
Abstract
Oligodendrocytes, the myelin-forming cells in the central nervous system, were visualized with excellent resolution at the light microscopic level using in situ hybridization (ISH). Digoxigenin (Dig)-tagged probes were synthesized and efficiently labeled by PCR. Specific probes to myelin genes were made by RT from brain total RNAs, followed by PCR with designed specific primers in the presence of Dig-11-dUTP. Probes specific to proteolipid protein (PLP), PLP and its isoform DM20 (PLP/DM20), and myelin oligodendrocyte glycoprotein (MOG) were synthesized and labeled. ISH was then applied on vibratomed tissue sections from mouse brains. Despite a low expression of MOG-specific and PLP-specific mRNAs in adult and newborn mouse brains, an oligodendrocyte population was detected. The specificity of Dig-labeled probes was confirmed with the double labeling of carbonic anhydrase II (CA II) and glial fibrillary acidic protein (GFAP) immunocytochemistry and ISH. This versatile and easy method for synthesis and labeling of specific probes to oligodendrocytes can be also applied to detect many other mRNAs in the nervous system and in other tissues.
Keywords
A
The present study describes a simple and rapid method for the synthesis and labeling by the polymerase chain reaction (PCR) of desired probes for ISH. Proteolipid protein of the myelin and myelin oligodendrocyte glycoprotein (MOG) cDNA probes were synthesized and labeled with digoxigenin by PCR. These probes were used for ISH on vibratomed sections to visualize the myelin-forming cells in the central nervous system, the oligodendrocytes.
PLP is highly expressed in neural tissue, whereas MOG is considered a minor constituent of myelin (for reviews see Campagnoni and Macklin 1988; Campagnoni and Skoff 2001). In the nervous system, the developmental appearance of DM20 precedes that of PLP (Ikenaka et al. 1992; Timsit et al. 1995). The MOG transcripts were detected during postnatal development only in myelinating oligodendrocytes (Solly et al. 1996).
The detection of the targeted mRNAs was compared to the localization obtained with other ISH techniques described in the literature. A double-labeling technique using Dig and tetramethyl rhodamine isothiocyanate (TRITC)-labeled probes for ISH in conjunction with immunohistochemistry (IHC) of GFAP, an astrocyte marker, and carbonic anhydrase II (CA II), an oligodendrocyte marker (Ghandour et al. 1979,1992,2000), was also described.
Materials and Methods
RNA Isolation
Total RNA was isolated from C57BL/6 mouse brains using TRIzol reagent according to manufacturer instructions (GIBCO BRL; Cergy Pantoise, France). To eliminate any eventual contamination with genomic DNA, isolated RNAs were then treated with DNase I, RNase free (Amersham Pharmacia Biotech; Orsay, France). In addition, before reverse transcription (RT), and the mRNA, we checked for genomic contamination by amplifying the RNA in a PCR reaction with primers that detect genomic PLP/DM20 (Klugmann et al. 1997).
Probe Preparation by RT-PCR Dig or TRITC Labeling
cDNAs were prepared from total mouse brain RNA using a Qiagen Omniscript kit (Qiagen; Courtaboeuf, France). Briefly, total RNA (1–2 μg/reaction) in the presence of Oligo dT15 primers (Promega; Charbonnieres, France), and Omniscript Reverse Transcriptase was incubated at 37C for 1 hr. Three μl of cDNA from the RT reaction was then subjected to PCR using Ready-to-Go PCR beads (Amersham Pharmacia Biotech) in the presence of digoxigenin-11-dUTP or TRITC-5-dUTP (Roche; Meylan, France) and 25 ng of each specific primer to PLP, PLP/DM20, or MOG. PLP/DM20, (Feutz et al. 2001) was amplified with forward exon 2 primer (5'-GCTAATTGAGACCTATTTCTCC) and the reverse exon 6 primer (5'-AGCAATAAACAGGTGGAAGGTC) (GIBCO BRL). PLP was amplified using specifically designed primers, forward exon 3 primer (5'-AAGGGGAGGGGTTCCAGAGG) and the reverse exon 3 primer (5'-AGGGCATAGGTGATGCCCAC) (GIBCO BRL). MOG probes were amplified with forward exon 1 primer (5'-ATGGCCTGTTTGTGGAGCTTC) and the reverse exon 7 primer (5'-TTAGCTCTTCAAGAAACTGTCC) (GIBCO BRL). Amplification was performed using Genius (Techne; Cambridge, UK) and Hybaid PCR Express (Franklin, MA) thermocyclers. The cycling conditions for PCR were 1 cycle of initial denaturation at 95C for 5 min and then for 40 cycles, each at 95C for 0.5 min, 63C for 1 min, and then at 72C for 0.5 min. The expected sizes of the probe products were checked by electrophoresis after migration on 1.5% agarose gel. The expected sizes of the probes were 730, 604/ 499, and 130 bp for MOG-, PLP/DM20-, and PLP-specific probes, respectively.
Tissue Preparation
Mice at postnatal days 1, 20, and 60 and one 1-year-old mouse were anesthetized with 9% chloral hydrate and the brains were fixed by intracardiac perfusion with a fresh solution of 4% paraformaldehyde (Electron Microscopy Sciences; Fort Washington, PA) in 0.1 M phosphate buffer, pH 7.4, containing 0.9% NaCl (PBS). Brains and spinal cords were removed and then postfixed in the same fixative for 24 hr at 4C. Fifty- and 100-μm sagittal sections were cut with a vibratome (Technical Products International; St Louis, MO). Sections were then kept at 4C overnight in PBS or 2 × SSC buffer.
Dot-blot Probes
Dig-labeled probes were tested by the dot-blot method using a nitrocellulose membrane (Millipore; Saint Quentin, France). The denatured probes were serially diluted and spotted onto a nitrocellulose membrane. Spotted probes were then incubated with alkaline phosphatase (AP)-tagged anti-Dig antibody and detected as described below.
In Situ Hybridization
Sections were washed in 0.1 M Tris-buffered saline, pH 7.5 (TBS) twice for 10 min at room temperature (RT), treated with 0.5% Triton X-100 (Sigma–Aldrich; Saint Quentin Fallavier, France) for 15 min at RT, and then briefly washed in the same buffer. Sections were treated with proteinase K (GIBCO BRL) 10 (μg/ml in 0.1 M Tris buffer, pH 8.0, 0.05 M EDTA for 10 min at 37C, immersed in 0.1 M triethanolamine (TEA) for 10 min, in 0.25% acetic anhydride in 0.1 M TEA for 15 min, and then washed in TBS. One μl of 1:2 diluted labeled RT-PCR product was added to 100 μl prehybridization solution (50% formamide, 2 × SSC, 5% dextran sulfate, 1 × Denhardt's, and 0.1 mg/ml salmon testis DNA), boiled for 10 min, and snap-cooled on ice.
Sections were incubated in the above hybridization solution overnight at 40C. Next day the sections were briefly washed in 4 × SSC, followed by two sequential washes of 5 min in 2 × SSC, 1 × SSC, and 0.5 × SSC. After three washes of 10 min in 0.1 M Tris buffer, tissue nonspecific binding was blocked by incubation of sections in 3% DNA blocking solution (Roche) for 1 hour at RT. Sections were rinsed in TBS and incubated with AP-labeled anti-DIG antibodies (anti-digoxigenin–AP, Fab fragments; Roche) at 1:200 dilution in 1 × Denhardt's in TBS at RT for 1 hr. Sections were then washed in TBS three times for 10 min and finally with 0.1 M Tris-HCl, 0.1 M NaCl, 0.05 M MgCl2, pH 9.5 buffer (TBS 9.5). The color was developed in TBS 9.5 in the presence of nitroblue tetrazolium (NBT) and 5-bromo-4-chloro-3-indolyl phosphate (BCIP) (Roche) for 30 min to 1 hr.
Sections were rinsed several times in TBS and then mounted on glass slides with AquaPoly/Mount (Polysciences; Warrington, PA). Controls for ISH consisted of sections incubated in prehybridization solution without addition of cDNA probes. Other control experiments were performed using sense Dig-labeled probes.
Double Labeling for PLP/DM20 ISH and Immunolocalization of CA II and GFAP
One-month-old mice were used. Tissue sections labeled for ISH with AP for PLP/DM20 or MOG were then treated for IHC. After several washes in TBS, pH 7.5, sections were incubated with 0.3% Triton X-100 for 15 min, then incubated with 1 × DNA blocking solution (Roche) for 30 min at RT and then with a rabbit polyclonal antibody to GFAP at 1:200 dilution (Dako; Glostrup, Denmark). Peroxidase-labeled secondary antibody to rabbit IgG at 1:200 dilution was used and peroxidase activity was detected using 3,3’ diaminobenzidine–4HCl (DAB) and hydrogen peroxidase. All substances if not otherwise stated were purchased from Sigma. A similar protocol was used for the double ISH with PLP/DM20–TRITC probes and CA II immunofluorescence. CAII antibodies bound to tissue sections were revealed with fluorescein (FITC)-tagged anti-rabbit IgG.
Results
Probe Synthesis and Labeling
cDNA probes to a major myelin protein in the CNS, PLP and its isoform DM20, were synthesized by RT-PCR from 20-day-old brain total RNA (Feutz et al. 2001). Two other probes, one to MOG, a minor protein of the central myelin (Johns and Bernard 1999), and the second to the PLP isoform only (but not to DM20) were also used. MOG RT-PCR was performed using the sense and antisense primers corresponding to the sequences 60–80 and 767–789, respectively, from the mouse MOG cDNA sequence (Ballenthin and Gardinier 1996). For the PLP-specific isoform, the sense primer corresponds to nucleotides 524–543 and the antisense primer to 612–635 nucleotides from exon 3 of the nucleotide sequence of the mouse proteolipid protein gene (Macklin et al. 1987).
The PCR products were separated on a 1.5% agarose gel after electrophoresis. The bands revealed with ethidium bromide correspond to the predicted sizes of the amplified fragments. The theoretically estimated sizes of the probes were 730, 604/499, and 113 bp for MOG, PLP/DM20, and PLP, respectively (Figure 1A). RT-PCR products for PLP/DM20 and MOG from 1-, 20-, and 130-day-old mice total brain RNAs (Figure 1B) showed high expression at postnatal day 20, whereas the expression was much lower in 1- and 130-day-old mice. To label the cDNA probes, Dig-11-dUTP was introduced to the reaction mixture before PCR cycling. The efficiency of cDNA labeling was checked using a series of dilutions of PCR products on dot-blots (not shown). The probes were then used without purification after the PCR step. However, a purification step of PCR products did not further improve the ISH technique used in this study.
In Situ Hybridization
We used the probes Dig-labeled by RT-PCR for ISH. To determine the specificity of the method, we used three probes to assess PLP/DM20, PLP, and MOG mRNAs in oligodendrocytes. Oligodendrocytes were stained in brain and spinal cord throughout different stages of mouse postnatal development. Of the three probes, PLP/DM20 gave the most intense staining in brain (Figures 2A–2D). Higher magnification of the corpus callosum at day 20 showed typical perinuclear cytoplasmic staining. Other authors (Verity et al. 1990; Ghandour and Skoff 1991; Feutz et al. 1995; Bessert and Skoff 1999; Thomson et al. 1999) described this pattern of staining of oligodendrocytes using ISH with full-length cDNA or cRNA antisense probes (Breitschopf et al. 1992; Spassky et al. 1998). A reduction in PLP/DM20 messages was observed as the animal aged (Figure 2D), which is in accordance with previous reports (Macklin et al. 1987; LeVine et al. 1990; Ikenaka et al. 1992).
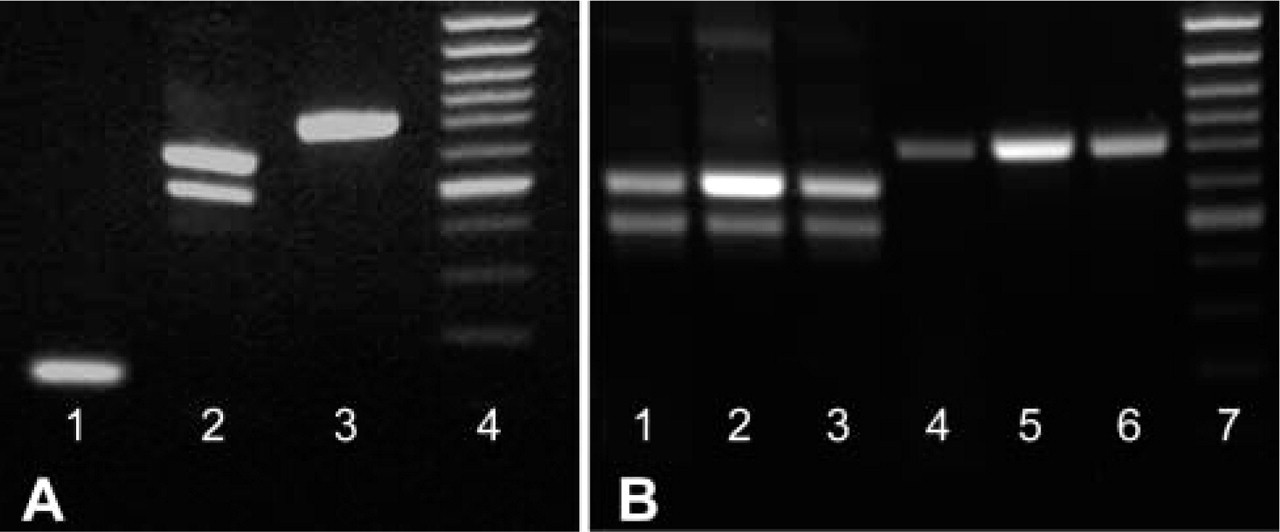
RT-PCR final products were separated on 1.5% agarose gel. The bands revealed with ethidium bromide correspond to the predicted sizes of amplified DNA fragments. Lane 1, PLP specific (113 bp); Lane 2, PLP/DM20 (604/499 bp); Lane 3, MOG (730 bp) probes; Lane 4, molecular weight markers (100 bp). (
The proteolipid protein gene produces two alternatively spliced proteins: DM20 and PLP. Our primers specific for PLP only were chosen mainly from the latter half of exon 3, which is missing in the splice form that produces mRNA for DM20. ISH using this probe labeled oligodendrocytes in brain and spinal cord from day 1 (Figures 2E–2G), day 20 (Figure 2H), and adult mice (Figure 2I). Higher magnification (Figures 2G and 2I) showed the same pattern of staining as with PLP/DM20 probes, which was predicted. Control experiments using hybridization buffer without cDNA (Figure 2J) or with labeled sense probes (not shown) did not show any significant staining in brain sections.
PLP and DM20 are highly expressed in oligodendrocytes, accounting for approximately 40% of total protein. To determine the sensitivity of our method, we assessed expression of MOG mRNA. MOG is a minor component of myelin, representing 0.05–0.1% of total myelin protein (Johns and Bernard 1999). MOG transcripts were detected in oligodendrocytes before myelin formation in 1-day-old mice (Figures 3A and 3B). The expression of MOG was also drastically downregulated in 2-month-old mice (Figure 3D) and was further decreased at 12 months (data not shown).
Combined IHC and ISH
We also assessed the possibility of using this method in combination with IHC. We used the PLP/DM20 probe for ISH and for IHC we used a classical marker for astrocytes, GFAP. The hybridization of the Dig-cDNA labeled probes and the detection of AP activity with BCIP and NBT were restricted to oligodendrocytes, while the typical astrocyte morphology was revealed with GFAP using immunoperoxidase IHC. No overlapping was observed between the two cell populations (Figures 4A and 4B). Furthermore, we combined ISH of PLP/DM20 using cDNA probes labeled with dUTP–TRITC and the immunofluorescence of CA II, a marker for oligodendrocytes (Ghandour et al. 1979,1992,2000) detected with FITC-tagged secondary antibodies (Figures 4C–4F). Complete overlapping between green oligodendrocytes (CA II-positive cells) and red PLP/DM20 mRNA-positive cells was found.
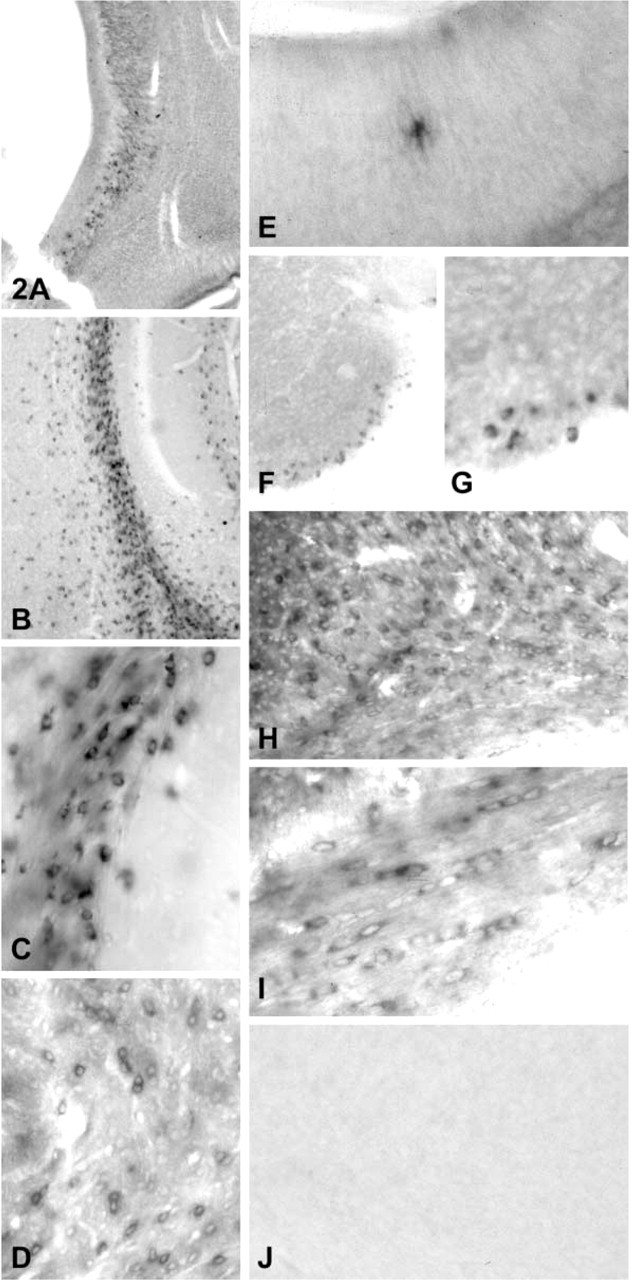
PLP/DM20- and PLP-specific transcripts visualized by ISH in brain and spinal cord tissue sections. PLP/DM20 transcripts were detected in cells in the subventricular layer of 1-day-old mouse brain (
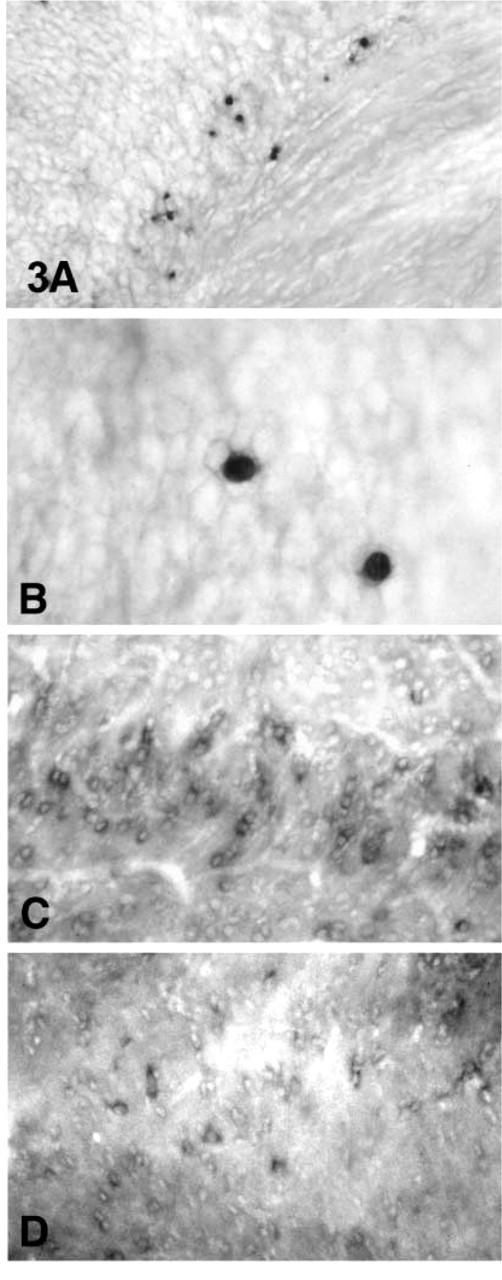
MOG probes were hybridized to brain tissue sections in 1-day-old (
Discussion
PCR represents the most common and widespread method for direct amplification of specific sequences of nucleic acid target molecules. The most sensitive method uses single-stranded cRNA riboprobes. Although these probes give little background, their synthesis requires elaborate techniques. The use of non-radioactive probes synthesized by PCR provides a significantly more rapid procedure in comparison with the preparation of probes using plasmid vectors. The use of crude Dig-labeled probes by PCR is less time-consuming because plasmid amplification, cutting, separation, and cleaning of DNA are excluded and concentration of probe by amplification and labeling takes place in one step in the PCR reaction. Dig-labeled DNA probes using RT-PCR have been used to assess mRNA in bone tissue (Fujii et al. 1999; Kitazawa et al. 1999a) and in liver (Kitazawa et al. 1999b), but fundamental differences exist between our present method and that described in those three references. Our method reported here is very simple, one-step PCR and no need for purification of PCR products. In contrast, the reported method (Fujii et al. 1999; Kitazawa et al. 1999a,b) is very laborious, consisting of a first unlabeled PCR, electrophoresis, gel cutting, second PCR with sense primers, and finally a second purification step.
Another advantage of the PCR-generated Dig-labeled probes is that the size and sequence can be easily selected. In addition, several sequences from the same mRNA can be easily generated and compared. We found that the size of the Dig probes between 400 and 700 bp gives the most intense staining for ISH. However, low sizes of ∼100 bp, which are hard to construct for RNA–RNA ISH, can be successfully labeled by PCR. We demonstrated this by using primers of 113 bp to detect the expression of PLP-specific mRNA. However, fewer cells were stained with the PLP-specific probe compared with PLP/DM20 probes. This observation is in accordance with previous studies that detected expression of DM20 before PLP in brain as early as E9.5 (Timsit et al. 1995; Peyron et al. 1997). Using IHC with an antibody raised against the 35 amino acids specific for PLP only, PLP expression was demonstrated to start later than DM20. Very low levels were found in rat premyelination oligodendrocytes at P7, during early stages of myelination (Trapp et al. 1997). However, expression of PLP-specific mRNA was reported in P3 rat subcortical white matter using radiolabeled probes (LeVine et al. 1990). Radiolabeled PLP probes were initially used to detect the specific mRNA but they then generated high background, and the morphology of the labeled cells was difficult to ascertain (LeVine et al. 1990; Woodruff and Franklin 1998; Thomson et al. 1999). In contrast, nonradioactive PCR Dig-labeled probes used in the present study for ISH in vibratome-fixed floating sections gave a lower background and the cell morphology was well defined.
In addition, the sensitivity of the ISH technique used in this study was proved by detection of low-expression MOG transcripts in oligodendrocytes. Published data (Solly et al. 1996) showed that the detection of MOG transcripts is limited to myelinated brain oligodendrocytes from 7-day-old postnatal mice but not at earlier time points. However, the present data showed that both MOG and PLP were detected in premyelinating oligodendrocytes from 1-day-old mouse brains and spinal cords. Our laboratory has been using ISH with plasmid full-length cDNA for more than 3 years (Bessert and Skoff 1999), and we see similar staining intensity, with maybe less background, in ISH with PCR-labeled probes.
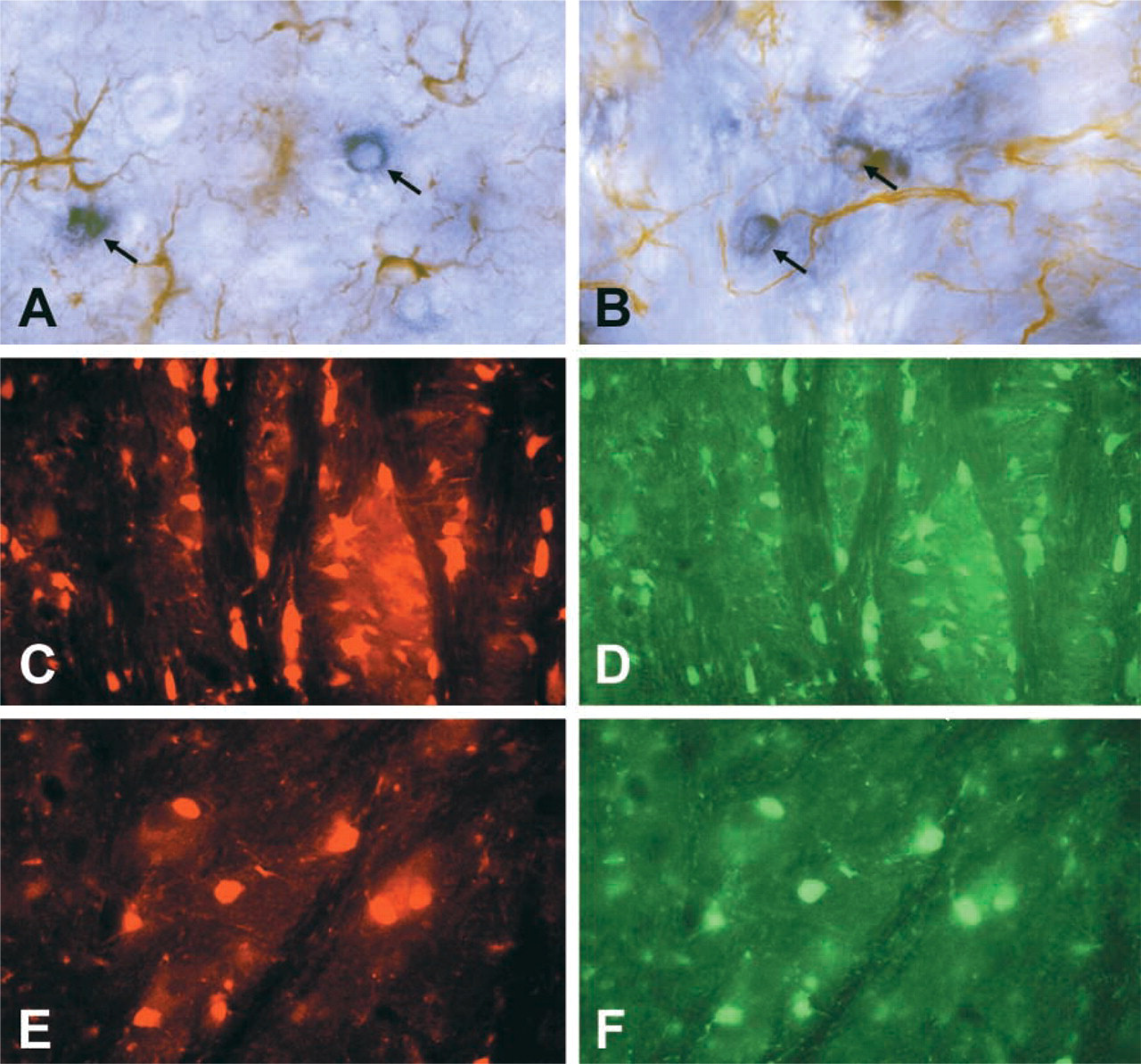
Double labeling for PLP/DM20 ISH and GFAP IHC in 30-day-old mouse brain. Hybridized PLP/DM20 Dig probes to tissue sections were revealed with AP-labeled anti-DIG antibody using BCIP and NBT. Antibodies to GFAP in the same tissue sections were detected using a peroxidase ICC technique with DAB. Blue oligodendrocytes in corpus callosum (arrows) were stained for PLP/DM20 mRNA, while the brown astrocyte population was stained for GFAP (
We also combined the ISH method with IHC to detect two types of cells, oligodendrocytes and astrocytes, in the same tissue section and to visualize the same cell population detected for the mRNA content and for antibodies to the cell markers. The combined ISH and immunohistofluorescence technique used in the present study provides evidence for the specificity of the cDNA probes and the antibodies alike. The present report shows that in situ hybridization with Dig-labeled cDNA by RT-PCR provides a simple and sensitive method to detect myelin mRNAs.
Footnotes
Acknowledgements
Supported by Association Française contre les Myopathies (AFM) to MSG and NINDS 38236 to RPS. We wish to thank Prof D. Grucker for support and encouragement and Ms D. Bessert and E. Scherrer for technical assistance.