
Abstract
Transduction of proteins and other macromolecules constitutes a potent technology to analyze cell functions and to achieve therapeutic interventions. In general, fusion proteins with protein transduction domains, such as TAT, are produced in a bacterial expression system. Here we describe the generation of a mammalian expression vector coding for TAT-EGFP fusion protein. Transfection of CHO-K1 cells by this vector and subsequent selection by Zeocin resulted in cell lines that express and secrete EGFP, a variant of the green fluorescent protein GFP. The ultimate cell line was produced by first cloning the stable integrants and subsequent selection of EGFP-expressing cells by flow cytometric sorting. In the resulting cell line approximately 98% of cells express EGFP. Using the same methodology, we generated cell lines that express DsRed fluorescent protein. The advantages of using such a mammalian expression system include the ease of generating TAT fusion proteins and the potential for sustained production of such proteins in vitro and, potentially, in vivo.
Keywords
T
Studies of the mechanism of transduction of proteins led to the identification of protein transduction domains (PTDs). The most widely studied of these sequences, in addition to TAT, are the Drosophila antennapedia peptide (Derossi et al. 1994) and the Herpes simplex virus VP22 protein (Elliott and O'Hare 1997). However, it is now recognized that other proteins and synthetic peptides have similar translocating properties (Derossi et al. 1998; Lindgren et al. 2000; Schwartz and Zhang 2000; Ford et al. 2001; Ho et al. 2001; Bogoyevitch et al. 2002; Lindsay 2002; Mai et al. 2002; Park et al. 2002; Futaki et al. 2003), some of which (e.g., the 9-mer of D-arginine) (Wender et al. 2000) or polylysine (Mai et al. 2002), are more effective in cellular uptake than Tat49-57.
The technology of generating TAT fusion proteins (Becker-Hapak et al. 2001) requires the synthesis of the fusion protein in which the TAT transduction domain (amino acids 47-57 of HIV Tat, termed TAT) is linked to the molecule of interest, using a bacterial expression vector. In general, the TAT peptide is also linked to a tag to facilitate its subsequent purification. The purified recombinant fusion protein can be added directly to mammalian cells in culture or injected in vivo into an animal (Schwarze et al. 1999). We have shown that retrograde ductal injection of a fusion protein (TAT-β-galactosidase) into rat salivary glands provides targeted delivery and that epithelial and mesenchymal cells of developing mouse submandibular glands in organ culture can be transduced by the same fusion protein (Barka et al. 2000). The technique outlined is generally successful but laborious. Furthermore, if a sustained effect of the TAT fusion protein is required, repeated exposure in vitro or administration in vivo is necessary.
We conceived an alternative technology that offers certain advantages in applying transduction techniques mediated by TAT and other PTDs. To this end, we constructed mammalian expression vectors expressing and secreting TAT fusion proteins, transfected cultured cells with such vectors, and established stable transformed cell lines. The TAT fusion protein is secreted by such cells into the culture medium, which can be added directly to other cells in culture. Cells can also be co-cultured with secreting transformed cells, thus exposing them continuously to the TAT fusion protein. If required, the TAT fusion protein can also be purified from the spent culture medium by conventional techniques. Furthermore, we foresee that such transformed cells could be a sustained source of PTD fusion peptides and other macromolecules in vivo. To demonstrate the feasibility of this approach, we describe, as a prototype, the construction of a mammalian expression vector designed for stable expression and secretion of TAT-green fluorescent protein (TAT-EGFP) in mammalian hosts and the use of this vector in transduction experiments.
Materials and Methods
Construction of TAT-EGFP Vector
First, two oligonucleotides were synthesized and annealed to generate a double-stranded oligonucleotide with restriction sites for AscI and HindIII restriction enzymes and encoding 11 amino acids (YGRKKRRQRRR, given in single-letter amino acid code) from the basic domain of HIV Tat. The sequences were: 5′-CGCGCC
Transfection
We transfected CHO-K1 cells using FuGENE 6 Transfection Reagent (Roche Applied Science; Indianapolis, IN) in serum-containing medium according to the manufacturer's instructions. The transfection efficiencies with FuGENE Reagent: DNA ratios (μl and μg, respectively) of 3:1 and 6:1 were similar and were about twice as great as that obtained with a ratio of 3:2. We carried out transfections with three plasmids: pEGFP-N1, pCMV-DsRed-Express (BD Biosciences Clontech; Palo Alto, CA), and pTAT-GFP. We selected stable transformed cell lines, designated as GFPN, CHO-RED and CHO-GFP, respectively, using 1 mg/ml Geneticin (GFPN and CHO-RED) or 400 μg/ml of Zeocin (CHO-GFP) starting 48 hr after transfection. Zeocin and Geneticin sensitivities of the parent CHO-K1 cells were determined in preliminary experiments. The transformed cells were maintained in the same concentrations of the antibiotics.
Tissue Cultures and Cell Lines
CHO-K1 Chinese hamster ovary cells (ATCC, #CCL-61) were grown in F12-K medium (American Type Culture Collection; Manassas, VA) supplemented with 10% fetal bovine serum (FBS), 50 U/ml penicillin, and 50 μg/ml streptomycin. CHO-GFP cells were grown in the same medium supplemented with 400 μg/ml of Zeocin. In the case of CHO-RED cells the medium was supplemented with 1 mg/ml of Geneticin. CHO-GFP and CHO-RED cells were cloned using cloning cylinders or by limiting dilutions. Clones were selected on the basis of expression of the fluorescent protein. For serum-free culture, CD CHO A Medium (GIBCO Invitrogen; Grand Island, NY), supplemented with 4 mM
Viability Assay
We assayed the viabilty of CHG-GFP-POS cells using the LIVE/DEAD Viability/Cytotoxicity Assay Kit (L3224) (Molecular Probes; Eugene, OR) following the protocol of the manufacturer. The two-color fluorescence assay is based on the enzymatic conversion by ubiquitous esterases of the virtually non-fluorescent cell-permeant calcein AM to the intensely fluorescent calcein, and the permeability of dead but not living cells by ethidium bromide. We grew the cells on coverslips and incubated with the reagent for 30 min at room temperature. According to our preliminary experiments, for this cell line the optimal concentrations of ethidium bromide and calcein AM are both 2 μM. The cells were killed by 30-min treatment with 70% ethanol. We viewed the preparations with a Zeiss Axioskop microscope and photographed them using the same exposure time for all preparations.
Exposure of Cultured Cells to Spent Media of TAT-GFP-secreting Cells
CHO-GFP-Clone 6 cells were cultured in 20 ml F12-K medium supplemented with 10% FBS, penicillin/streptomycin, and 400 μg/ml Zeocin in 10-cm petri dishes. When the cultures were near confluence, the medium was replaced with 2-4 ml fresh medium without Zeocin. The medium was collected after 3 hr of incubation at 37C and centrifuged at 3000 rpm for 5 min. The supernatant was used immediately or kept at −20C.
To test the transduction of TAT-GFP fusion protein, we plated CHO-RED-Clone 15 cells onto coverglasses in 35-mm dishes, 2-2.5 × 105 cells per dish in 2 ml medium. Next day, we replaced the medium with 2 ml spent medium prepared as described above. In general, we have examined the cells under a fluorescence or confocal microscope without fixation. However, we also fixed some cultures after 30 min, 1, 2, 4, or 24 hr of exposure and examined them under a fluorescence microscope.
Western Blots
For Western blots, we cultured CHO-GFP-C16 or CHO-GFPPOS cells in serum-free medium. We harvested the medium from cultures at near confluence, concentrated it by Amicon Ultra Filter Devices (30,000 MWCO), separated the proteins by SDS-PAGE (10%), and electroblotted them onto PVDF membranes. The Western blots also included recombinant EGFP (BD Biosciences Clontech). For immunostaining, we used a monoclonal antibody to EGFP [BD Living Colors A.v. Monoclonal Antibody (JL-8); BD Biosciences Clontech] at a dilution of 1:2500 and a peroxidase chemiluminescent method (ECL Advance Western Blotting Detection Kit; Amersham Biosciences, Piscataway, NJ) with the secondary antibody, peroxidase-labeled anti-mouse IgG, at a dilution of 1:10,000. For blocking and for the dilutions of the antibodies we used 1 × TBS/Casein Blocker (Bio-Rad; Hercules, CA).
Fluorescent Microscopy
We examined most cultures without fixation with a confocal microscope [Leica TCS-SP (UV)] or a Zeiss Axiphot fluorescence microscope. For observations of living cells, they were grown in Glass Bottom Microwell Dishes (MatTek; Ashland, MA). However, some cultures were observed after fixation. To this end, cultured cells were rinsed twice with PBS, fixed in 2% formaldehyde [formaldehyde, (methanol free) 10% ultrapure EM grade; Polysciences, Warrington, PA, diluted with PBS)] for 30 min, and washed in PBS twice for 5 min. The cultures were mounted in Vectashield Hard Set Mounting Medium (Vector Laboratories; Burlingame, CA). The microscopic images of living and fixed cells were comparable.
Flow Cytometry
For sorting of cells expressing GFP or DsRed we used a FACSVantage SE high-speed cell sorter (Becton Dickinson; Mountain View, CA) (Flow Cytometry Shared Research Facility, Mount Sinai School of Medicine). Cells positive for GFP or DsRed were then cultured in F12-K1 medium supplemented with 10% FSB and antibiotics.
Results
Transfection and Establishment of Cell Lines Expressing the Green or Red Fluorescent Protein and Those That Express and Secrete TAT-EGFP
For our experiments we used three vectors: (a) pEGFP-N1, which encodes a red-shifted variant of wild-type GFP and is optimized for enhanced fluorescence and high expression in mammalian cells. Expression is under the control of the early promoter of CMV; (b) pCMV-DsRed-Express, which encodes a variant of Discosoma sp. red fluorescent protein (DsRed) and is codon-optimized for high expression in mammalian cells; and (c) pTAT-TAG. The parent vector, pSecTag2 A, used for the construction of pTAT-TAG, is designed for high-level stable and transient expression in mammalian hosts and for secretion of proteins fused at the N-terminus to the murine IgK-chain leader sequence.
CHO-K1 cells were transfected with pEGFP-N1 or pCMV-DsRed-Express at high efficiency (Figures 1A and 1D). The transfection efficiency with pTAT-GFP was less. We established stable transformed cell lines expressing EGFP, DsRed, or TAT-EGFP, respectively. In the case of cells transfected with pEGFP-N1 or pCMV-DsRed-Express, the selective medium included 1 mg/ml of Geneticin. In the case of cells transfected with pTAT-TAG, the selection was based on resistence to 400 μg/ml of Zeocin. However, in further experiments we used only two cell lines expressing TAT-EGFP, designated CHO-GFP, or DsRed, designated CHO-RED, respectively. We cloned both CHO-GFP and CHO-RED cells and selected clones on the basis of the percentage of cells expressing the fluorescent protein and the level of expression. However, we applied these criteria without accurate quantitation. In cultures of the two clones selected, CHO-GFP-C16 and CHO-RED-Cl15, not all cells express the corresponding fluorescent protein and the level of expression varies greatly among the cells (Figures 1G and 1I). The reason for this variation is not known. These clones were maintained in the selective medium for several months without apparent change in the expression of EGFP or DsRed.
Fluorescence and phase-contrast (
Sorting of Cells Expressing GFP or DsRed Using a Flow Cytometer
We estimated that only approximately 20% of cloned CHO-GFP-Cl6 cells express the EGFP (Figures 2A-2C). To increase the percentage of cells expressing TAT-EGFP, we sorted GFP+ (positive) and GFP (negative) cells using a flow cytometer. The result of cell sorting confirmed that 20.2% of CHO-GFP-Cl6 cells express EGFP and that 98% of the cells in the GFP+ fraction express GFP. Both GFP+ and GFP cell populations were expanded and examined, without fixation, under a fluorescent or confocal microscope. Whereas in the culture derived from the sorted GFP- cells very few cells expressed GFP (data not shown), in the culture derived from the GFP+ fraction, hence designated CHO-GFP-POS, most cells showed green fluorescence (Figures 2D-2F). In GFP+ cells, GFP was localized to both the nucleus and cytoplasm, although the concentration appeared to be higher in the nucleus. The nucleoli did not contain GFP. The expression of GFP was similar in sparse (Figures 3A-3C) and confluent cultures (Figures 3D-3F) (Figures 3C and 3F are merged images).
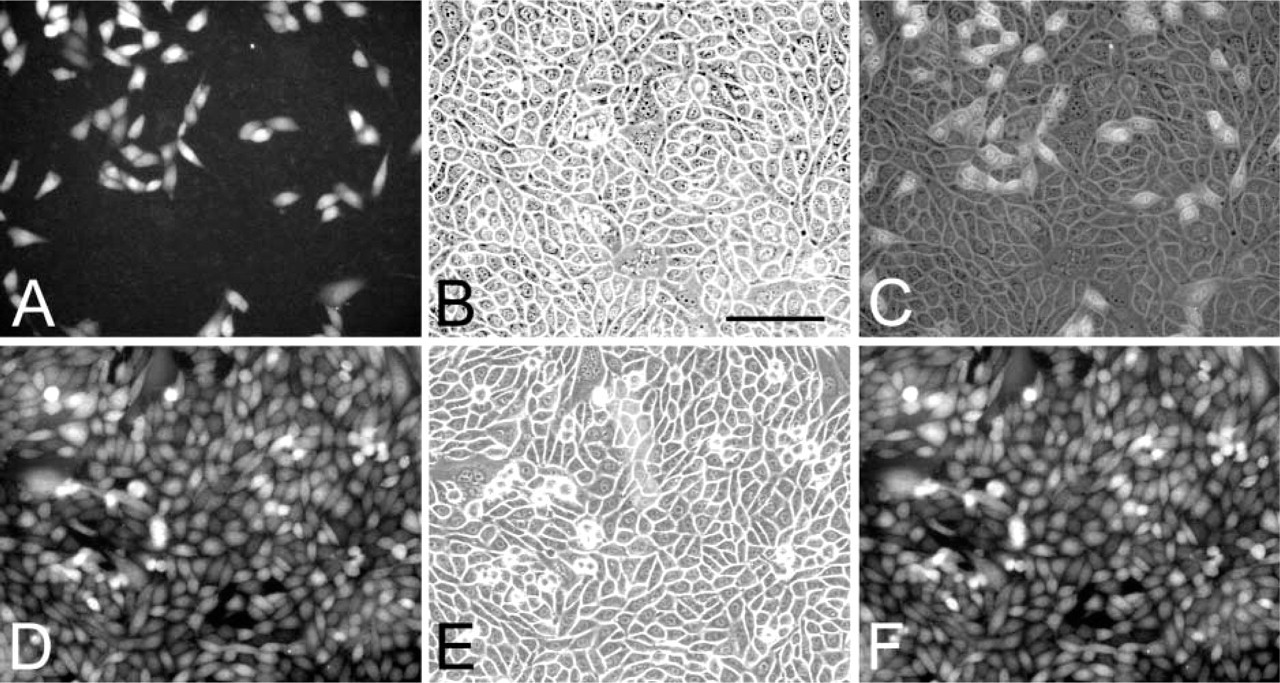
Comparison of GFP expression in CHO-GFP-Cl6 and CHO-GFP-POS cells. In the stable transformed and cloned cell line CHO-GFP-Cl6, about 20% of cells express GFP. In cultures of cells sorted for the expression of GFP, CHO-GFP-POS (fourth passage), >98% of the cells express GFP. (
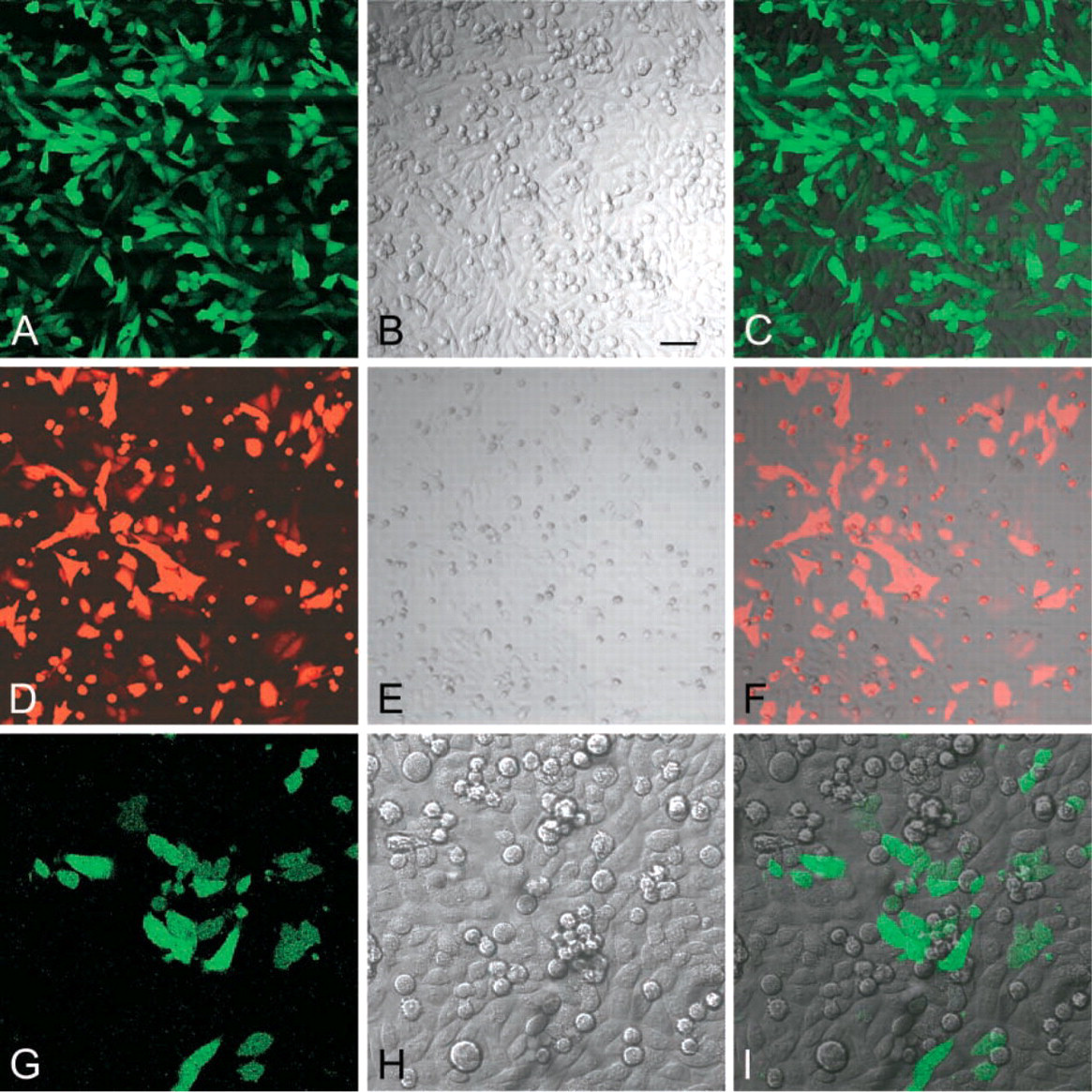
Confocal images of living CHO-GFP-POS and CHO-RED-POS cells. Sparse (
Similarly, we sorted cells that express DsRed protein in the cell line CHO-RED-Clone15. According to the flow cytometeric data, 40% of cells in this cell line express the red fluorescent protein. Sorting resulted in a cell population expressing DsRED by 99.5% of the cells. We expanded this cell population, hence designated CHO-RED-POS, for future experiments. There was a great variation in the intensity of red fluorescence among the cells. DsRed was localized mostly in the cytoplasm. However, confocal images revealed the presence of the fluorescent protein also in the nuclei. The nucleoli were devoid of DsRed (Figures 3G-3I) (Figure 3I is a merged image).
Viability Assays of CHO-GFP-POS Cells
By using a two-color fluorescence assay, we estimated the viabilty of CHO-GFP-POS cells used to collect spent media. In sparse cultures and in cultures near confluence, >99% of the cells were viable. When cells killed by fixation with 70% ethanol were stained, all cells showed intense red fluorescence in the nuclei and only faint fluorescence in the cytoplasm, an indication of the proper concentration (2 μM) of ethidium bromide in the assay reagent (Figure 4A). In such preparations, no green fluorescence was detected, again indicating the appropriateness of the reagent's calcein AC concentration, 2 μM (Figure 4B). Cultures of CHO-GFP-POS cells showed intense green fluorescence of living cells (Figure 4C), and only occasional dead cells in the red channel (Figure 4D). In overgrown cultures, the proportion of dead cells increased but was still less than 5% (data not shown).
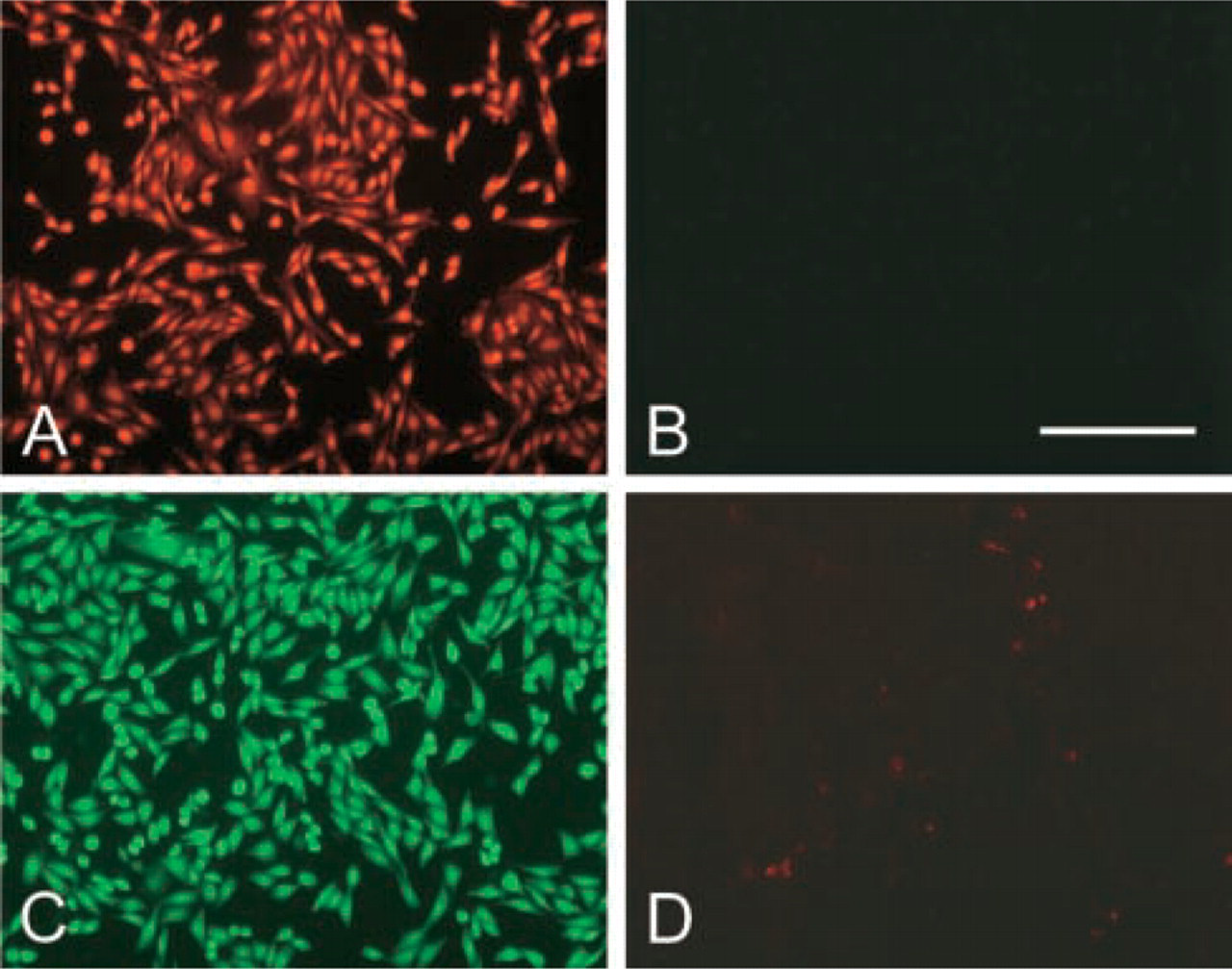
Viabilty assay, as described in Materials and Methods, of CHO-GFP-POS cells. Fluorescence images of dead, ethanol-fixed cells. (
Secretion of TAT-EGFP by CHO-GFP-Cl6 and CHO-GFP-POS Cells
CHO-GFP cells secrete TAT-GFP into the culture medium. This was established by Western blots of spent culture media and by exposing cells to media harvested from CHO-GFP-Cl6 cultures. For the latter experiments, we used stable transformed cells expressing DsRed as recipient, thus avoiding any ambiguity that might be caused by green autofluorescence in some cells.
Western blots of concentrated serum-free spent media of CHO-GFP cells, prepared by using a monoclonal antibody to EGFP, revealed a single band of protein with the same migration in SDS-PAGE as rEGFP (Figure 5). The apparent molecular weights of rEGFP and the reacting protein of the concentrate were approximately 60 kD, suggesting that they may represent dimers of EGFP. However, the nature of these bands was not investigated.

SDS-PAGE and Western blot of recombinant EGFP and concentrated spent medium of CHO-GFP-Cl6 cells. The migrations of rEGFP and the protein of the spent medium were similar and indicated an apparent molecular weight of about 60 kD, suggesting that they represent dimers of EGFP. CHO-GFP-Cl6 cells were cultured in F12-K medium supplemented with 10% FBS to near confluence in 10-cm dishes. The medium was replaced by 4 ml serum-free medium which was collected 14 hr later, centrifuged, and concentrated. SDS-PAGE and immunoblots were prepared as described in Materials and Methods. Lanes 1-3, Coomassie blue staining. Lanes 4 and 5, immunoblot using a chemiluminescence detection method. Lane 1, molecular weight markers; Lanes 2 and 4, 10 ng of recombinant EGFP; Lanes 3 and 5, 10 μl of concentrated spent medium.
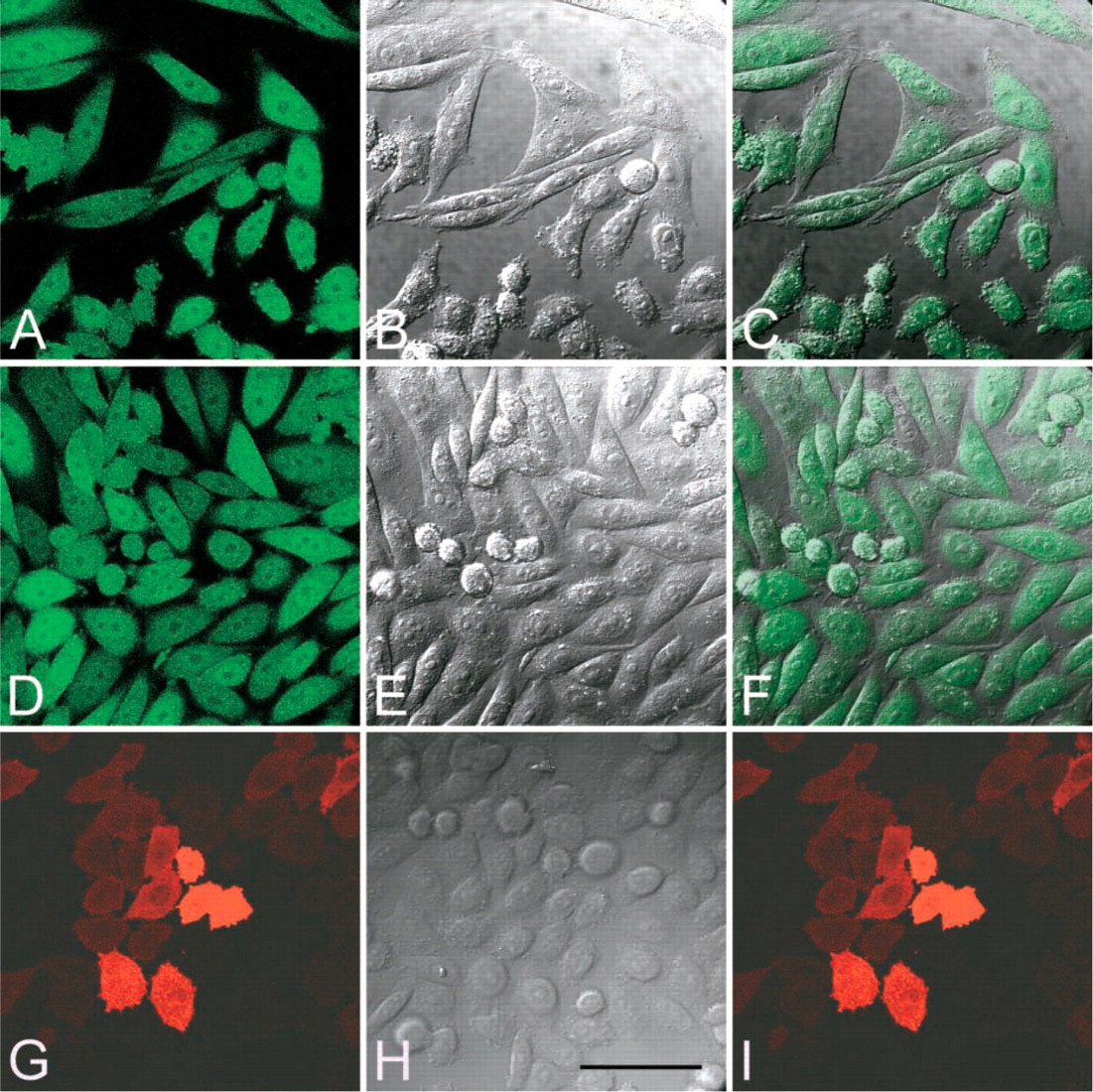
We exposed CHO-RED-C115 cells, grown on coverglass, to spent media of TAT-GFP-secreting CHO-GFP cells for 30 min, 1, 2, 4, and 24 hr. Microscopic examination of randomly selected fields of these live cultures revealed that most CHO-RED cells expressed the DsRed, albeit with different intensity, and that the GFP was co-localized in most cells (Figure 6). However, cells showing very intense red fluorescence revealed no green fluorescence (Figure 6C, merged image). There were no obvious differences in the percentage of cells showing green fluorescence or in the intensity of green fluorescence of CHO-RED cells exposed to TAT-GFP for 30 min to 24 hr (data not shown). This indicates that CHO-GFP cells secrete TAT-EGFP and that the uptake of TAT-GFP is rapid and essentially complete after 30 min of incubation. This is in accord with previous findings reported in the literature using different TAT fusion proteins. In addition to CHO-K1 cells, we have successfully transduced several other cell types with the secreted TAT-GFP (data not shown).
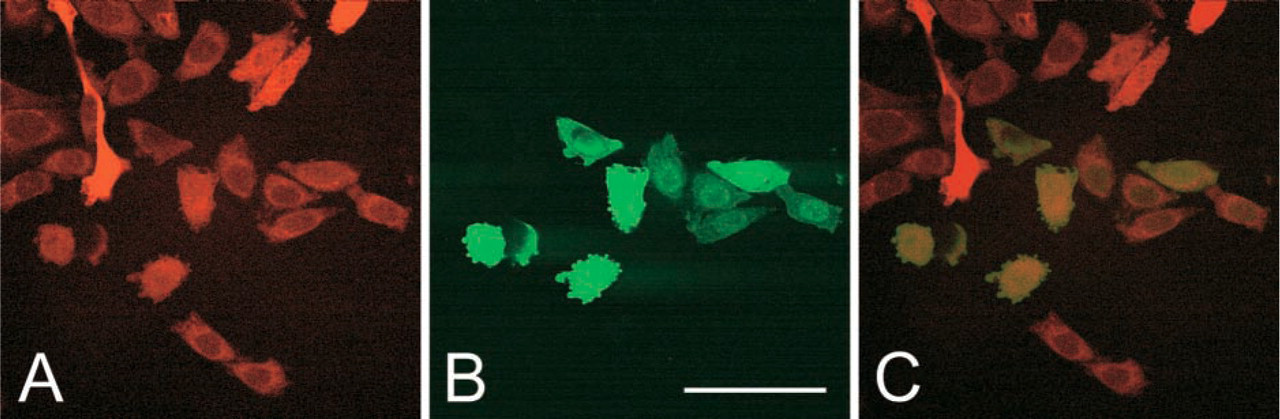
Transduction of CHO-RED-Cl15 cells by TAT-GFP secreted by CHO-GFP-Cl6 cells. Confocal images of living cells. Clones of cells expressing DsRed (CHO-RED-Cl15) were plated on coverglasses and exposed for 1 hr to the spent medium of CHO-GFP-Cl6 cells that express and secrete TAT-GFP. Most of the cloned recipient CHO-RED-Cl15 cells express the DsRed (
Discussion
Transduction of proteins and other macromolecules, such as DNA and cancer chemotherapeutic agents, into cells both in vitro and in vivo is now widely applied to investigate cell functions and for therapeutic purposes. Peptide-mediated cell delivery systems are greatly expanded by the recognition of protein transduction domains (PTDs) and synthetic peptides with translocating properties (reviewed by Watson and Edwards 1999; Lindgren et al. 2000; Schwartz and Zhang 2000; Ford et al. 2001; Lindsay 2002; Denicourt and Dowdy 2003). Protein transduction is claimed to have advantages over virus-mediated gene delivery systems and may be applicable for genetic intervention, including gene therapy (Schwartz and Zhang 2000; Ford et al. 2001). One of the most intriguing developments of protein transduction technology is its application in therapy of brain ischemia (reviewed by Denicourt and Dowdy 2003).
The technique of producing TAT fusion proteins, and PTD fusion proteins in general, requires the synthesis and purification of such proteins using bacterial expression systems (Vocero-Akbani et al. 2000; Becker-Hapak et al. 2001). As an alternative, we have developed a methodology based on the use of a mammalian expression vector, pSecTag2. This vector is designed for high-level expression and secretion of proteins by transient or stable integrants. As a model, we developed stable transformed cell lines, derived from CHO cells, expressing and secreting TAT-EGFP. Although we have applied the same technology to generate cell lines that express and secrete TAT-β-galactosidase, GFP offers high sensitivity and ease of detection in fixed and living cells to illustrate the advantages of this technique.
CHO-K1 cells transfected with pEGFP-N1 or pTAT-GFP express EGFP. Similarly, cells transfected with pCMV-DsRed-Express express the red fluorescent protein DsRed. We have obtained cell lines of stable integrants by exposing the transfected cells to Zeocin (CHO-GFP) or Geneticin (CHO-RED). Cloning of Zeocin-resistant CHO-GFP cells resulted in clones in which only a fraction of the cells expressed EGFP. Of 10 clones examined, clone 6 showed the highest percentage of cells, approximately 20%, expressing GFP. The level of expression varied from cell to cell. The reason for this variation in expression of the stable transgene is not known but is probably, at least in part, caused by the inherently stochastic nature of gene expression. Stochastic mechanisms in gene expression operate in both prokaryotes and eukaryotes and may explain the phenotypic variations in isogenic populations of cells (McAdams and Arkin 1997; Elowitz et al. 2002; Ozbudak et al. 2002; Blake et al. 2003). Other mechanisms, such as the conversion of GFP to the fluorescent form (Heim et al. 1994) and the equilibrium between synthesis of the protein and its secretion, are probably also operational. Non-uniform production of GFP among HEK293 cells transfected with a vector coding for a mutant of GFP has been described (Malek and Khaledi 1999).
Analysis by flow cytometry confirmed that only approximately 20% of CHO-GFP-Cl6 cells expressed GFP. Sorting of GFP+ cells and their expansion led to a cell line in which approximately 98% of the cells expressed EGFP. Whether the strategy of first cloning stable transformed cells by conventional cloning technique and subsequent “cloning” by sorting of cells expressing the transgene is more efficient than cloning by flow cytometry alone remains to be investigated.
A similar cell-to-cell variation was evident in CHO-RED cells. This conspicuous cell variation in the level of DsRed fluorescence existed even in the CHO-RED-POS cell line that was generated by sorting DsRed-expressing cells.
CHO-GFP cells not only express but also secrete GFP. This was established by Western analysis of media in which the cells were cultured and by exposing cells to spent media. For the latter experiment, we used a cell line (CHO-RED) expressing Ds-Red fluorescent protein. This strategy provided unequivocal proof of secretion and transduction of TAT-EGFP by excluding the presence of autofluorescent material in the recipient cells. Viability studies indicated that approximately 99% of CHO-GFP-POS cells that provided the spent media were viable, obviating the possibility that TAT-GFP was released from dead cells and not secreted by living cells.
In summary, the use of a mammalian secretory system to generate PTD fusion proteins offers certain advantages: relative ease of preparing the PTD fusion proteins in soluble form, which can be added directly to cultured cells, and potential post-translational modifications, such as glycosylation and formation of disulfide bonds. However, such proteins can also be purified by conventional techniques for in vitro or in vivo applications, providing a stable source of such proteins in vitro and possibly in vivo. We foresee therapeutic applications of this technique in which the parent cells transfected with constructs coding for the PTD fusion proteins could conceivably be the patient's own cells. This system is also adaptable to the study of autocrine regulatory mechanisms by transfecting cells with constructs coding for PTD-fused putative autocrine peptides.
Footnotes
Acknowledgements
Supported by NIH grant RO1 DE10858 to Dr E. Gresik. Microscopy was performed at the MSSM-Microscopy Shared Research Facility, supported in part by funding from NIH-NCI shared resources grant (1 R24 CA095823-01).
We are grateful to Ms H. van der Noen for excellent assistance, Dr I. Karpichev for advice in cloning, Dr C. Iomini for stimulating discussions, and Mr P.T. Carman for assistance with microscopy.