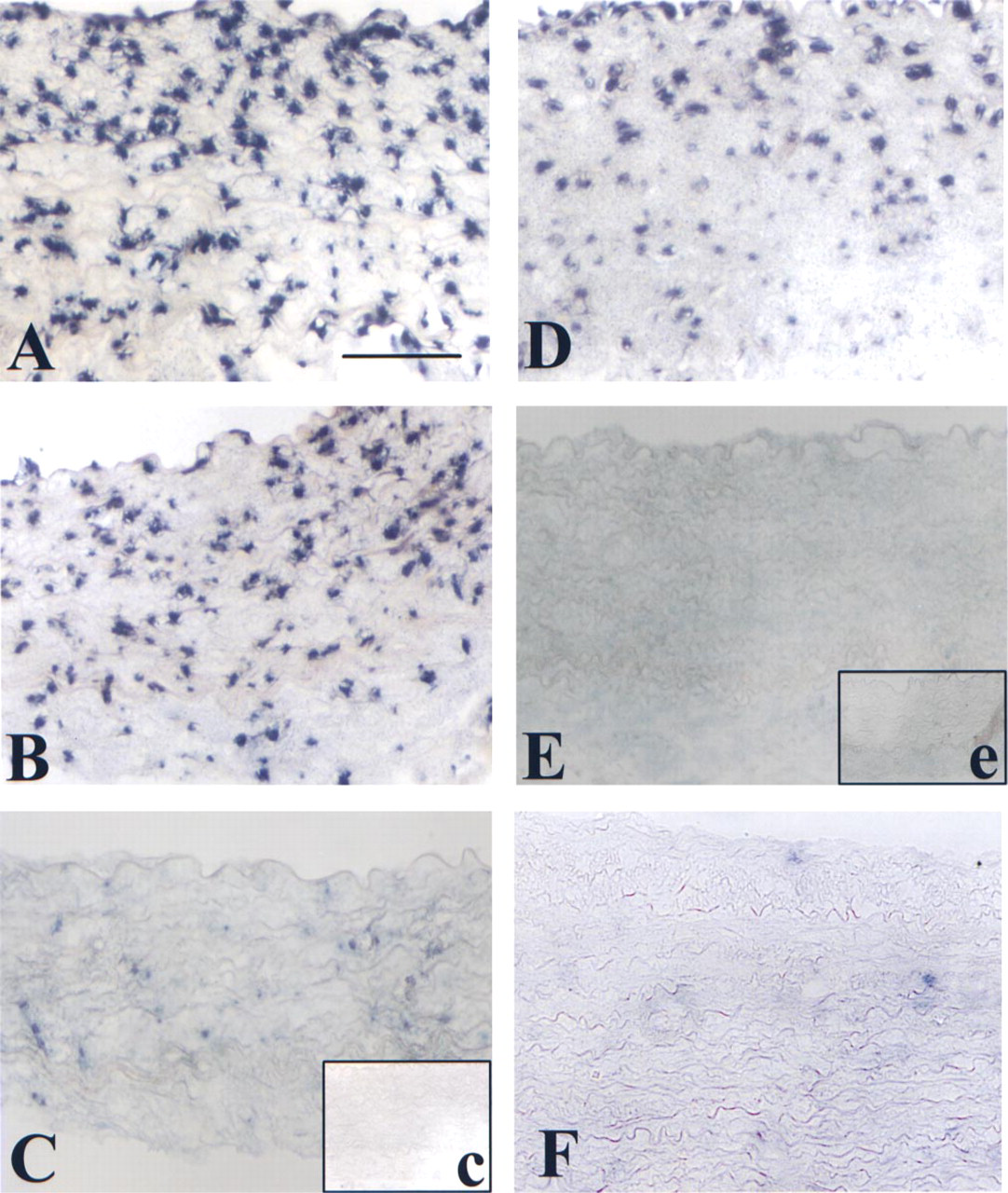
VCAM-1 mRNA is detected by nonradioactive ISH as described in Materials and Methods in normal rabbit aorta after LPS stimulation for 4 hr. Hybridization was performed with various dilutions of antisense probe: 100 ng (
We performed ISH under the same conditions for each tissue, using an sm-MHC probe that is highly expressed in normal aorta. We compared different tissue preparations to obtain a significant signal for mRNA. In cryosections and freeze-dried tissues, mRNA was detected in a very sensitive way. Both techniques yielded essentially similar staining results in terms of number of positive cells (cryosections 0.25 ± 0.15 vs freeze-drying 0.28 ± 0.16; p=0.0017) as shown in Figure 1 and Table 1. However, no signal was obtained in PFA-fixed tissues with identical probe concentration and incubation conditions (Figure 1A). To obtain a signal with PFA-fixed tissues, we increased the proteinase K concentration (1 μg/ml for freeze-dried tissues vs 20 μg/ml for PFA-fixed tissues) and the time of treatment from 15 to 30 min. Moreover, we added a step at 90C, at the beginning of hybridization. Under these conditions (Figure 4B and 4C; Table 1) we could detect and measure some mRNA hybridization.
We compared the sensitivity of the mRNA detection by ISH. Because we compared ISH feasibility for normal tissues (Figure 1) we compared the sensitivity of the response after tissue stimulation by treatment with LPS, which induces a proinflammatory response for arterial smooth muscle cells. Because after PFA fixation we did not obtain valuable detection for ISH, we tested methanol fixation. Figure 2 shows VCAM-1 probe hybridization at different dilutions on freeze-dried tissues and methanolprefixed tissues embedded in paraffin. After freeze-drying we found an excellent labeling close to dose response. Even at 1 ng probe dilution we were able to detect the presence of the targeted RNA (Figures 2A-2C). However, after methanol fixation and paraffin embedding the tissue was no longer sensitive for high dilutions of the probe (Figures 2D and 2E) but was better than after PFA fixation. As a negative control we hybridized the VCAM-1 riboprobe (100 ng) on normal rabbit aorta (Figure 2F). The absence of hybridization demonstrated the specificity of this riboprobe.
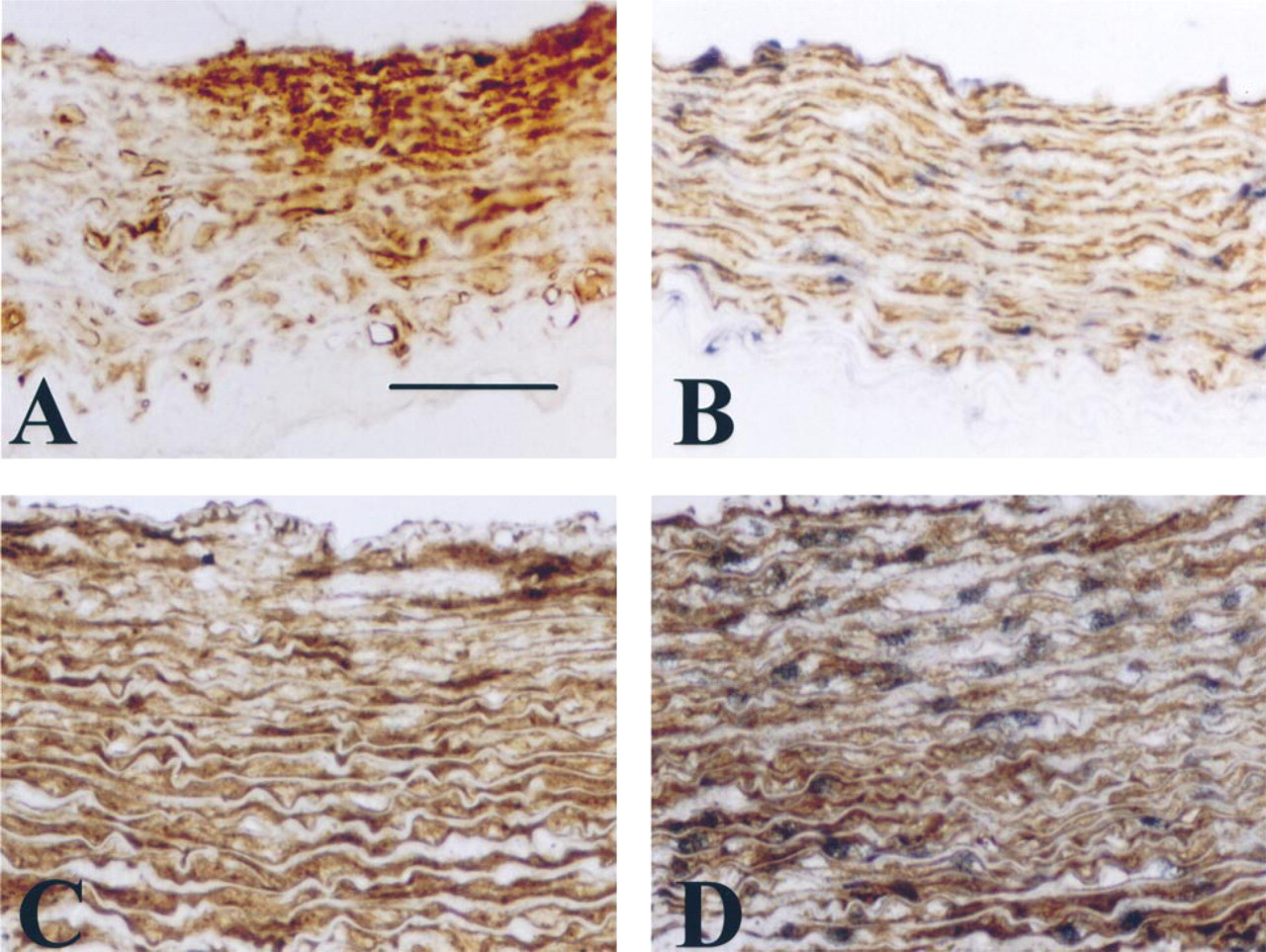
Immunohistochemical staining and nonradioactive ISH for sm-MHC. (
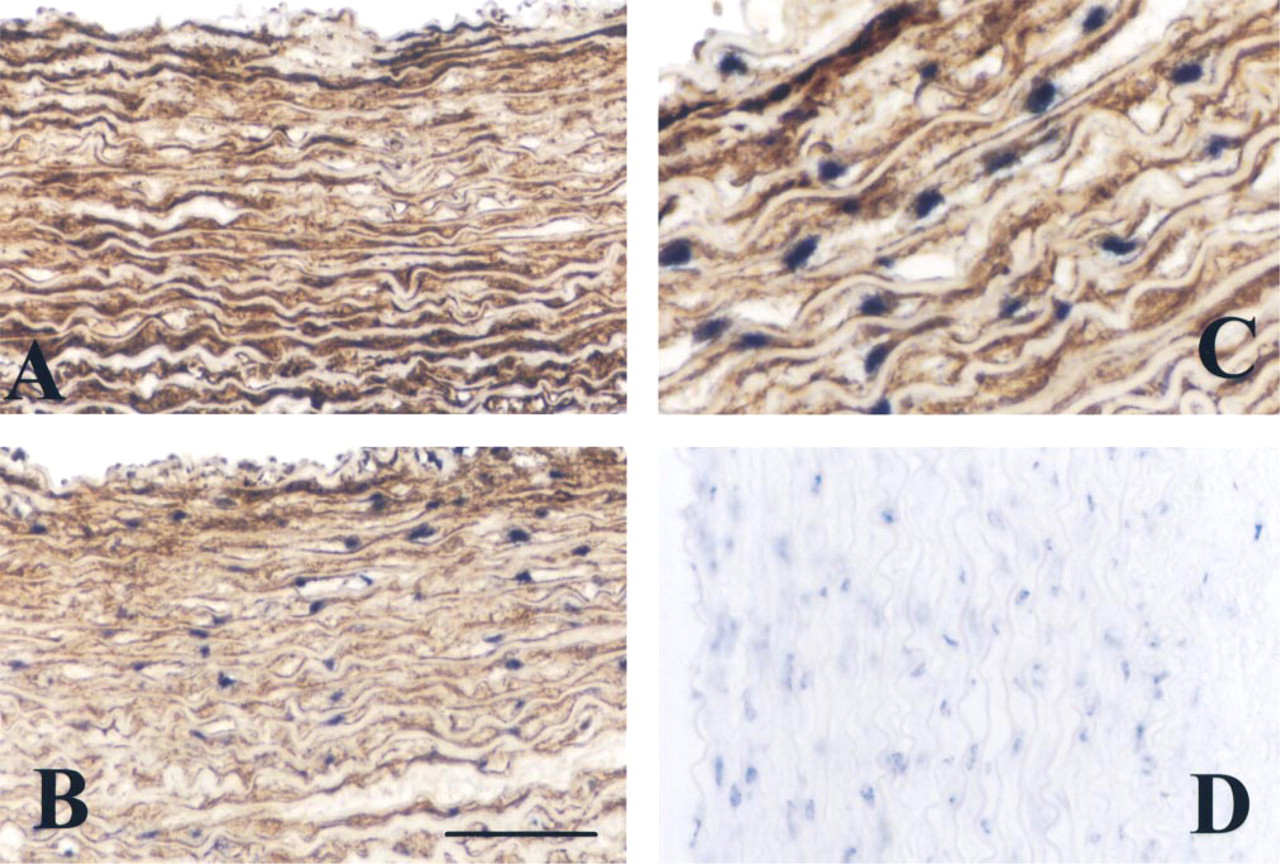
Nonradioactive ISH for sm-MHC mRNA after treatment with 20 μg/ml proteinase K and a step at 90C before hybridization, and sm-α-actin antigen detection (
Double Labeling: ISH Immunostaining
Histological examination is enhanced by the development of a double detection technique, which allows identification of different markers in the same area. For this purpose, we compared the three tissue preparations for RNA detection and antigen distribution. We chose to detect sm-MHC or VCAM-1, both very sensitive antigens that are present only on cryosections (Cybulsky and Gimbrone 1991; Glukhova et al. 1991).
Figure 3 shows the double detection of sm-MHC mRNA and the immunochemical labeling of the same protein. We compared cryosections and freeze-dried tissue preparation, and in both cases we were able to detect the antigen after ISH. However, with freeze-drying one can observe the quality of the tissue response (Figure 3C). Both antigenic and ISH labeling yielded very thin and well-contrasted staining. Moreover, in cryosection preparations it is more difficult to preserve tissue integrity in terms of stability of ISH staining which decreases over time and is associated with loss of antigenic and cellular morphological integrity (Figures 1 and 3). Moreover, freeze-drying did not affect antigenic quantitation even after ISH (without ISH 8576 ± 1193 vs after ISH 7939 ± 2962; p=0.59). However, after ISH, in cryosections we noted loss of the antigenic content, which is highly significant (without ISH 6699 ± 1087 vs after ISH 3707 ± 1069; p=1.46 × 106).
Double staining can also be performed for prefixed tissues with very stable antigens (Figure 4). Because the sm-MHC MAb does not work on prefixed tissues (Figure 4D), as positive control we used the sm-α-actin, which is not sensitive to tissue fixation conditions. Figure 4 shows double staining of sm-MHC mRNA by ISH and sm-α-actin antigenic detection. This was possible only after tissue section treatment with proteinase K (20 μg/ml). This treatment affects the quantity of the sm-α-actin antigenic detection (8307 ± 1036 before ISH vs 6205 ± 905 after ISH), which can be a handicap for detection of sensitive antigens.
Discussion
Histological techniques enable the pathologist to investigate and observe the distribution of cells or molecules in a defined tissue. A number of techniques can be applied to preserve tissue organization and antigen conformational properties. With the advent of monoclonal antibody technology, one can note the development of many approaches to increase antigenic detection in fixed tissues (Katoh et al. 1997; Pileri et al. 1997; Hopwood 1998). In parallel the detection of mRNA in fixed tissues opens a new era of localizing protein synthesis in a particular area. The main problem of ISH is to preserve the availability of the RNA to the complementary probe associated in an environment free of RNase (Urieli-Shoval et al. 1992).
In this study we used an easy and rapid technique that enables detection of RNA along with the ability to preserve sensitive antigens and to detect them. Freeze-drying has been described as a very potent strategy for detection of very sensitive antigens (Stein et al. 1985; Meredith et al. 1996). However, this approach was never applied for ISH. Detection of mRNA of sm-MHC was possible in classical tissue fixation with PFA and paraffin embedding (Urieli-Shoval et al. 1992). However, for this detection we had to increase proteinase K digestion (from 1 μg/ml for freeze-dried tissue to 20 μg/ml for PFA-fixed tissue). The hybridization conditions were also optimized by a step of 20 min at 90C at the beginning of hybridization, which is unnecessary for either cryosections or freeze-dried sections. All these steps provide further randon antigenic detection. Moreover, unlike Urieli-Shoval et al. (1992), we found that methanol fixation is better than paraformaldehyde fixation. This might be explained by the fact that Carnoy's reagent contains chloroform and acetic acid in addition to methanol.
ISH can localize effective synthesis of a given molecule in a cell, but in histological studies it is usually important to define exactly the origin or nature of cells. Moreover, double staining for ISH followed by immunohistochemical labeling on the same slide provides a more precise technique for such analysis. Tissues prepared by freeze-drying remain very suitable for antigen detection even after ISH. For example, Figure 3 and Table 1 show that the different treatment used for ISH does not alter the integrity of antigen compared to sections treated only for immunochemistry. Figure 4 shows the results of double staining after paraformaldehyde fixation. Because we were unable to detect sm-MHC antigen, to characterize cells we detected sm-α-actin antigen, which is more stable. Tissue freeze-drying is a very convenient and easy technique compared to cryosectioning. For routine work, the price of the equipment is comparable. However, it is easier to cut paraffin-embedded freeze-dried than tissues to cut frozen tissues. The quality of the tissues is better preserved, and section adhesion under the slides is better. Moreover antigenic detection depends on the quality of the fixative, which is not affected for freeze-dried tissues.
The development of nonradioactive ISH detection has facilitated observation of mRNA hybridization more easily and more rapidly than radioactive labeling. Another point is the quality of the observation of localization of the hybridization, because radioactive visualization is not possible in the same plane of the tissue section. We show here that freeze-dried tissue does not affect mRNA localization or further antigenic detection. Freeze-drying eliminates water without modifying protein folding in the absence of any further chemical or proteinase treatment. However, most methods of conventional tissue preservation use chemical agents for dehydration (alcohol, acetone, xylene), which can modify tertiary and quaternary protein structure. Freeze-drying associated with ISH tagged with digoxigenin opens new and easy possibilities to perform double antigen detection, even with very sensitive antigens.
Acknowledgments
Supported by Université Bordeaux 2 “Victor Segalen” and by l'Établissement Public Régional d'Aquitaine.