
Abstract
We have studied the transduction of TAT-HA-β-galactosidase fusion protein into two cell lines of rat salivary gland origin, A5 and C6–21, into cells of fetal mouse submandibular glands in organ culture, and into rat submandibular gland after retrograde duct injection, using a histochemical method to demonstrate β-galactosidase activity. Transduction of the fusion protein into A5 and C6–21 cells was concentration- and time-dependent. Therefore, the intensity of the β-galactosidase staining, which was cytoplasmic, was less after 1 hr of exposure compared to exposures up to 24 hr. However, the fusion protein was transduced into 100% of both types of cultured cells. When explants of mouse fetuses at 13 days of gestation were exposed to the fusion proteins, both epithelial and mesenchymal cells were stained for the enzyme, with a conspicuous accumulation of the reaction product at perinuclear cytoplasmic regions. The histochemical staining of the mesenchymal cells was more intense compared to that seen in epithelial cells. TAT-HA-β-galactosidase fusion protein was also delivered to rat submandibular glands by retrograde duct injection. Histochemical staining for β-galactosidase activity of cryostat sections prepared from the injected glands revealed that the transduction of the fusion protein was also time- and dose-dependent. In the glands of rats sacrificed from 10 min to 1 hr after the retrograde injection, essentially all acinar and duct cells showed cytoplasmic staining. The intensity of the staining then declined, and was not seen in the glands of rats killed 24 hr after the injection of the fusion proteins. These results indicate that a full-length, active TAT fusion protein can be targeted to salivary gland cells both in vitro and in vivo to analyze physiological, developmental, and pathophysiological processes.
Keywords
T
As a continuation of our work on transduction of salivary gland cells in vivo by retroviral vectors (Barka and van der Noen 1996, 1997), we describe here the transduction of TAT-HA-β-galactosidase fusion protein into cells of submandibular gland (SMG) origin and into fetal SMG, as well as the targeted delivery of the fusion protein into rat SMG by retrograde duct injection. These findings open up the possibility of experimental cellular manipulations of developing and adult salivary glands by using constructs of fusion proteins.
Materials and Methods
Purification of TAT-HA-β-gal and HA-β-gal Fusion Proteins
Bacteria [BL-21(CD3)pLysS] expressing the vectors pTAT-HA-β-gal and pRSET-HA-β-gal were the generous gift of Dr. S. F. Dowdy. The vector pTAT-HA has an N-terminal 6-histidine leader followed by the 11-amino acid TAT protein transduction domain, glycine residues, the hemaglutinin tag (HA), and a polylinker (Nagahara et al. 1998). Into this was cloned the β-gal to produce the in-frame fusion protein pTAT-HA-β-gal. In the construct pRSET-HA-β-gal the TAT domain was removed.
Purification of TAT-HA-β-gal was done under native conditions using Ni-NTA metal-affinity chromatography according to the manufacturer's (QIAGEN; Valencia, CA) protocols, with modifications. For preliminary experiments Ni-NTA Spin Columns and for large-scale purifications the batch method was used. Briefly, the method was as follows. The bacteria were grown in LB medium (Sigma–Aldrich; St Louis, MO), and expression was induced by 1 mM isopropyl-β-
This purification protocol was adapted after preliminary experiments had established that about 50% of TAT-HA-β-gal is soluble under non-denaturing native conditions. Furthermore, in our hands, the fusion protein prepared under denaturing conditions (8 M urea) and further purified by ion exchange chromatography showed poor transduction efficiency.
HA β-gal fusion protein was purified according to the protocol of Dr. S. F. Dowdy (personal communication). This differed from the protocol of purification of TAT-HA-β-gal by including protease inhibitors (1 μg/ml each of aprotinin and leupeptin and 5 μg/ml phenylmethylsulfonyl fluoride) (Sigma–Aldrich) and omitting the lysozyme treatment. Most of the HA-β-gal fusion protein was eluted from the Ni-NTA matrix by 100 mM imidazole.
Cell Cultures and Transduction of Cultured Cells with the Fusion Proteins
The epithelial duct cell line of rat SMG origin, A5 (Brown et al. 1989), was a gift of Dr. B.J. Baum. A5 cells were grown in DMEM supplemented with 10% FBS and antibiotics in a Nunc Lab-Tek Chamber Slide System (Fisher Scientific; Pittsburgh, PA). One day before the addition of the fusion protein, 25,000–50,000 cells/well were plated. The medium was then removed and replaced with 0.4 ml DMEM, 5% FBS containing about 20 μg/ml fusion protein TAT-HA β-gal or HA-β-gal. The approximate concentrations of the fusion protein preparations were estimated by comparing the intensity of the specific β-gal bands with that of molecular weight standards after SDS-PAGE and Coomassie blue staining. Control cultures were fed by the same medium without the fusion protein. After 1–24-hr exposure, the cultures were processed for the histochemical demonstration of β-gal activity.
Experiments of the same design were also done using rat SMG-derived C6–21 cells (Quissell et al. 1997) (a gift of Dr. Quissell). These cells display several characteristics of acinar cells. C6–21 cells were grown in DMEM:F12 1:1, 5% FBS, and 1 μl/ml MITO. MITO SERUM EXTENDER is lyophilized from a solution of Dulbecco's PBS containing EGF, transferrin, insulin, endothelial cell growth supplement, triiodothyronine, hydrocortisone, progesterone, testosterone, estradiol-17, selenious acid and O-phosphorylethanolamine (concentrations are not given) (Becton–Dickinson; Fisher Scientific, Pittsburgh, PA). We have found that MITO could replace the supplements described by Quissell and co-workers (1997) to grow C6–21 cells. These cells do not grow unless MITO is added to the medium.
Organ Culture and Transduction
SMGs were isolated from CD-1 mouse fetuses at E13 (E0 = day of vaginal plug) and collected into BGJb medium containing TAT-HA-β-gal, incubated for 24 hr at 37C in 5% CO2 and 95% humidity. The glands were rinsed in PBS, fixed in 0.5% glutaraldehyde for 20 min, and processed for histochemical demonstration of β-gal activity. They were then photographed, postfixed in formalin, embedded in Historesin, and sectioned at 6 μm. This experiment was repeated using several explants.
Retrograde Duct Injection of the Fusion Proteins
Female Sprague–Dawley rats weighing 190–200 g were used. The fusion proteins in 0.1 ml of DMEM were injected retrogradely into the left lobe of the SMG by cannulating the main excretory duct (Omnell and Qwarnström 1983) as in our previous studies (Barka and van der Noen 1996, 1997). The rats were anesthetized (Ketamine 60 mg/kg and xylazine 8 mg/kg IM) and received 6 mg/kg pilocarpine-HCl SC 45–60 min before cannulation to deplete the gland of its secretory products. After insertion of the cannula, 1 mg/kg atropine was injected SC to reduce residual saliva flow. The rats were sacrificed by CO2. Ten min (one rat), 15 min (two rats), 1 hr (two rats), 4 hr, and 19 hr (one rat each) after administration of the fusion protein, both lobes of the SMG and cervical lymph nodes were removed for histochemical analyses. In carrying out the animal experiments, institutional guidelines were observed.
Histochemical Methods
The cultures were rinsed in PBS, fixed in 0.5% glutaraldehyde in PBS for 20 min at room temperature, washed 3 × 1 min in PBS, and incubated for the demonstration of β-gal activity (Sanes et al. 1986). In general, incubation was overnight at room temperature in the dark. The same method was used to demonstrate β-gal activity in cultured fetal glands in toto or in sections and cryostat sections of SMGs and lymph nodes.
Results
Cell Culture Experiments
Before carrying out the in vivo experiments, we studied the transduction of the TAT-HA-β-gal fusion protein into salivary gland-derived cell lines in vitro. For most experiments, we used the A5 cell line which was generated from fragments of rat SMGs exposed to 3-methylcholanthrene (Brown et al. 1989). In addition, the C6–21 cell line was also employed, which derived from rat SMG. C6–21 cells have several characteristics of acinar cells.
A5 cells were grown in tissue culture chambers and were exposed to the fusion proteins in DMEM medium containing 5% FBS for 1–24 hr. The cultures were then fixed and stained for β-gal. Cells not exposed to TAT-HA-β-gal revealed no β-gal activity (Figure 1). All cells exposed to the fusion protein showed enzyme activity. Both the frequency of cells stained and the intensity of staining were concentration- and time-dependent (Figures 2–4). After exposure to low concentrations of the fusion protein for 1 hr, only scattered cells were stained intensely. However, after exposure for 2 hr most cells were stained, and by 6 hr all cells stained but with differing intensity. By 18 or 24 hr, the staining, which was cytoplasmic, became uniform. Such a uniform staining was obvious in cells grown as a monolayer, but scattered cells at the periphery of the culture also displayed a strong enzymatic activity. With higher concentration of TAT-HA-β-gal, all cells revealed enzyme activity after 1 hr of exposure.
As shown earlier, the TAT sequence is required for penetration of proteins into the cells. Cells exposed to HA-β-gal stained faintly compared to cells exposed to TAT-HA-β-gal (data not shown). This staining may be caused by β-gal adsorbed to cell surfaces and not removed by the rinsing performed before fixation. However, when the cells were exposed to high concentrations of HA-β-gal, the staining was more intense but apparently not intracellular.
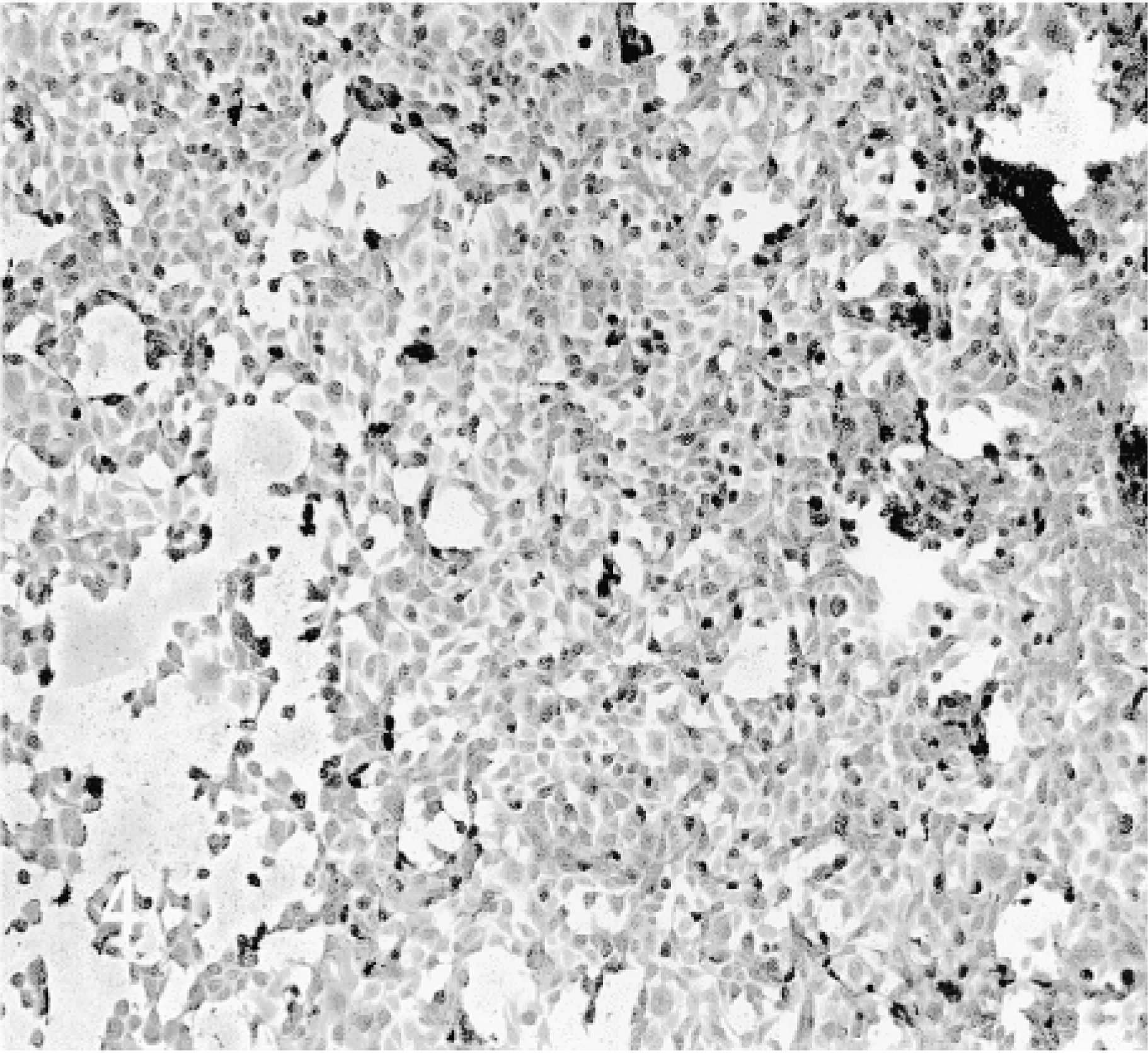
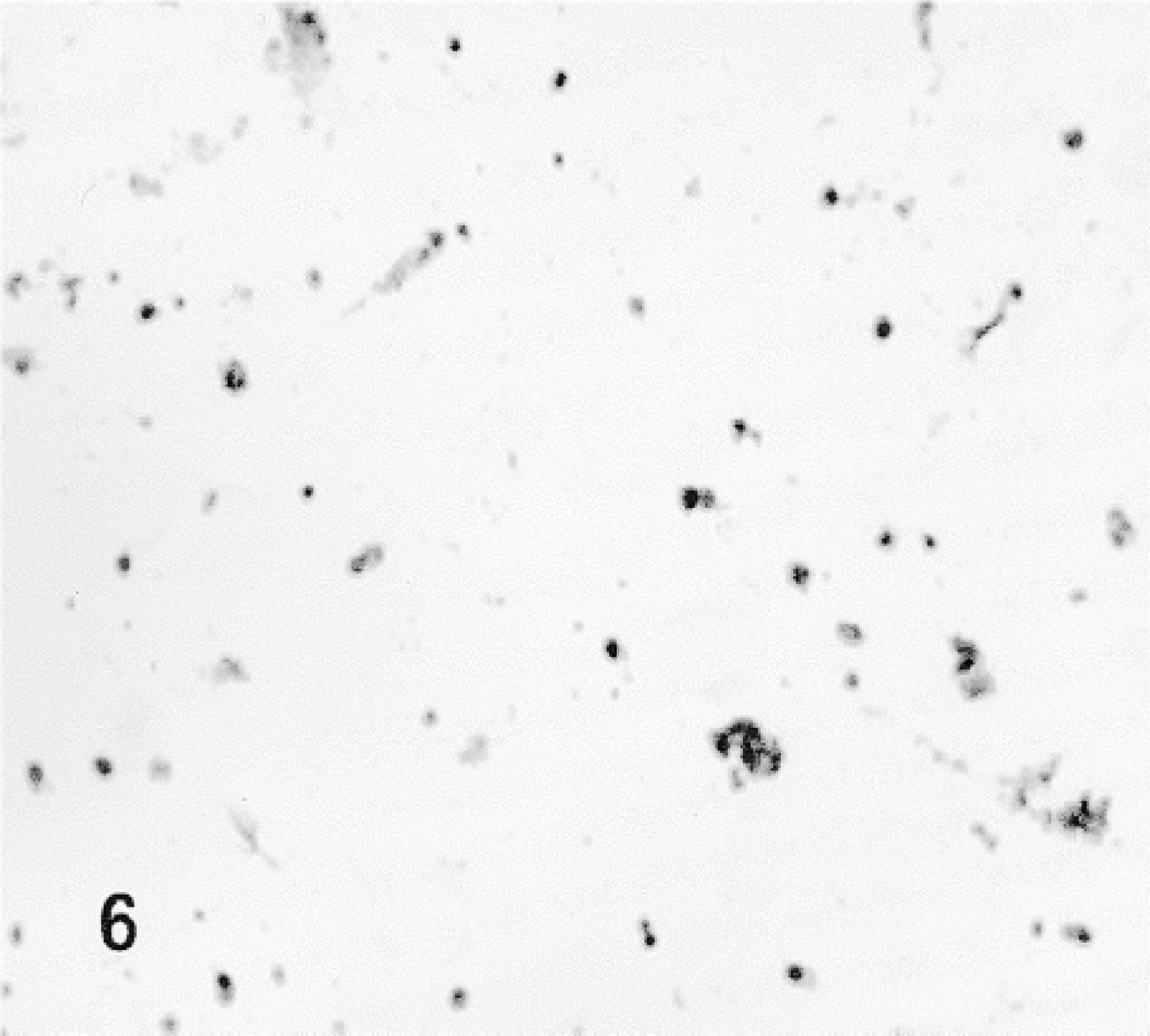
C6–21 cells revealed no β-gal activity (Figure 5). Cells exposed to the TAT fusion protein for 18–24 hr were strongly stained (Figure 6). Shorter exposure times with this cell line were not studied.
Organ Culture Experiments
SMGs isolated from E13 fetuses were exposed to DMEM medium containing 5% FBS and about 20 μg/ml TAT-HA-β-gal fusion protein. After an incubation period of 24 hr, the glands were processed for demonstration of β-gal activity both in toto and in sections after embedding in Historesin. Staining for β-gal was seen throughout the explant, but the reaction was more intense in the mesenchyme than in the epithelium (Figure 7). Microscopically, distinct staining of the membrane, but not the contents, of the nuclei of both epithelial and mesenchymal cells was observed (Figure 8). Cytoplasmic staining was weak in epithelial cells but was stronger and distinctly granular in the mesenchymal cells. We observed no endogenous β-gal activity in fetal SMGs (data not shown).
In Vivo Experiments
Fusion proteins TAT-HA-β-gal and HA-β-gal were injected by retrograde duct injection into the SMG, and both lobes, injected and contralateral non-injected, as well as cervical lymph nodes, were examined for the localization of β-gal activity. No β-gal activity was detected in the non-injected lobes of the glands (Figure 9) or in the sublingual gland (Figures 10 and 12), which is attached to the SMG but has an independent excretory duct. A few scattered cells revealed enzyme activity in the lymph nodes. The nature of these cells was not investigated.
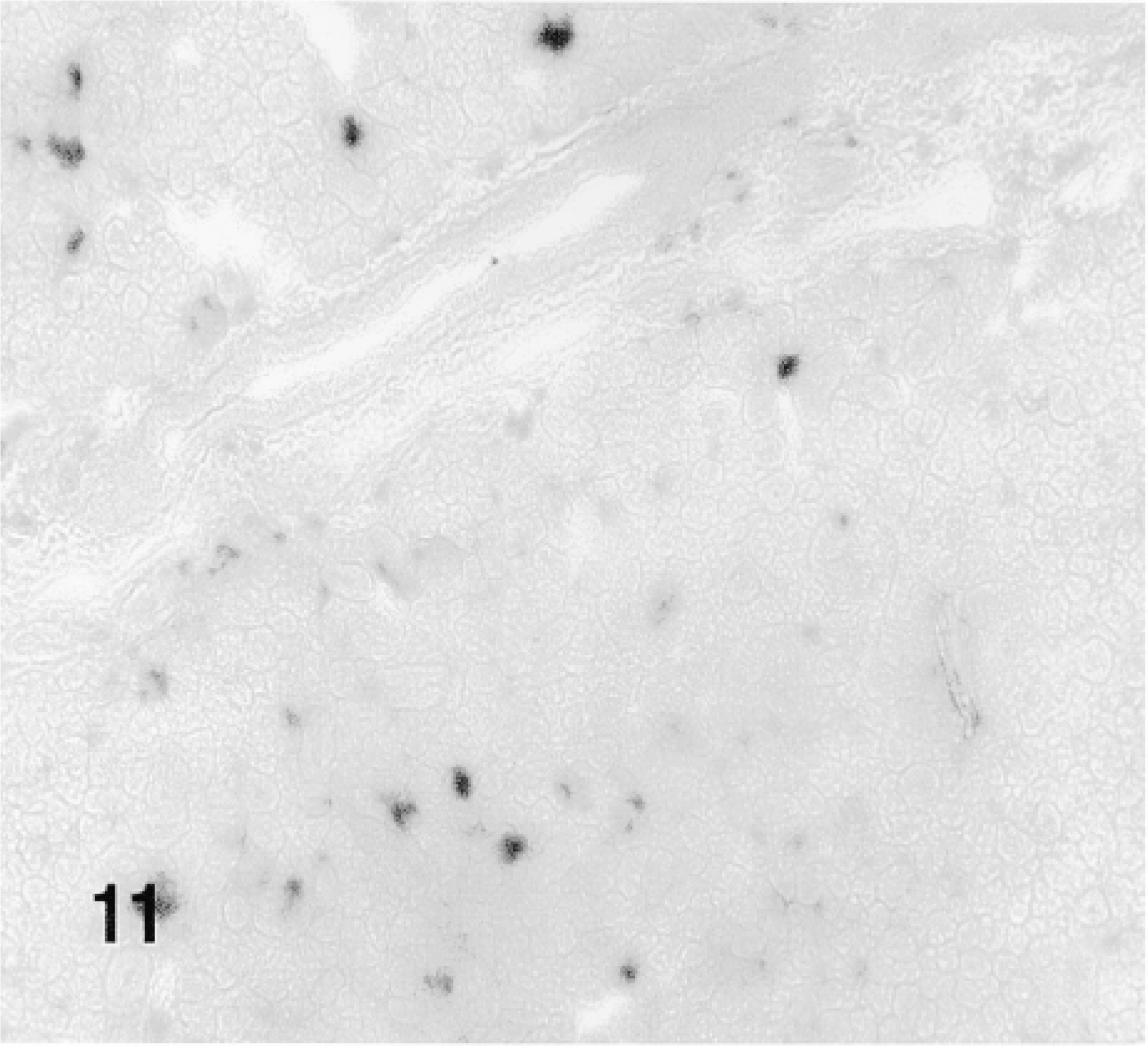
β-Gal activity was detected in the glands of rats sacrificed 10 min to 4 hr after the administration of TAT-HA-β-gal (Figures 10–13) but not in the gland injected 19 hr earlier. In the glands of rats sacrificed 10 or 15 min or 1 hr after injection of the fusion protein, most if not all cells showed enzyme activity. These included both duct and acinar cells. The staining was intense, but with some variation among the lobules. This variation was probably caused by the uneven distribution of the retrogradely injected solution. In the glands of rats sacrificed 1 or 4 hr after the retrograde injection of the fusion protein, all cells were stained intensely. Although not quantified, the intensity of the staining appeared to decline with time. A comparison of the intensity of staining 1 hr after the injection of the fusion protein, using two different preparations, suggests that the transduction is dose-dependent. Occasionally, reaction product was seen in the lumen of small ducts and in connective tissue cells. The latter may be caused by escape of the injected material, perhaps because of the pressure exerted by the injection. One hour after the retrograde duct injection of HA-β-gal, in approximately the same amount as that of TAT-HA-β-gal, there was no specific staining in the gland (Figure 14).

Culture of A5 cells stained for β-gal.
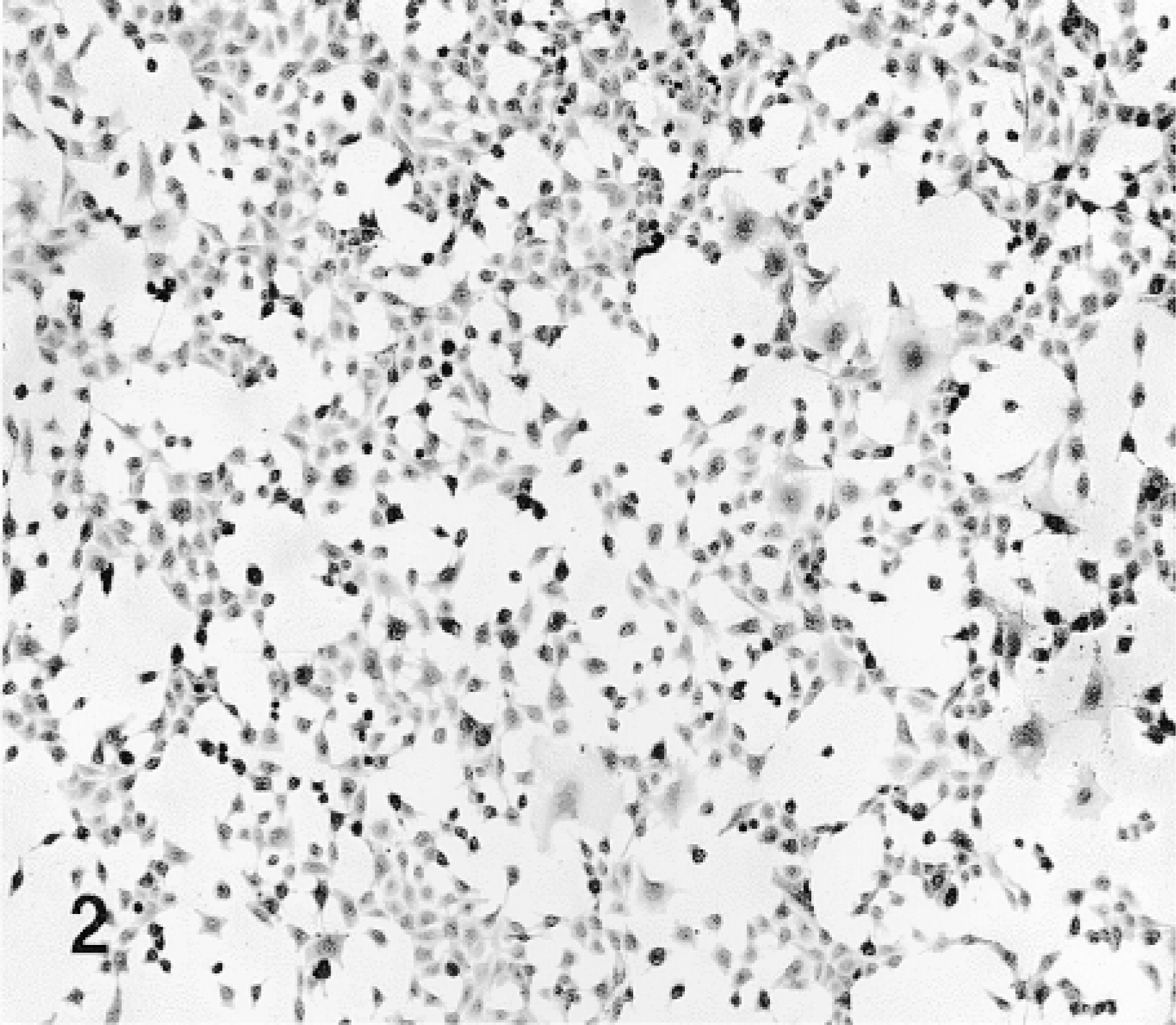
A5 cells exposed to TAT-HA-β-gal for 1 hr.
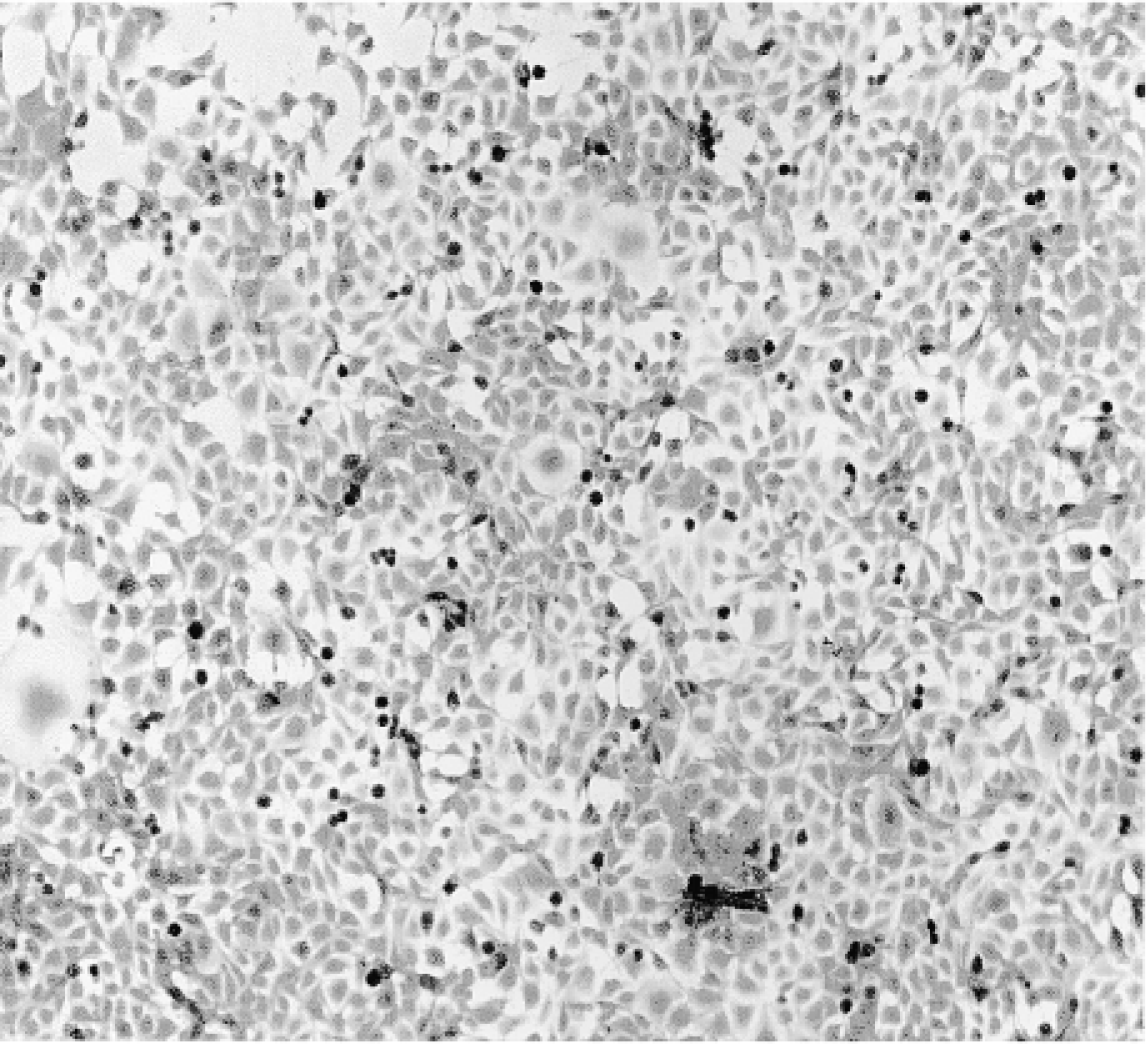
A5 cells exposed to TAT-HA-β-gal for 4 hr.
A5 cells exposed to TAT-HA-β-gal for 24 hr.

C6–21 cells stained for β-gal.
C6–21 cells exposed to TAT-HA-β-gal for 18 hr.
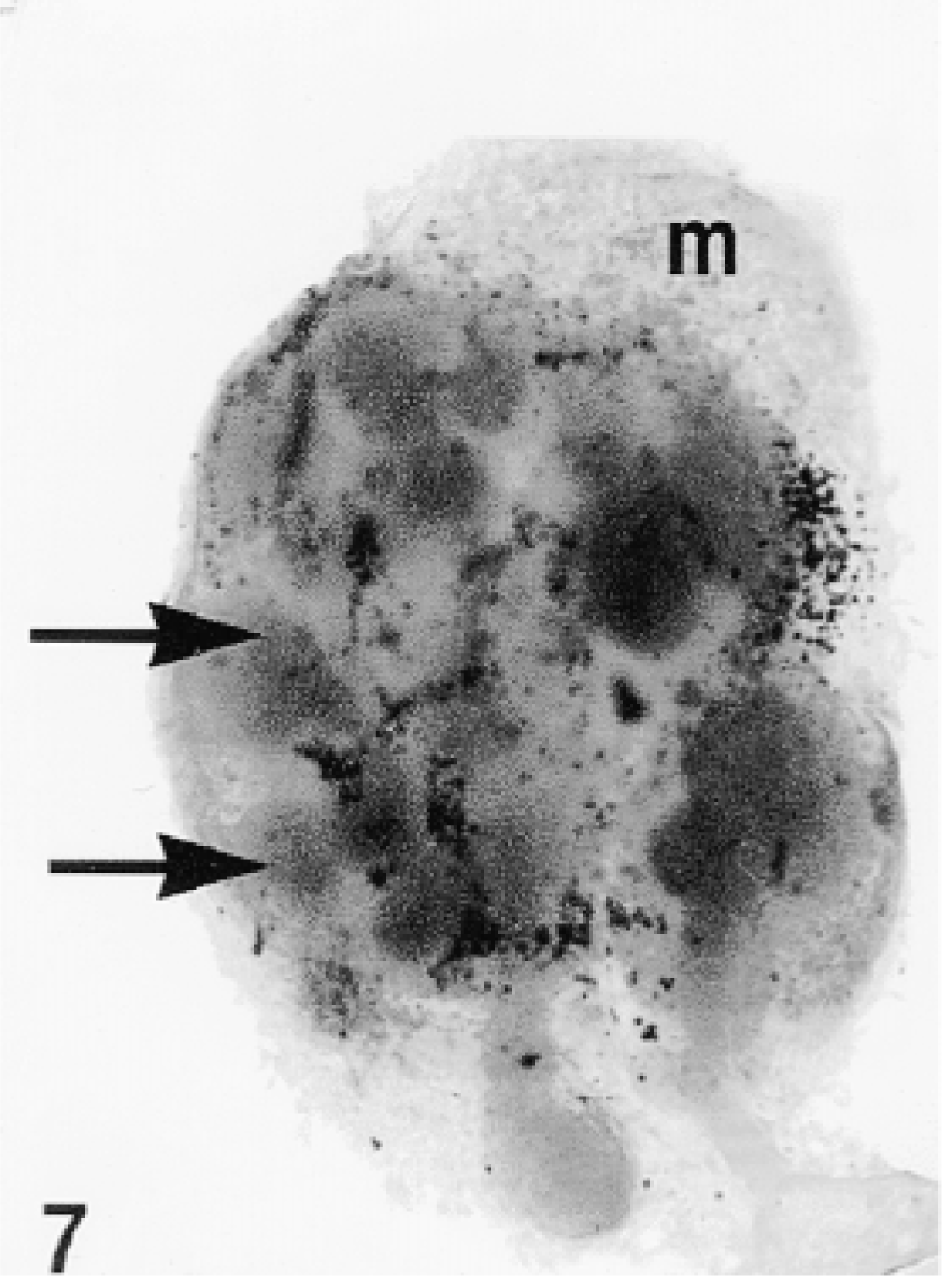
Total gland, organ culture. The original photograph was taken at ×50 magnification.
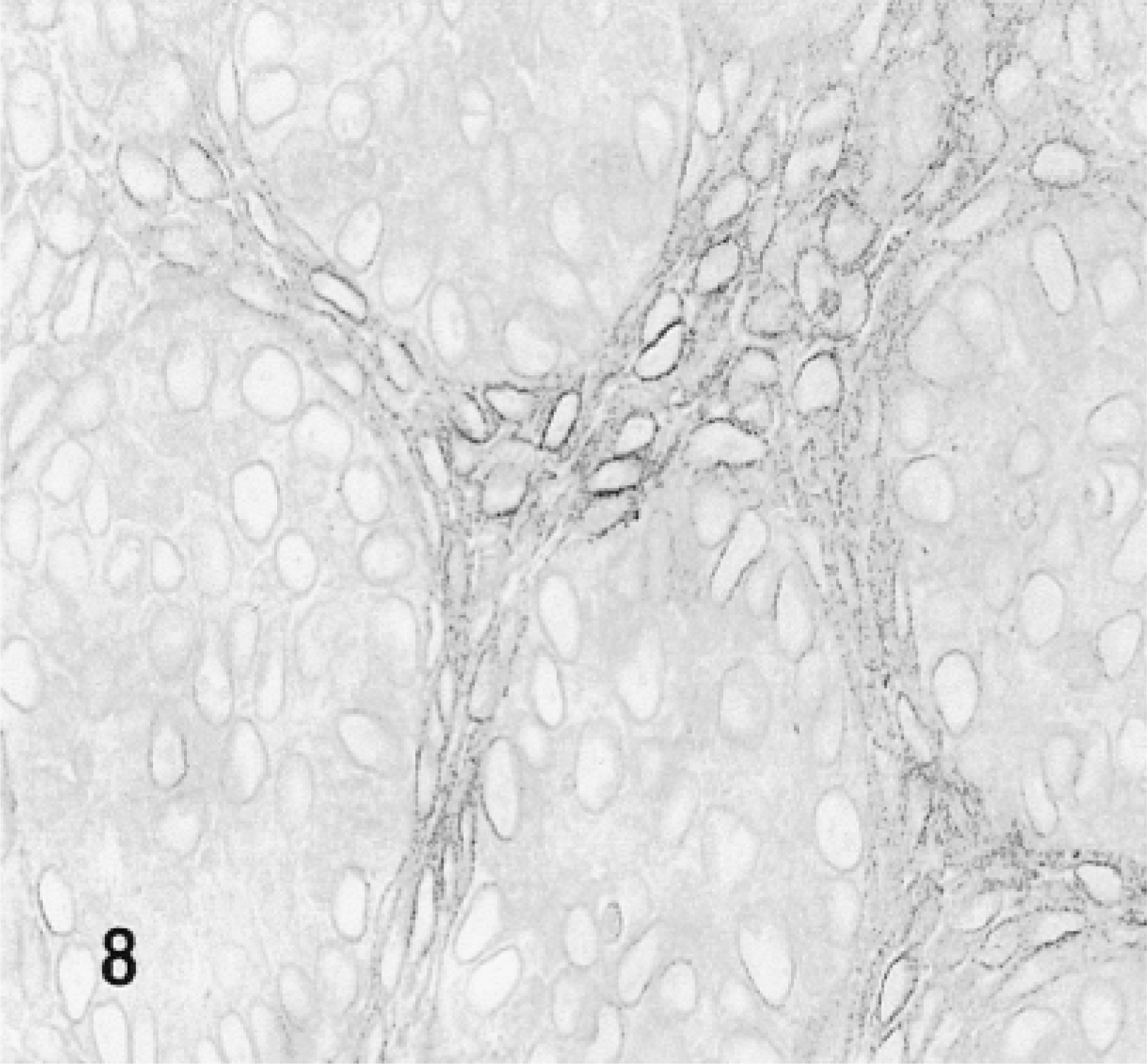
Light micrograph of a section prepared from the gland shown in Figure 7. Portions of cross-sections of five endpieces and intervening mesenchyme are shown. All epithelial and mesenchymal cells are stained for β-gal with an accumulation of the reaction product in the perinuclear regions. Arrows indicate endpieces; m, mesenchyme.
Discussion
HIV TAT-mediated protein transduction, originally described in 1988 (Green and Loewenstein 1988; Frankel and Pabo 1998), is a rapidly expanding field. The methodology now allows the generation of full-length TAT fusion proteins and their introduction into primary and transformed cultured cells with an efficiency approaching 100%. Because TAT domain-mediated penetration of fusion proteins does not involve cell receptors or transporters (Mann and Frankel 1991; Vives et al. 1997), essentially any cell type, including neurons, can be transduced. Hence, that the salivary gland-derived cell lines A5 and C6–21 were transduced by the fusion protein TAT-HA-β-gal was not unexpected. The transduction of these cell lines was time- and concentration-dependent. When cultures of A5 cells were exposed to TAT-HA-β-gal for 18–24 hr, 100% of cells stained for β-gal, with higher intensity than cells exposed for 1–2 hr. Whether the increase in transduction efficiency was caused by continuous uptake of the fusion protein from the culture medium and/or by activation of the enzyme by structural changes has not been established. However, because we used non-denaturing conditions for the isolation of TAT-HA-β-gal, no refolding by chaperones has to be involved in the increase of enzymatic activity. We chose the salivary gland-derived cell lines because of our interest in transducing salivary gland parenchymal cells in vivo.

Cryostat section stained for β-gal of the non-injected (control) lobe of the SMG of a rat.
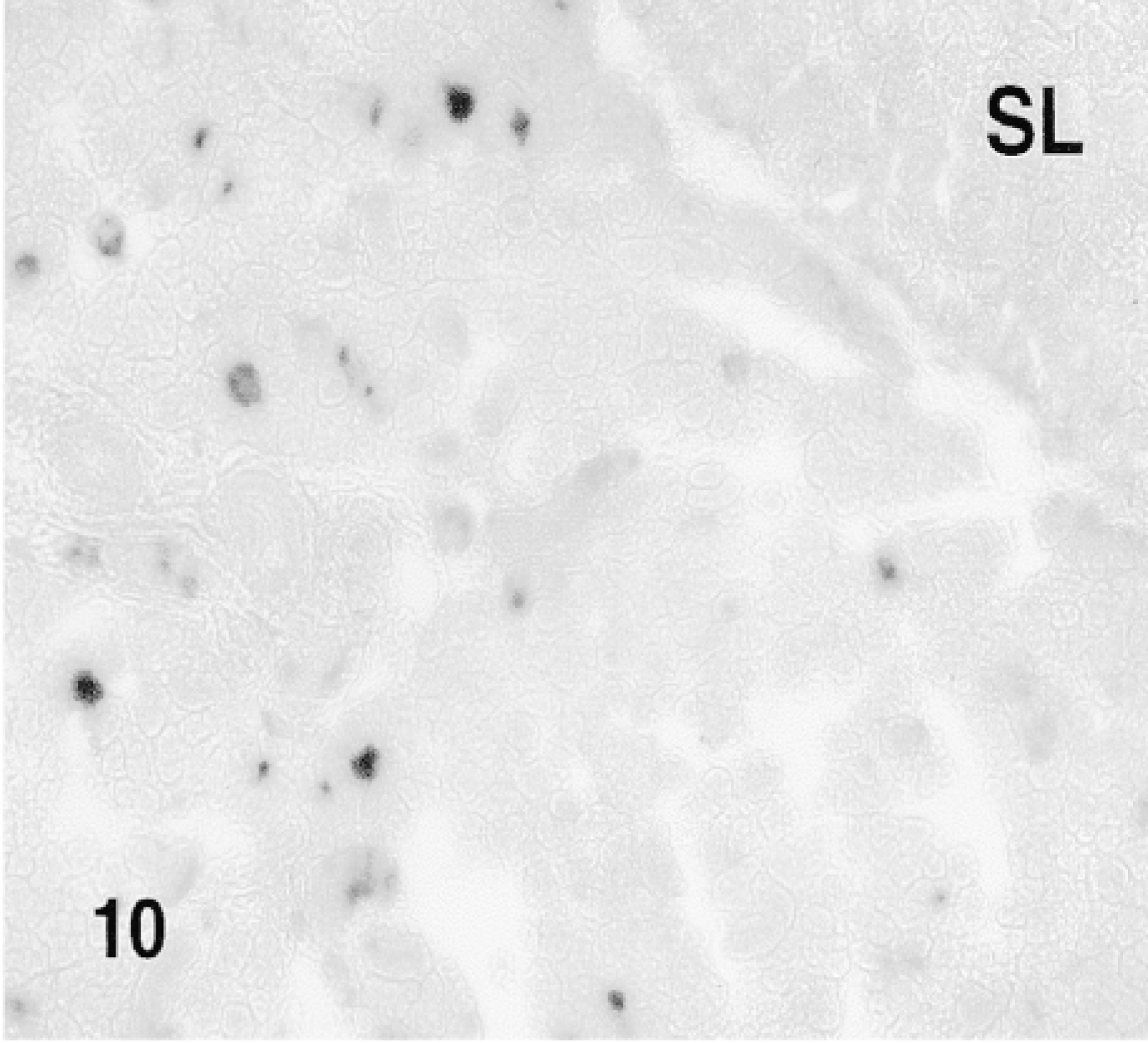
10 min. after the injection of the fusion protein. SL: sublingual gland.
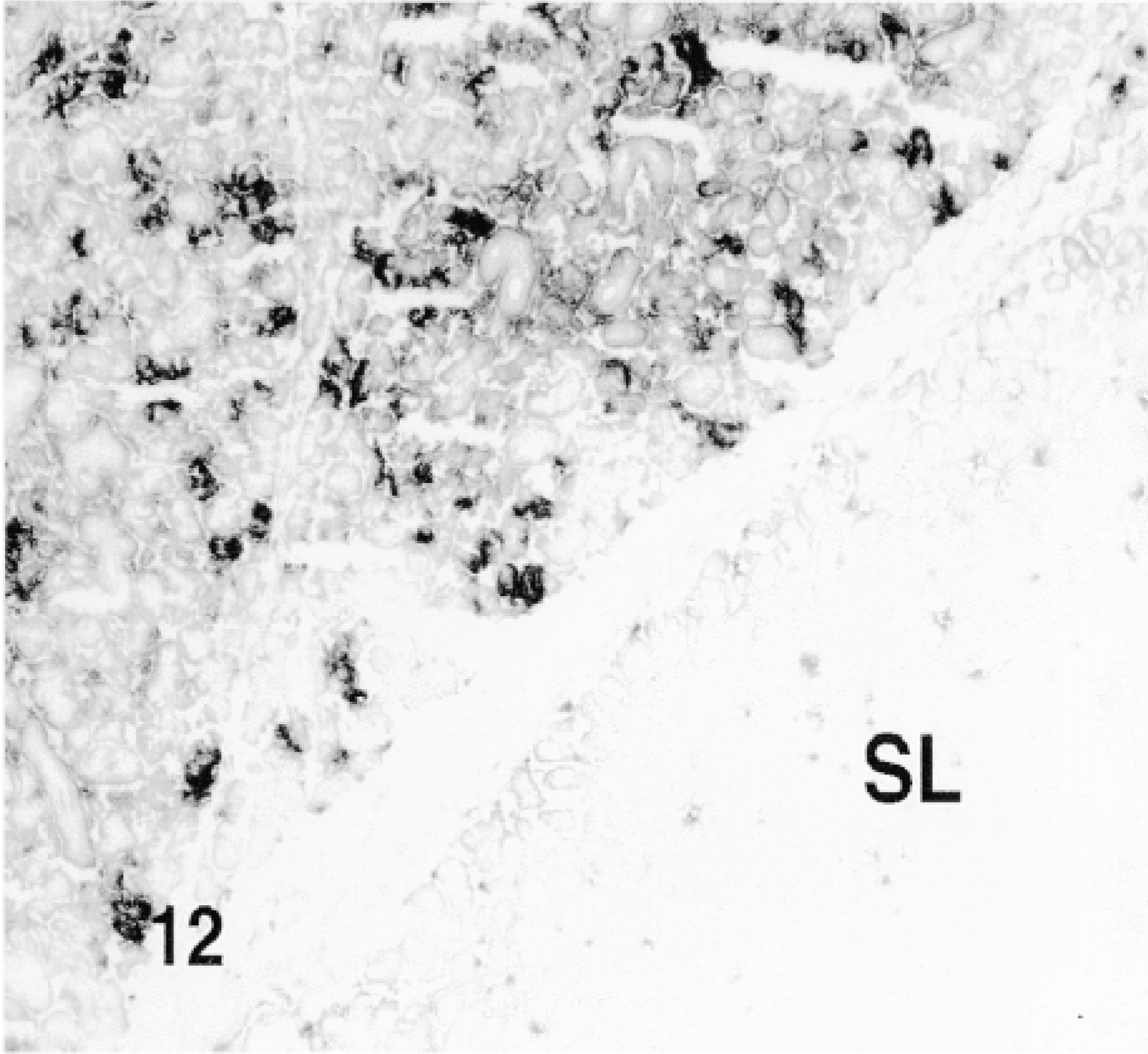
1 hr after the injection of the fusion protein. SL: sublingual gland.
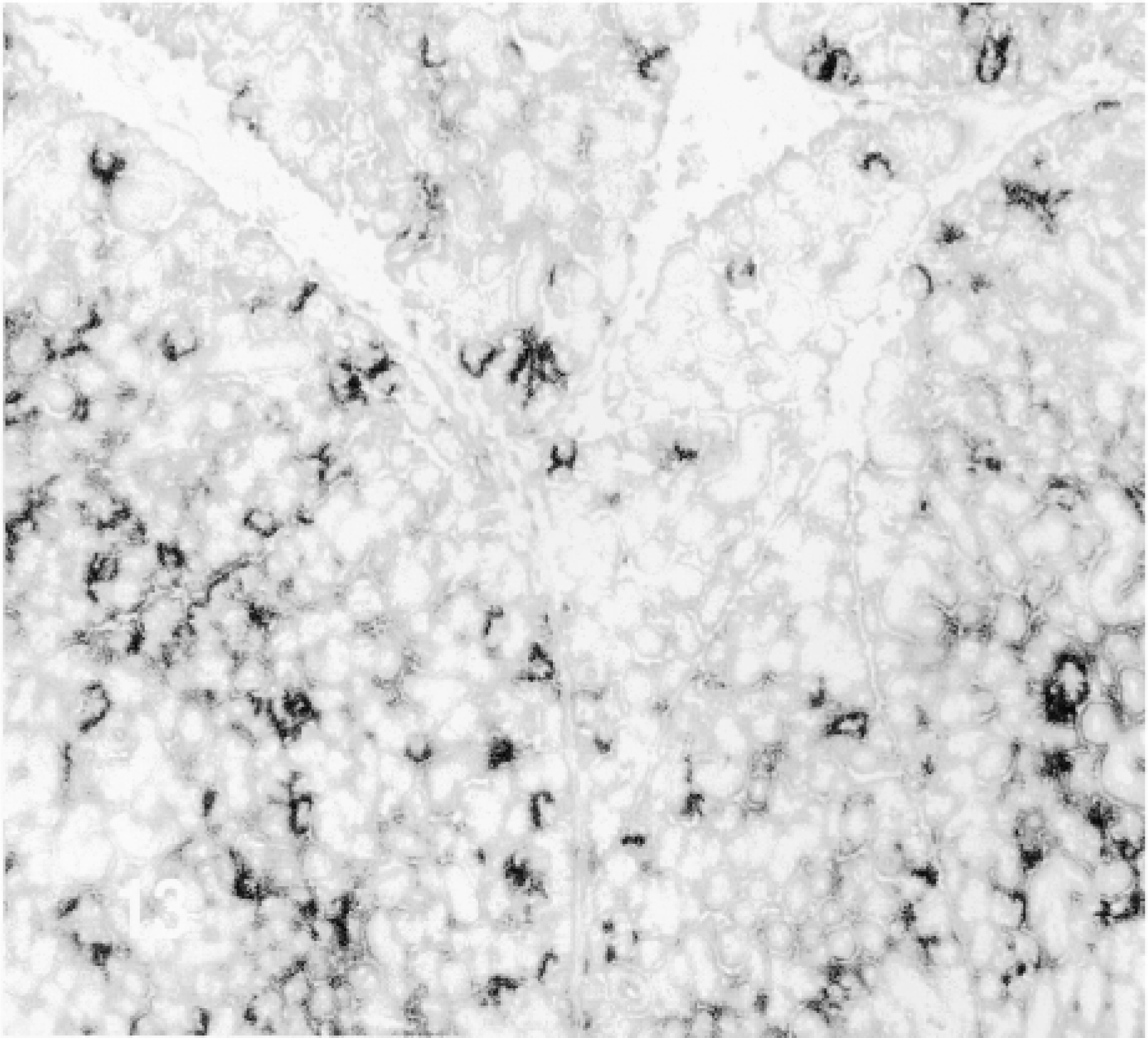
1 hr after the injection of the fusion protein.
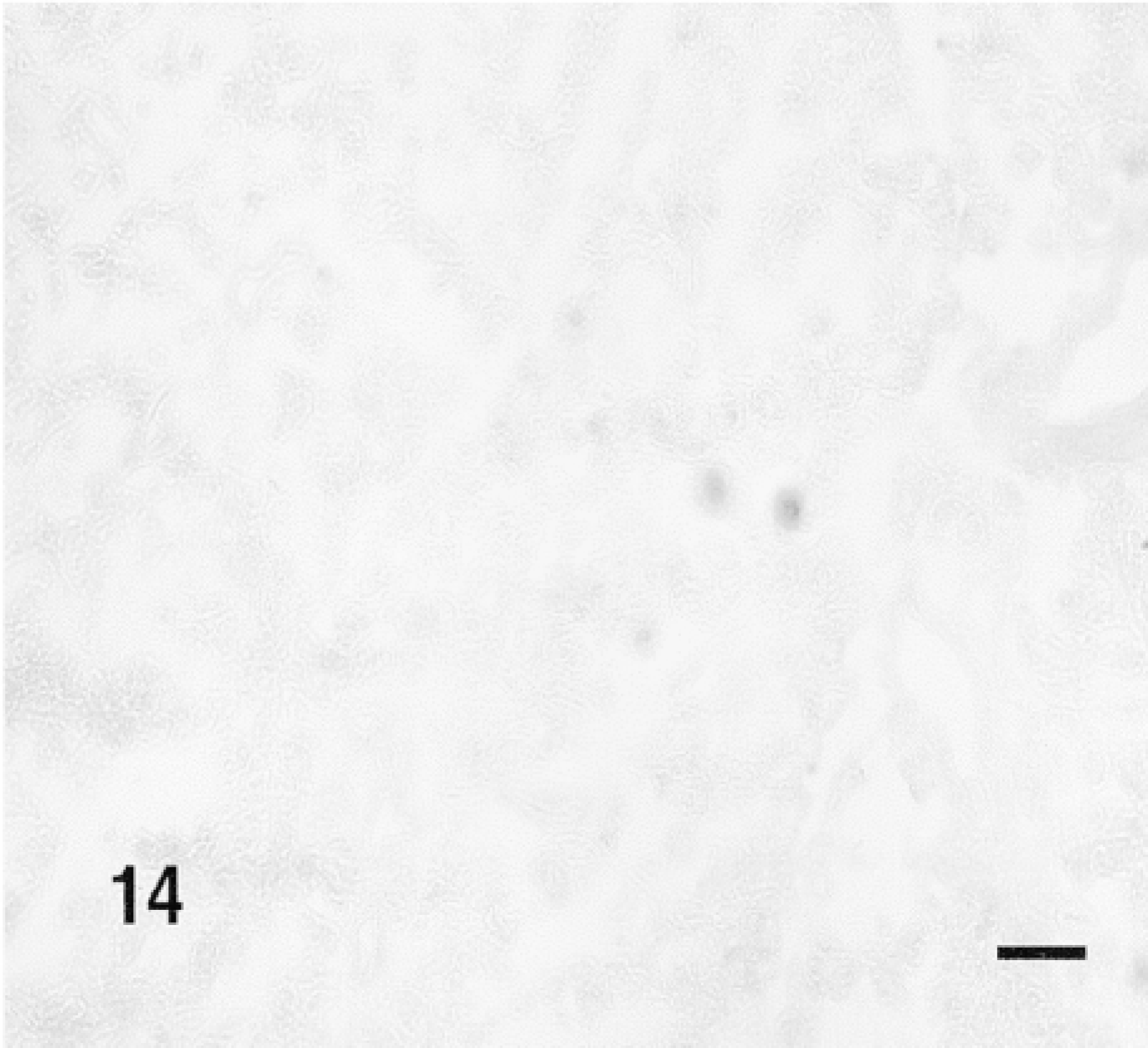
Cryostat section stained for β-gal of the SMG of a rat that received retrograde duct injection of HA-β-gal 1 hr earlier. Bar = 100 μm.
We have shown for the first time that TAT-HA-β-gal fusion protein is transduced into both epithelial and mesenchymal cells of an explanted fetal organ, the mouse SMG. The potential of using TAT fusion proteins to deliver full-length functional proteins into the cells of explanted organs holds great promise for the analysis of developmental mechanisms. This is particularly true because other methods, such as introduction of expression vectors, peptidyl mimetics, and antisense oligonucleotides into cultured organs is notoriously difficult.
Schwarze and co-workers (1999) have already established that, by IP injection TAT-HA-β-gal can be transduced into a variety of murine tissues, including liver, kidney, heart muscle, lung, spleen, and brain. The fusion protein injected into the peritoneum reaches all tissues via the systemic circulation. Such a universal distribution somewhat limits the applicability of fusion proteins for studies of the physiological or pathophysiological processes involving a single organ or tissue. By cannulating the main excretory duct of the SMG and injecting the fusion protein retrogradely into the SMG, we have achieved a targeted delivery to acinar and duct cells. A similar approach can be used to deliver fusion proteins to the parenchymal cells of the parotid gland.
After the retrograde duct injection, the acinar and duct cells showed β-gal activity at 10 min to 6 hr. However, no enzyme activity was detected at 19 hr. This is in contrast to the in vitro situation where maximum transduction was seen after 24 hr of exposure. A more detailed analysis of the time course of β-gal expression is needed to establish whether this decline is cell- or organ-dependent.
In summary, the targeted delivery of fusion proteins by retrograde duct injection into salivary glands will enable analyses of cellular and molecular regulatory mechanisms, including regulation of cell replication and growth, of the salivary glands. In addition, transduction of fusion proteins into developing glands in an organ culture will provide a valuable experimental approach to study the mechanism of branching morphogenesis of salivary glands and of organ development in general.
Footnotes
Acknowledgements
Supported by grants DE11729 (TB) and DE10858 (EG) from the National Institute of Dental and Craniofacial Research.
We are grateful to Dr Steven F. Dowdy for his advice.