
Abstract
At the epithelial/mesenchymal interface of most tissues lies the basement membrane (BM). These thin sheets of highly specialized extracellular matrix vary in composition in a tissue-specific manner, and during development and repair. For about two decades it has been apparent that all BMs contain laminins, entactin-1/nidogen-1, Type IV collagen, and proteoglycans. However, within the past few years this complexity has increased as new components are described. The entactin/nidogen (E/N) family has expanded with the recent description of a new isoform, E/N-2/osteonidogen. Agrin and Type XVIII collagen have been reclassified as heparan sulfate proteoglycans (HSPGs), expanding the repertoire of HSPGs in the BM. The laminin family has become more diverse as new α-chains have been characterized, increasing the number of laminin isoforms. Interactions between BM components are now appreciated to be regulated through multiple, mostly domain-specific mechanisms. Understanding the functions of individual BM components and their assembly into macromolecular complexes is a considerable challenge that may increase as further BM and cell surface ligands are discovered for these proteins.
B
A significant number of interactions contribute to the supramolecular assembly of BMs. The current BM model proposes two networks, one consisting of collagen Type IV and the second made up of multiple laminins, interconnected via entactin-1 (Yurchenco and O'Rear 1994; Timpl and Brown 1996). In vitro studies indicate that perlecan interacts with the other three major components through either its core protein, in the case of entactin-1 and Type IV collagen, or its heparan sulfate glycosaminoglycan chains, as is the case for laminin-1 (Battaglia et al. 1992; Reinhardt et al. 1993). Other minor components, such as BM40/SPARC/osteonectin and fibulin-1 and −2, interact with one or more of the major constituents, and these interactions may be tissue-specific or developmentally regulated (Aumailley 1995). The macromolecular nature of the BM has become more complex within the past few years as more components are characterized. Recent developments in BM composition and biology include the description of entactin-2/nidogen-2, characterization of agrin and collagen Type XVIII as heparan sulfate proteoglycans, and expansion of the laminin family.
Entactin-2/Nidogen-2/Osteonidogen
During the early 1980s, two groups described a novel BM glycoprotein now known as entactin or nidogen. Entactin-1/Nidogen-1 was initially isolated from murine EHS tumor and cell cultures (Carlin et al. 1981; Timpl et al. 1983; Paulsson et al. 1986a). Non-mammalian E/N has been described in the ascidian Halocynthia roretzi (Nakae et al. 1993), Caenorhabditis elegans (Lee and Cheung 1996), Drosophila melanogaster (Kumagai et al. 1999), and Danio rerio zebrafish, (Gong et al. 1997; Clark et al. unpublished observations; Figure 1). Human and mouse sequences have 85% homology at the amino acid level, which suggests that this protein has been highly conserved during evolution. Since the initial discovery, E/N has been found to promote cell attachment (Chakravarti et al. 1990), neutrophil chemotaxis (Senior et al. 1992), trophoblast outgrowth (Yelian et al. 1993), and angiogenesis (Nicosia et al. 1994). In addition, E/N is believed to play a crucial role in BM formation owing to its ability to form complexes with laminin, Type IV collagen, perlecan, and the fibulins. E/N importance in BM formation was highlighted when antibodies against laminin fragments, which blocked binding of E/N, were shown to perturb organogenesis and the formation of BMs, and led to cell necrosis in embryonic kidney and lung organ cultures (Ekblom et al. 1994). Because E/N appears to play such an important role in BM formation, it was perhaps not surprising that further isoforms were recently discovered.
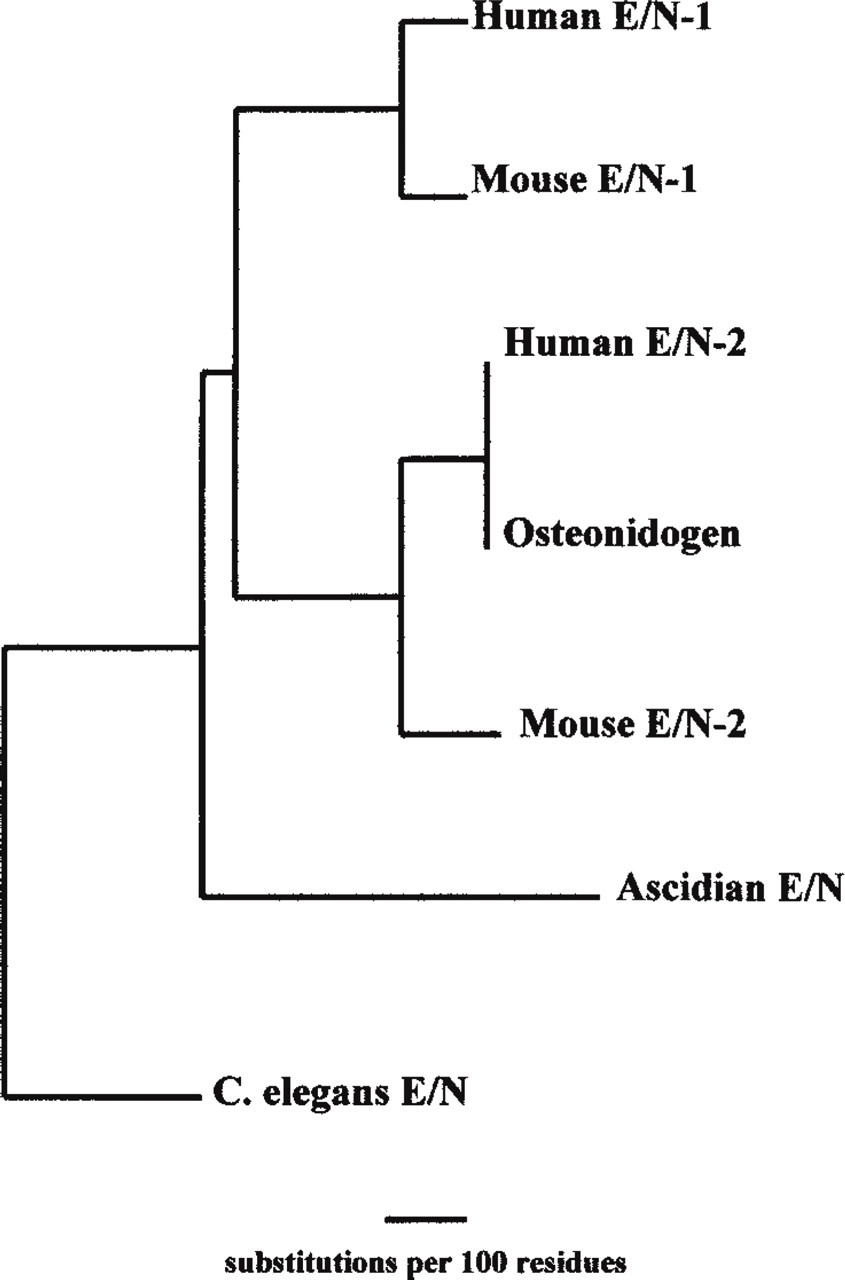
Phylogenetic tree of E/N-1 and E/N-2 amino acid sequences.
Kimura et al. (1998) were the first to describe an E/N-related glycoprotein. They were studying novel factors secreted from KUSA cells, an osteoblast-like cell line, that would affect bone formation. A novel protein with homology to E/N-1 was cloned and was named entactin-2. A human counterpart to this mouse clone, osteonidogen, was already present in the database and shared 71% similarity (Gen Bank D86425 accession). Another human sequence was published shortly after entactin-2 by Timpl's group and, in keeping with tradition in the entactin family, it was named nidogen-2 (Kohfeldt et al. 1998).
Mouse E/N-2 has only 27% amino acid similarity compared to mouse E/N-1, whereas human E/N-2 shares 46% amino acid similarity with human E/N-1. The human sequence of nidogen-2 is identical to osteonidogen except for 121 amino acid positions at the amino terminus (Kohfeldt et al. 1998). This new, emerging entactin family is not based on sequence similarity alone. Despite low sequence homology, the structural conservation between E/N-1 and E/N-2 is striking because E/N-2 maintains the three-globular-domain structure separated by a link region between G1 and G2 and a rod between G2 and G3. A structural comparison of each of the domains of mouse E/N-2 with mouse E/N-1 shows 31.2%, 38.4%, and 33.7% homology between the G2, G3, and rod domains, respectively. The G1 and link domains are less conserved, with 24% homology for G1 and 8% in the link region (Kimura et al. 1998; Figure 2). Rotary shadowing reveals that the average length of E/N-2 is 40 nm, a little larger than that seen for recombinant mouse and human E/N-1, which measures 30 nm on average (Kohfeldt et al. 1998).
Recombinant human E/N-2 expressed in EBNA-293 cells was purified by metal chelation chromatography and yielded a major polypeptide of 200 kD, with a smaller one of 170 kD by SDS-PAGE electrophoresis (Kohfeldt et al. 1998). It is therefore larger than E/N-1 (150 kD) and the 170-kD band was determined, by Edman degradation, to begin within the link region, suggesting that E/N-2 may share the proteolytic sensitivity of the E/N-1 isoform (Mayer et al. 1995). Hexosamine analysis of human E/N-2 demonstrated 25 ± 2 glucosamine and 19 ± 2 galactosamine residues, indicating that all five predicted N-glycosylation sites and that many of the O-glycosylation sites are substituted, and that recombinant E/N-1 contains fewer oligosaccharides, which is a partial explanation for the difference in mobility on SDS-PAGE (Kohfeldt et al. 1998).
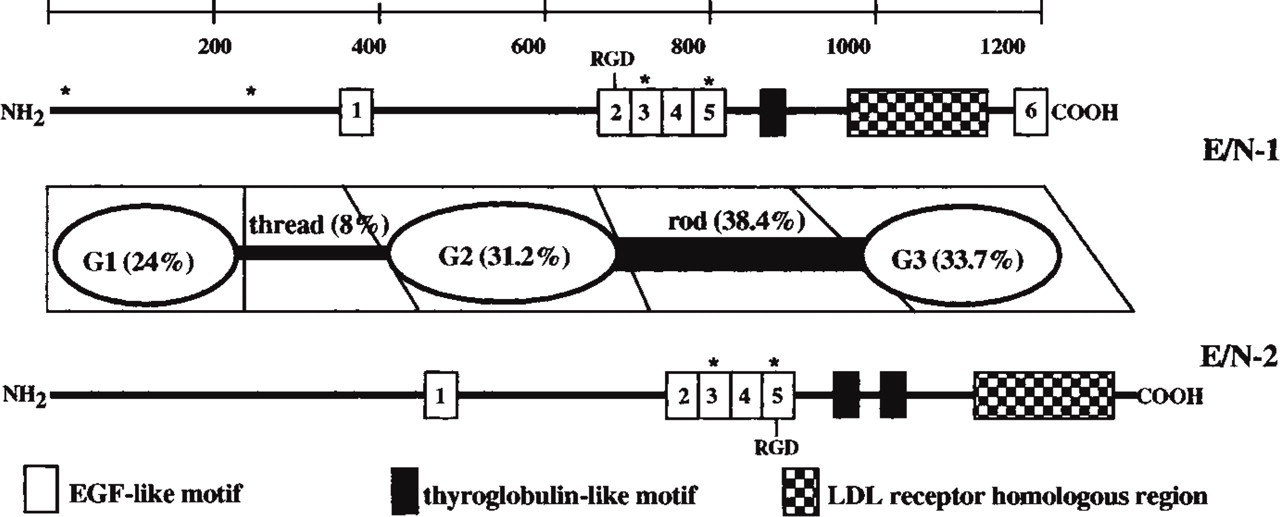
Schematic structure of E/N-1 and E/N-2. Homologies represented are for the murine amino acid sequences. The potential Ca2+-binding sites are marked by a star.
Structural motifs are conserved between E/N-1 and E/N-2. The E/N-2 G2 domain contains EGF repeats 1 and 2 at its N-terminal, but with four additional EGF-like motifs in the rod domain. The G3 domain contains an EGF precursor/LDL receptor-homology region, and there are consensus sequences for calcium binding within the third and fifth EGF-like motifs. Differences between E/N-1 and E/N-2 include the presence of two thyroglobulin-like motifs in the rod domain of E/N-2 as opposed to one found in E/N-1. E/N-1 has an additional EGF-like motif at the carboxy terminal and two additional potential calcium binding sites at its amino terminal.
E/N-2 is as widespread as E/N-1. Northern blot analysis showed strong expression of E/N-2 transcript in heart, lung, skeletal muscle, kidney, liver, and testis, with lower levels in the brain and spleen, very similar to E/N-1. E/N-1 transcripts were more abundant than those of E/N-2 in liver, lung, and pancreas (Kimura et al. 1998; Kohfeldt et al. 1998), and higher protein levels of E/N-1 in fibroblasts and two mesenchyme-derived tumor cells were described. Similar levels of the two isoforms were seen in heart, skeletal muscle, and kidney. The placenta contained more E/N-2 protein.
Double immunofluoresence microscopy revealed co-localization in kidney, skin, and testis. More specifically, in kidney both E/N isoforms were mainly localized in the BM zones of proximal and distal tubuli, glomeruli, and Bowman's capsule. Co-localization was also seen in skin, including the dermal–epidermal junction, and in BMs of appendages and vessels of the dermis. In the testis, staining was present around seminiferous tubules and in deposits around Leydig cells. Two tissues in which co-localization did not occur were cardiac and skeletal muscle. E/N-1 staining was found around cardiocytes in the endomysium and perimysium and around blood vessels and nerve bundles. E/N-2 was equally prominent around vessels and nerves but was considerably weaker in the BM surrounding cardiocytes and myotubes (Kohfeldt et al. 1998). Therefore, although E/N-2 may have similar roles to E/N-1, the tissue distributions in heart and skeletal muscle suggested the possibility of isoform-specific functions.
To address this, Timpl and colleagues evaluated the protein–protein interactions of the two E/N isoforms (Kohfeldt et al. 1998). A kinetic assay using the laminin-1 P1 fragment, which contained the E/N-1 binding site, showed an affinity about 1000-fold lower for E/N-2 than for E/N-1. E/N-2 also had a 100-fold less affinity than E/N-1 in a solid-phase binding assay with P1 or recombinant laminin γ1III3 5, possessing the only high-affinity binding site for E/N-1. Deletion of the E/N-1-binding module in recombinant γ1III3–5 abolished binding of both E/N-1 and E/N-2, indicating that both proteins were interacting with the same region. However, when laminin-1 was saturated with E/N-1, E/N-2 was still able to bind, indicating a second site for E/N-2 interaction. E/N-2 bound collagen Types I and IV, as well as perlecan, with the same affinity as E/N-1, indicating that these binding sites are conserved between the two proteins. In contrast, E/N-2 was unable to bind fibulin-1 and −2 or tropoelastin (Sasaki et al. 1999), unlike E/N-1.
Both mouse isoforms have an RGD sequence present in their rod domains suggestive of interactions with integrins. It occurs within the second EGF-like repeat of E/N-1 and the fifth EGF-like repeat of E/N-2. However, human E/N-2 has a YGD motif that may not be active in cell adhesion. E/N-1 and E/N-2 differ in cell adhesion properties (Kohfeldt et al. 1998). Some cell lines adhere and spread on both human E/N-1 and −2, but in all cases more cells adhered to E/N-2. This interaction could not be inhibited by synthetic RGD peptide or blocking antibodies to β1 and α3 integrins; these reagents do block adhesion to E/N-1. Therefore, the data indicate that different integrins and/or non-integrin receptors are involved in cell adhesion to the two isoforms. These differences also suggest that the two isoforms have separate roles. Specific and redundant roles of these isoforms in BM architecture and function remain to be elucidated, and gene ablation studies in mice are eagerly awaited.
Basement Membrane Proteoglycans: Agrin, Type XVIII Collagen, and Leprecan
The presence of BM proteoglycans was originally indicated in the kidney glomerulus by regular arrays of polyanionic binding sites for ruthenium red and cationized ferritin, as well as their sensitivity to heparitinase and nitrous acid (Kanwar and Farquhar 1979; Simionescu et al. 1984). Heparan sulfate (HS) and small amounts of chondroitin sulfate (CS) PGs have been isolated from many in vivo sources, such as the glomerular BM (GBM), placenta, extra-embryonic membranes, and tumors (EHS), or in vitro sources such as PYS-2 and L-2 yolk sac tumor cell lines (Kanwar et al. 1981; Fenger et al. 1984; Fujiwara et al. 1984; Parthasarathy and Spiro 1984; Paulsson et al. 1986b; Couchman 1987). PGs in the basement membrane putatively play a structural role (Iozzo 1998) in maintaining tissue histoarchitecture (Reinhardt et al. 1993; Fosang and Hardingham 1996; Costell et al. 1999; Hopf et al. 1999), aid in selective filtration processes (Miettinen et al. 1986), sequester growth factors (Roberts et al. 1988; Aviezer et al. 1994; Gohring et al. 1998; Sharma et al. 1998) and extracellular ions (Lerner and Torchia 1986), and help regulate cell differentiation (Li et al. 1987). Some of these roles have been shown for perlecan, but not much is known about the functions of other BM proteoglycans. Perlecan was the only large BM heparan sulfate proteoglycan described for many years. Perlecan null mice show no evidence of abnormalities until embryonic Day 10 (E10), and between E10 and 12 most embryos die with evidence of bleeding into the pericardial sac. Some null mice survive further but perish around birth with severe defects in the brain and skeleton (Arikawa–Hirasawa et al. 1999; Costell et al. 1999). Because perlecan is known to first be expressed at the two-cell stage, implications of perlecan's role in the initial embryonic attachment to the uterine wall (Smith et al. 1997) can be questioned because normal Mendelian ratios were present in the litters producing the perlecan null mice. In addition, most BMs appeared to be normal in the homozygous embryos, suggesting either that perlecan does not have a role in BM assembly and/or that other PGs may substitute for perlecan (Olsen 1999). Possible candidates are two new BM HSPGs, agrin and Type XVIII collagen. Interestingly, the core proteins of these newfound HSPGs were described some years ago; only recently has their proteoglycan nature been exposed.
Agrin
McMahan and colleagues first identified agrin as a 150-kD polypeptide found in extracts of Torpedo electric organ (Nitkin et al. 1987). Addition of agrin to cultured muscle cells triggered formation of acetylcholine receptor (AChR) aggregates in the myotube membrane (Godfrey et al. 1984), resembling those seen in nerve-induced clusters, hence the origin of its name from the Greek word “ageirein,” which means to assemble.
Signals that regulate muscle contraction, as well as some other aspects of muscle function, are conveyed from moto neurons to muscle at the neuromuscular junction (NMJ). Nerve-derived agrin is crucial for NMJ organization because of its ability to induce clustering of AChRs with postsynaptic membranes. AChRs regulate the electrical activity and contractile state of the muscle (Bowe and Fallon 1995). Clustering of AChRs is accompanied by ECM, membrane, and cytoskeletal components of the postsynaptic apparatus (Wallace 1989). Not only is agrin important for muscle contractility throughout life but it is also important in triggering the formation of postsynaptic specialization in the developing synapse (McMahan 1990). Mice null for the neural isoform (z-agrin) of agrin developed normally until the last fetal day, E18, but died in utero or were stillborn (Gautam et al. 1996). No mutant embryos were seen to move, suggesting disruption of the neuromuscular function, although histological analysis of E15 and E18 embryos failed to detect gross abnormalities in any tissues. AChRs were synthesized at almost normal levels in the mutant mice, but the level and pattern of AChR clustering were abnormal, resulting in defective pre- and postsynaptic differentiation (Li et al. 1999). The absence of agrin impaired synapse formation but did not abolish it, suggesting that there are other synapse organizers. One candidate is the muscle-specific receptor tyrosine kinase (MuSK) because mice null for MuSK had a similar phenotype to the agrin null mutant (DeChiara et al. 1996; Glass et al. 1996; Gautam et al. 1999).
Neural HSPGs have been implicated in the stimulation of neurite outgrowth during development. HSPG binding to the neural cell adhesion molecule (NCAM) is required for NCAM-meditated cell adhesion (Cole and Glaser 1986; Cole et al. 1986; Riopelle and Dow 1990; Burg et al. 1995). When the HS binding domain was functionally ablated by site-directed mutagenesis, NCAM's adhesive function was abolished (Reyes et al. 1990). To elucidate the role of HSPGs in the development of the nervous system, Cole and colleagues characterized chick brain HSPGs and identified which one was responsible for modulating NCAM function (Tsen et al. 1995a). An HSPG with a 250-kD core protein, expressed in early periods of chick development, had the capability to interact with NCAM. Using core protein-specific antibodies, an E9 chick brain cDNA expression library screen produced clones that were homologous to chick agrin cDNA. Before this, agrin had not been described as an HSPG, nor had intact agrin (based on its predicted sequence) been isolated. Previous attempts yielded fragments of about 100 kD in chick (Godfrey 1991) and a protein slightly smaller than 200 kD in rat (Rupp et al. 1991). The polypeptide identified by Nitkin et al. (1987) was 150 kD, but once the cDNA was cloned it was evident that a piece was missing because the deduced molecular mass was greater than 200 kD. cDNAs from rat (Rupp et al. 1991), ray (Smith et al. 1992), chicken (Tsim et al. 1992), mouse (Rupp et al. 1992), and human (Groffen et al. 1998a) all predicted a mass greater than that first described. Proteolytic sensitivity and/or the failure to identify intact agrin might be explained by its PG nature, because the molecular mass of the glycanated agrin is in excess of 400 kD, which would perhaps have been too large to resolve on acrylamide gels used in previous studies. Tsen et al. (1995b) were able to detect agrin from brain and vitreous body on gradient gels, and Western blotting with a rabbit polyclonal antiserum to agrin revealed a heterogeneous product of over 400 kD, which shifted to a discrete polypeptide of 250 kD after heparitinase treatment. Furthermore, agrin bound tightly to Q-Sepharose and was eluted only at ionic strengths over 0.5 M NaCl because of the net negative charge of the GAG chains. Although Tsen et al. (1995b) were the first to show that agrin was indeed an HSPG, this possibility had arisen once before when tryptic peptides from bovine kidney tubule BM HSPGs showed high sequence similarity to rat agrin (Hagen et al. 1993; Groffen et al. 1997).
Human agrin has been mapped to chromosome 1 pter and mouse chromosome 4 (Kallunki et al. 1991; Rupp et al. 1992). Agrin has a modular structure similar to that found in other BM proteins. The amino terminal begins with a signal peptide, followed by nine follistatin-like domains. These domains share similarity with Kazal-type protease inhibitors, including pancreatic trypsin inhibitor, follistatin, ovoinhibitor, thrombin inhibitor, and elastase inhibitor. Two laminin-type EGF repeats, with laminin B-chain homology, separate the eighth and ninth follistatin domains. These EGF repeats may form a rodlike structure similar to those found on the short arms of laminin. More centrally located are two domains rich in proline, threonine, and serine, with particular conservation of the proline residues across species; there are no cysteine residues in these domains. An SEA module follows, with similar structures found in perlecan, sperm protein, enterokinase, and MUC1. This module is believed to play a role in secondary structure and has been suggested to be involved in O-glycosylation (Bork and Patthy 1995). The carboxy terminal has four EGF-like repeats and three laminin-G-like repeats (Rupp et al. 1991; Groffen et al. 1998a; Cotman et al. 1999). Agrin's modular structure is conserved across species, especially in the laminin binding domain and the SEA module.
There are six GAG attachment site consensus sequences, SGXG, in chick agrin cDNA (Figure 3). These regions contain additional SG sequences that are preceded or followed by acidic amino acids, which are conducive to HS addition (Zhang and Esko 1994; Esko and Zhang 1996). Some of these GAG attachment sites are found within the C-terminal region, but it has yet to be determined if HS chains are involved in AChR clustering (Tsen et al. 1995a). However, no specific serine residues have as yet been identified as sites of glycanation.
Agrin expression is widespread. A dot-blot evaluating mRNA levels in human tissues shows agrin to be most prominent in the adult kidney, lung, liver, and thyroid gland, with low amounts detected in all other tissues. Fetal tissues had low levels in all tissues tested, with most seen in the kidney and lung (Groffen et al. 1998a; Raats et al. 1998). In chick, agrin expression was most prominent in the brain. The function of agrin in the brain may range from promoting synaptogenesis to regulating cell adhesion mediated by heparin binding proteins in the central CNS that include NCAM, thrombospondin, laminin, and myelin-associated proteins. Indirect immunofluorescence microscopy of human tissues showed strong agrin localization in the lung and kidney, in addition to both the fetal and adult capillaries of the brain. Clusters of agrin antigenicity were also seen at the NMJs of human skeletal muscle. A commonality between the glomerular, alveolar, and neuromuscular BMs has been suggested, with the prevalence of agrin in these tissues being an example. They are all fused BMs assembled by two different cell types and they express the α3-,α4-, and α5-chains of collagen Type IV and laminin β2-chain, whereas most other BMs express collagen Type IV α1- and α2-chains. In the lung, agrin was evident at the alveolar and capillary BM, and its function in the alveolar BM could be related to the follistatin repeats that are linked to protease inhibition (Van de Lest et al. 1995; van Kuppevelt et al. 1997). Kidney agrin was most abundant in the GBM, where it is the major HSPG of this unique structure (Groffen et al. 1998a,b; Figure 4D). Bowman's capsule, tubule, and vascular BMs also contain some agrin. It is postulated that agrin, rather than perlecan, may be involved in charge-selective ultrafiltration, because perfusion of rat kidney with heparitinase resulted in an increase in permeability for anionic macromolecules and albuminuria (Rosenzweig and Kanwar 1982). The kidney glomerulus is also one of few in vivo sites at which agrin has been shown to be an HSPG (Groffen et al. 1998a,b). Agrin has recently been found to be a major HSPG in senile plaques and neurofibrillary tangles, hallmarks of Alzheimer-diseased brain. It is speculated that it may protect these protein aggregates from degradation because it contains nine protease-inhibiting domains (Verbeek et al. 1999).
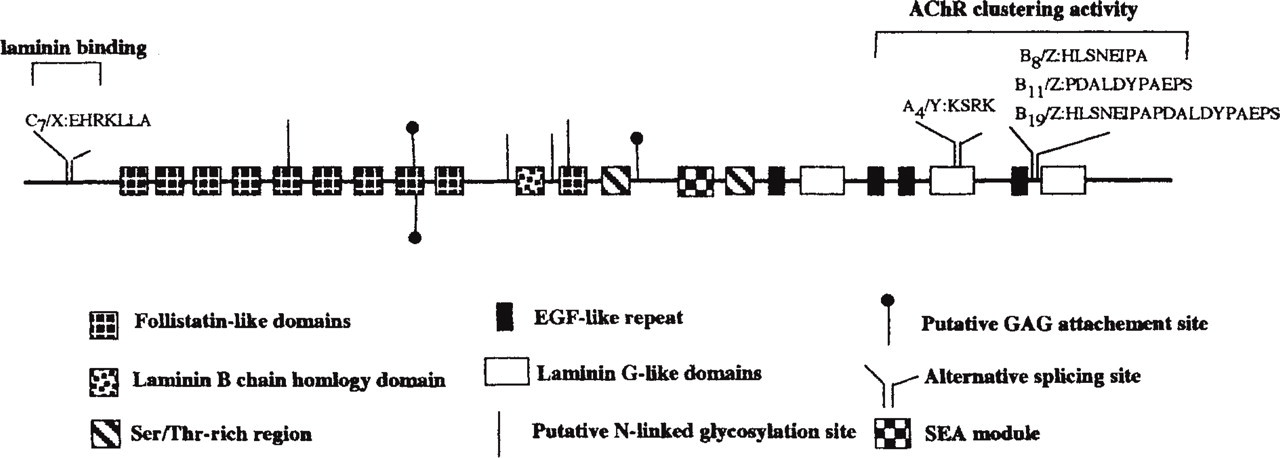
Structural model of agrin. Splice variants for chicken and human are indicated. The amino acid residue sequences and numbering pertain to chicken.
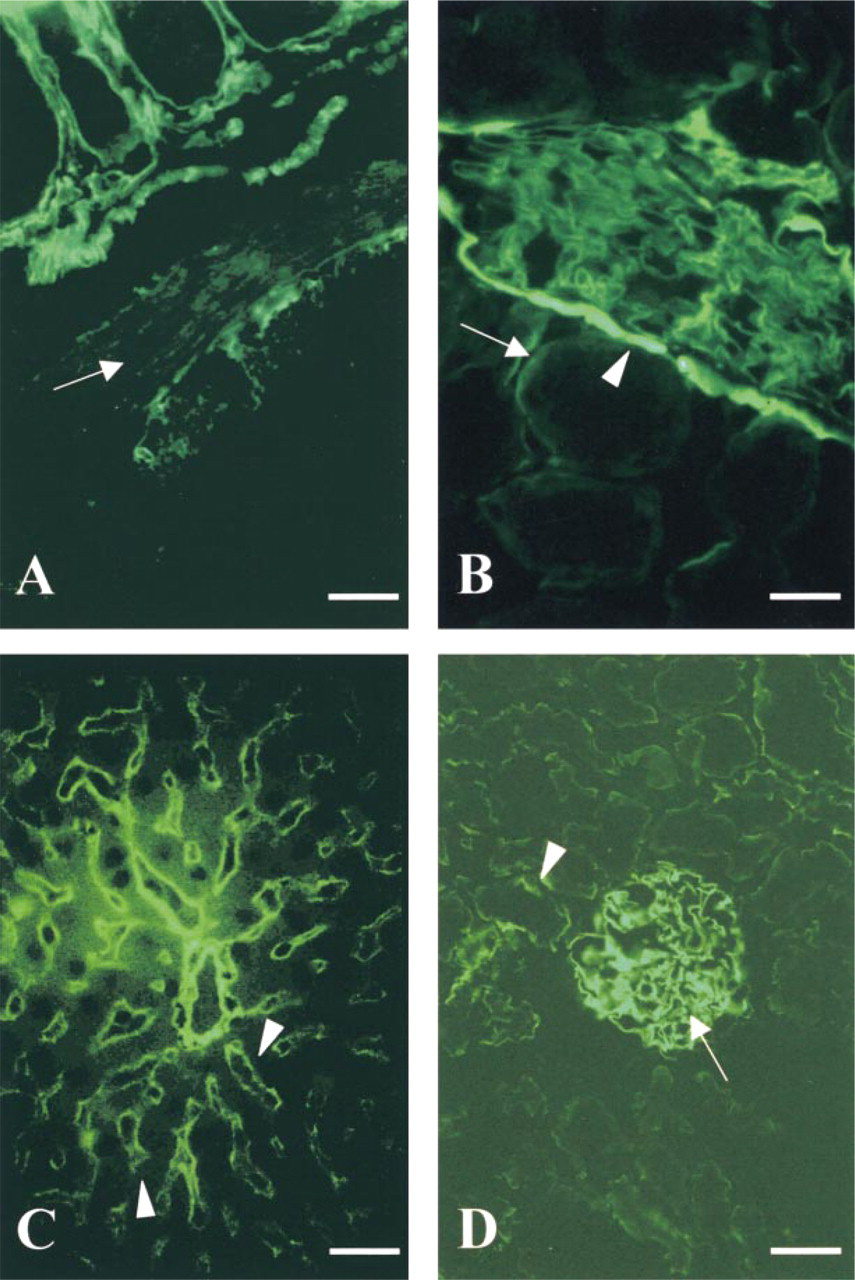
Indirect immunofluoresent staining for collagen Type XVIII (
Alternate splicing of the transcript, yielding differentially spliced forms of agrin core protein, is tissue-specific (Tsen et al. 1995a; Gesemann et al. 1996; O'Toole et al. 1996). In chicken, the alternate splice sites are at three locations, C, A, or B (X, Y, and Z are the mammalian designations for C, A, and B, respectively). Four known iso-forms are generated by alternate splicing: A4B0, A4B8, A4B11, and A4B19 (Figure 3). Agrin isoforms that have an insert at the B/Z site are expressed only in neurons and have clustering activity, whereas non-neural cells, including glia, and myotubules express agrin isoforms lacking the B/Z inserts and are unable to induce clustering of AChRs. A/Y isoforms lacking a B/Z insert are restricted to muscle; this four-amino-acid insert appears to be involved in binding of agrin to heparin and PGs (Ferns et al. 1993). These isoforms are developmentally regulated, e.g., in developing rat brain B11 and B19 are both expressed in E4 spinal cord, B11 is reduced by E20, and B 8 is detected at E14 and increases thereafter. This suggests that the multiple isoforms have different functions in nervous system development. The mechanisms that control the alternate splicing have yet to be elucidated but are tightly regulated, with as yet unclear functional consequences (Cohen et al. 1997; Dunlevy and Hassell 2000).
Further insight into agrin's functions may be implied from agrin interacting or binding proteins. The amino terminal binds the coiled-coil region of laminin-1 (Denzer et al. 1997), and plasmon resonance analysis has shown high binding affinity between agrin and laminin-2 (Cotman et al. 1999). Within the same region for laminin interaction, agrin can also bind α-dystroglycan (Gee et al. 1994; Gesemann et al. 1998). The dystroglycan complex is coupled to AChRs by the 43-kD cytoplasmic protein rapsyn, but the role of α-dystroglycan as an agrin receptor remains unclear. Furthermore, the clustering activity of agrin has been mapped to the carboxy terminal. Other putative agrin receptors include heparin binding growth association molecule (HB-GAM) (Daggett et al. 1996), neuregulins (Meier et al. 1998), integrin αvβ1 (Martin and Sanes 1997), and NCAM (Burg et al. 1995). Newer binding partners for agrin are FGF-2, thrombospondin, and tenascin-C (Cotman et al. 1999). Agrin, as well as having the potential to contain HS, is also capable of binding HS in its A/Y splice isoform (Cotman et al. 1999). There is clearly much to learn regarding the many isoforms of agrin, its role and distribution as an HSPG, and the receptor signaling that it mediates.
Collagen XVIII
By definition, all collagenous polypeptides possess at least one sequence of a repeated Gly-X-Y motif; this motif allows three polypeptides to fold into triple-helical domains which are rigid and inextensible. In fibrillar collagens, the triple-helical domains polymerize in a staggered fashion to form fibrils. Collagens classified as fibrillar contain a long, uninterrupted stretch of Gly-X-Y repeats; these include collagen Types I, II, III, V, and XI. Nonfibrillar collagens have interruptions within the Gly-X-Y motif, creating regions of flexibility and thus interfering with their ability to form fibrils. These include Types IV, VI, VII-X, and XII–XIX. The most recently described subfamily of nonfibrillar collagens are the multiplexins, for proteins with
In addition to multiple genes, multiple promoters, and alternative splicing, which provides heterogeneity in collagens, there are also post-translational modifications, including glycanation. Types IX and XII are two members of the collagen family that are part-time proteoglycans, sometimes having GAG side chains. In this case, the GAGs attached to each of these collagens are chondroitin sulfate (Fukai et al. 1994). Until recently, no collagenous HSPGs had been described, but Halfter et al. (1998) showed that collagen XVIII was a BM heparan sulfate proteoglycan.
The collagen α1 (XVIII) cDNA sequence contains an open reading frame of 3420 bp, including ten triple-helical (COL) domains, varying in length, separated by 11 non-collagenous (NC) regions. The two terminal NC regions, NC11 at the amino terminal and NC1 at the carboxy terminal, are approximately 300 residues in length, whereas the other NC regions are only 10–24 residues in length. Northern blot analyses indicate multiple RNA species and an abundance of message in kidney and liver. The human sequence was published in 1994 and showed 81.6% identity with that of the mouse (Oh et al. 1994b; Rehn et al. 1994). In addition, the α1(XVIII) collagen gene was characterized and localized to chromosome 10 for mouse and 21q22.3 for human (Oh et al. 1994b). An orthologue to vertebrate Type XVIII collagen, cle-1, has also recently been characterized in C. elegans (Ackley and Kramer 1999); this may be a precursor form of both Types XV and XVIII collagen.
Type XVIII collagen has a region of homology with a large heparin binding amino-terminal portion of thrombospondin-1, a multifunctional glycoprotein with affinity for several molecules. This domain has also been identified in collagens V, IX, XI, and the large amino termini of collagens XII and XIV. However, the sequences believed to be involved in heparin binding are not conserved in the collagens. Therefore, the significance of the thrombospondin homology is unknown (Rehn and Pihlajaniemi 1994). One RGD site is also found in the sequence, but thus far collagen XVIII has not been shown to function in cell adhesion (Halfter et al. 1998).
Some Gly-X-Y motifs of the collagenous domains are incomplete, and this is reminiscent of three other collagen chains α1 (XV), α1(XVI), and α1(XVII) (Abe et al. 1993; Rehn and Pihlajaniemi 1994). Type XVII collagen is also associated with some BMs. The α1(XVIII) collagen chain is most similar to the α 1 (XV) chain both in the lengths of some carboxy terminal triple-helical domains and the NC1 of collagen XVIII [60% amino acid identity of mouse Type XVIII with the human α 1 (XV) chain], with conservation of the four cysteine residues (Oh et al. 1994a; Rehn and Pihlajaniemi 1994; Rehn et al. 1994).
Since the initial publications, both mouse and human collagen XVIII have been shown to have more than one variant; three are reported in mice and two in human (Muragaki et al. 1995; Rehn and Pihlajaniemi 1995; Rehn et al. 1996; Saarela et al. 1998). All variants, however, contain a common region of 299 amino acids upstream of the ten interrupted triple-helical sequence domains; this region contains the thrombospondin homology (Figure 5). The differences are in the N-terminal region and signal peptide. The S form, or separate signal peptide, was described by Rehn and Pihlajaniemi (1994) and is also referred to as NC1–517. The CR form contains a domain with 10 Cys residues, which shares similarity to the extracellular domain of the Drosophila transmembrane receptor protein frizzled and two frizzled-like proteins from rat; this variant is also referred to as NC1–764. The third variant, the A form, contains only an acidic residue-rich region upstream of the common 299 amino acids and is also referred to as NC1–301 (Muragaki et al. 1995; Rehn and Pihlajaniemi 1995). The longer splice variant, CR/NC1–764, has not been described in humans (Saarela et al. 1998). The A/NC1–301 and CR/NC1–764 variants are splice variants transcribed from the same promoter and have the same signal peptide. The S/NC1–517 is transcribed from a separate promoter and has a different signal peptide (Rehn et al. 1996). The three forms of collagen XVIII have variable tissue distributions and expression levels. A/NC1–301 is constitutively expressed in all tissues but is more abundant in kidney and testis. The A/NC1–301 and CR/NC1–764 have a more diverse distribution than the S/NC1–507. Northern blot analysis using a chicken clone as a probe shows an abundance of transcript in the kidney and heart but little in the brain and liver, with similar results in the mouse. The mRNA was also found to be prominent in cultured astrocytes. In situ hybridization with chicken embryo sections shows an abundance of mRNA in the developing heart, kidney, and peripheral nervous system, with some in the meninges surrounding the spinal cord. There was strong labeling of the roof plate of spinal cord and small amounts in the ventricular lining of the CNS. Agrin, in contrast, is abundant in the CNS, and is prominent in radial glial cells and the motor neurons of the spinal cord. Agrin is less abundant in the peripheral nerves or muscle compared to collagen XVIII (Halfter et al. 1998).
Muragaki et al. (1995) first showed the immunohistochemical localization of collagen XVIII in vivo. Positive staining for collagen XVIII was seen along the BM zones of vessels in the intestinal villi, the choroid plexus, skin, liver (Figure 4C), and kidney. Halfter et al. (1998) carried out detailed immunofluoresent staining of chicken tissues with an antibody specific for collagen XVIII. In the vascular system, collagen XVIII appeared later in development compared to Type IV collagen, but in post-hatch birds the distribution of collagen XVIII and IV in the spinal vascular system was indistinguishable. Collagen XVIII was abundant in the retina, epidermis, pia, cardiac and striated muscle, kidney (Figure 4B), blood vessels, and lung. The distribution of collagen XVIII was similar to that of other BM HSPGs but was not completely identical. The most apparent difference was in the gut, where agrin, perlecan, and Type IV collagen decorated the inner and outer smooth muscle layer, whereas collagen XVIII was absent (Figure 4A). Collagen XVIII, therefore, is the second most abundant BM collagen next to collagen IV (Halfter et al. 1998).
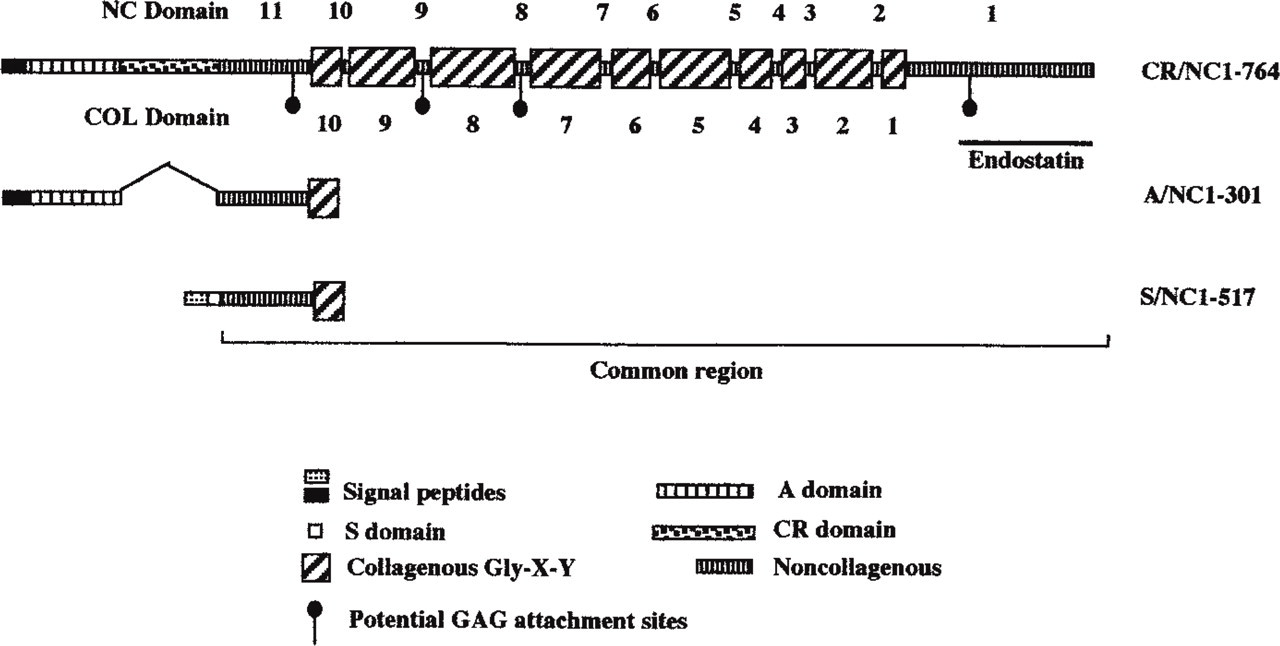
Schematic structures of the full-length variant polypeptides of the collagen XVIII chains. The non-collagenous domains are indicated above the structure and the collage-nous domains are indicated below.
Several Ser-Gly dipeptides suitable for GAG attachment are found in the collagen XVIII sequence, raising the suspicion that collagen XVIII was a PG. When the protein was digested with chondroitinase to remove CS/DS GAGs, however, there was no change in the electrophoretic mobility of the protein (Halfter et al. 1998). The characterization of collagen XVIII as a heparan sulfate proteoglycan was described in conjunction with the cloning of a portion of the chicken homologue (Halfter et al. 1998). Two monoclonal antibodies recognizing a heparitinase-sensitive proteoglycan from chick embryonic basal lamina were used to screen an E5 chick yolk sac cDNA expression library. Clones retrieved from the screen showed 90% homology with the ninth and tenth domains of human and mouse collagen XVIII.
Western blotting analysis of the vitreous body extracts showed a smear of 300 kD, the heterogeneity typical of a proteoglycan (Halfter et al. 1998). A polypeptide of about 180 kD appeared after heparitinase, but not chondroitinase ABC. The protein was completely susceptible to collagenase digestion as expected. Collagen XVIII was found in the urea extracts of meninges, amnion, and kidney. In contrast to agrin, collagen XVIII was not in a PBS extract and is solubilized only in the presence of chaotropes. All tissues digested with heparitinase resulted in a core protein of 180 kD. Mesangial tissues contained only a minor 180-kD polypeptide without heparitinase digestion, indicating that collagen XVIII may exist almost exclusively as a HSPG. Moreover, these data might suggest that all three chains of a single trimer bear HS chains. However, collagen XVIII can be expressed without HS in liver (Musso et al. 1998). Undenatured collagen XVIII is 700–1000 kD, suggesting that trimers of the 300-kD monomers are formed. Trimerization was independent of HS side chains because heparitinasetreated samples did not dissociate the complex. Collagen XVIII bound to anion exchange resins with affinity typical of PGs and similar to that of both agrin and collagen IX, two other PGs present in the vitreous body. Moreover, all three splice variants were synthesized as HSPGs (Halfter et al. 1998).
Collagen XVIII is of particular interest because of a 22-kD anti-angiogenic peptide with tumor-suppressing activity, known as endostatin, within its C terminal (O'Reilly et al. 1997; Dhanabal et al. 1999). The NC1 domain is composed of three subdomains: N-terminal association region (∼50 residues); a central protease-sensitive hinge region (∼70 residues); and a C-terminal endostatin domain (∼180 residues; Sasaki et al. 1998). Sasaki et al. (1998) compared the endostatin protein to that of the murine full-length NC1 monomer (38 kD) domain of collagen XVIII. Both recombinant proteins were isolated from 293 cells. The full-length NC1 was more sensitive to endogenous proteolysis compared to endostatin. Solid-phase assays showed that full-length NC1 had strong interactions with perlecan and laminin-1, whereas endostatin showed 100-fold weaker interaction. This suggests that, when released by cleavage, endostatin would be free to diffuse. However, both recombinant proteins had the ability to bind fibulin-1 and −2. This binding also occurred in the presence of EDTA, unlike other fibulin interactions, which are calcium-dependent. Interestingly, endostatin-like proteins ranging from endostatin (22 kD) to NC1 monomer (38 kD) were not detected in conditioned medium from 10 cell lines including fibroblasts, epithelial, and tumor cells. Low levels were detected in the medium of human aortic and microvascular endothelial cells. Tissue extracts from brain, skeletal muscle, heart, kidney, testis, and liver all contained endostatin epitopes, and serum levels were 120–300 ng/ml (Sasaki et al. 1998).
Immunohistology of embryonic tissues localized endostatin to many but not all blood vessels and to some other BM zones. Immnunoelectron microscopy revealed a strong association of endostatin with elastic fibers of arteries. In addition, there was a distinct co-localization of endostatin with fibulin-1, fibulin-2, and nidogen-2, which correlated with in vitro binding studies (Miosge et al. 1999). Endostatin has yielded anti-tumor responses in several in vivo models (O'Reilly et al. 1997; Boehm et al. 1997). The avascular nematode C. elegans may require endostatin for neurogenesis. Deletion of the endostatin domain in cle-1, the homologue of type XVIII collagen, caused egg-laying defects and slight un-coordination (Ackley and Kramer 1999). The mechanism behind endostatin's action is unknown, but its crystal structure predicts a prominent heparan sulfate binding site (Hohenester et al. 1998), suggesting that endostatin competitively inhibits heparin binding angiogenic factors (Chang et al. 1999). Binding to BM HSPGs (including collagen XVIII itself) is unknown but could provide a mechanism for storage similar to that proposed for members of the fibroblast growth factor family. Further investigation into both collagen XVIII and endostatin will help elucidate the mechanisms by which endostatin is released from its parent molecule and functions as an angiogenesis inhibitor.
The obvious growth of the BM HSPG family is under way, as is the family of BM chondroitin sulfate PGs (CSPGs). Surprisingly, perlecan can be included in this family because it can be secreted in some tissue and cell lines in culture with CS GAG substitution (Hassell et al. 1992; Couchman et al. 1996; Tapanadechopone et al. 1999).
New Additions to the Laminin Family
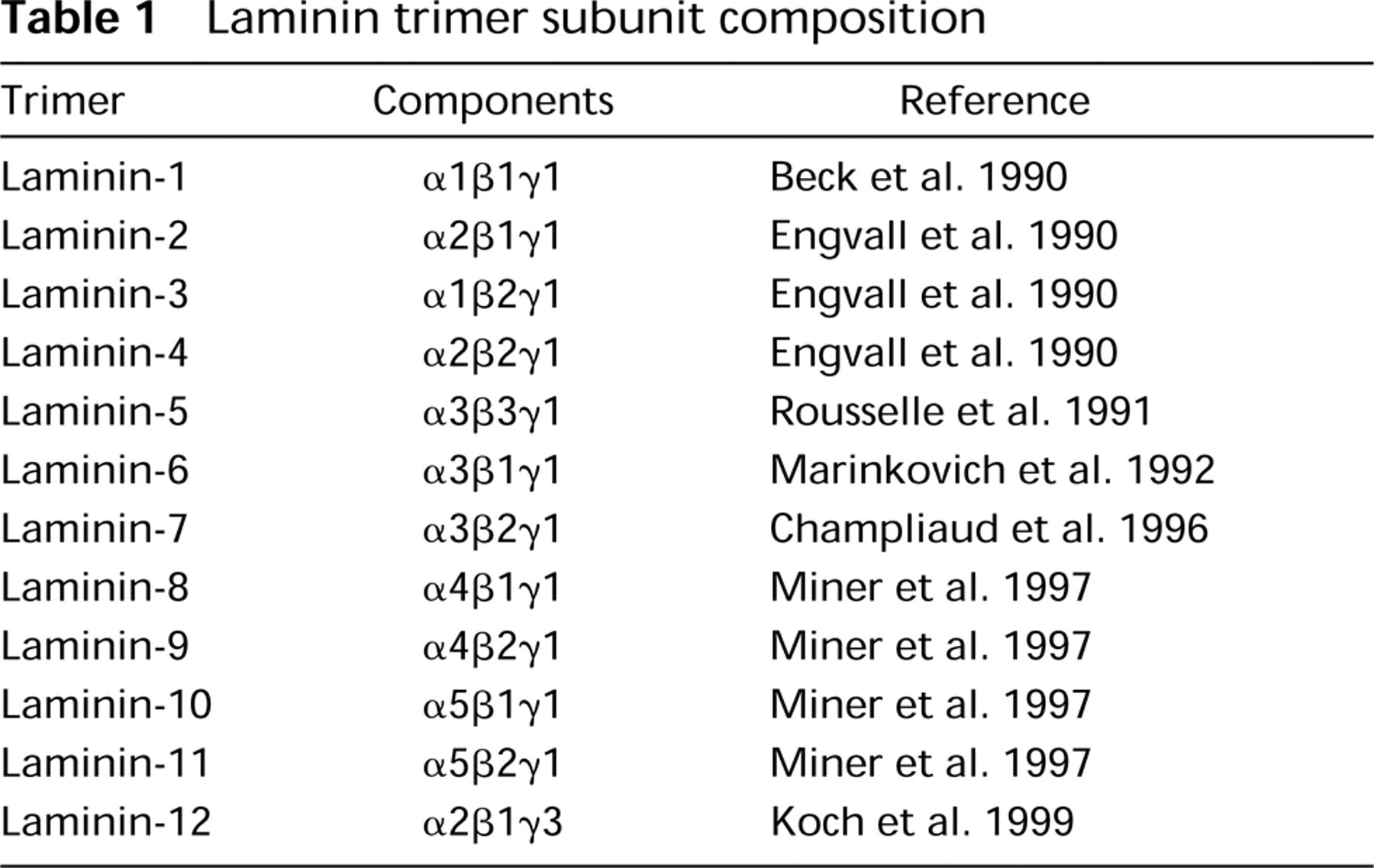
Laminin was first described as a component in the stroma of the EHS tumor (Timpl et al. 1979) and the extracellular deposit of murine parietal endoderm PYS cells (Chung et al. 1977, 1979). The purified large glycoprotein was named laminin, now referred to as laminin-1, and for several years was the only laminin isoform reported. Three different gene products encoding three separate polypeptides, A, B1, and B2, later renamed α, β, and γ, respectively (Burgeson et al. 1994), combine through ionic interactions and disulfide linkages to form the large cruciform shaped complex of laminin-1 (Beck et al. 1990). In its native state the complex is approximately 900 kD. When reduced and denatured, the α-chain polypeptide has a mass ∼400 kD; the smaller β- and γ-chains are both ∼200 kD (Martin and Timpl 1987; Nissinen et al. 1991). Antibodies recognizing the laminin-1 complex stained all BMs, and staining with β- and γ-chain specific antibodies was widespread. However, laminin α1-chain was reported to be absent or low in adult and embryonic tissues (Durbeej et al. 1996, and references therein), whereas other studies reported a widespread distribution of the α1 chain (Engvall et al. 1990). The absence of α1 in some BMs led to the possibility of other α-chains and additional laminin isoforms. To date, six α-, three β-, and three γ-laminin polypeptides have been described. Interestingly, γ1 is found in complexes with each of the α-chains, which explains why antibodies against α1 stained all BMs, leading to the erroneous impression that there was perhaps just a single laminin trimer, as found in the EHS tumor. Thus far, there is evidence of 12 different laminin isoforms in vivo (Miner et al. 1997), while only five intact trimers have currently been purified (Table 1).
The multiplicity of laminin isoforms provides for heterogeneity among BMs. The laminin family plays essential roles in structural integrity, cell adhesion, and signaling (Ekblom et al. 1998). Interaction of cells with laminins is mediated by a variety of cell surface receptors, including integrins, membrane-bound proteoglycans, and glycoproteins, such as dystroglycan (reviewed by Timpl and Brown 1996; Aumailley and Smyth 1998). Functions of the laminin family are revealed by naturally occurring mutations that result in genetic disorders and also by “knockout” models (Ryan et al. 1996). γ1 null mice are embryonically lethal; there is no survival beyond 5.5 days post coitum due to the lack of BM establishment and failure of parietal yolk sac development. Embryoid bodies from γ1 null cultured ES cells lacked BMs and produced disorganized extracellular deposits of collagen Type IV and perlecan. Entactin-1 and laminin α1-chain were secreted but did not become extracellular matrix-associated. Furthermore, the cells failed to differentiate into stable myotubes. The laminin γ1 subunit is therefore necessary for laminin assembly and organization of BM components (Smyth et al. 1999). The only other chain deletion that results in lethality is α5 (Miner et al. 1998). Invertebrates also require laminins. A null mutation in the Drosophila α3,5/lam A chain, the precursor of vertebrate α3 and α5 chains, is embryonically lethal, with visible defects in mesoderm-derived tissue (Yarnitzky and Volk 1995) and the NMJ (Prokop et al. 1998). The newly described Drosophila α1,2-chain, the precursor of vertebrate α1-and α2-chains, is essential for embryonic viability and is involved in processes that require cell migration and cell adhesion (Martin et al. 1999).
The most recently described BM laminin isoforms are laminins 8–11 (Miner et al. 1997; Miner and Patton 1999; Table 1). Being the largest subfamily of laminin chains, the α subfamily is comprised of six mammalian and two Drosophila members. The discovery of the α4 (Lentz et al. 1997) and α5 (Miner et al. 1995) chains led to the identification of laminins 8 (α4β1γ1), −9 (α4β2γ1), −10 (α5β1γ1), and −11 (α5β2γ1). Laminin α-chains vary in size; the α1 (∼400 kD), α2, α3b (∼360 kD) (Doliana et al. 1997; Miner et al. 1997), and α5 (∼450 kD) chains are full-length chains, α5 being the largest, whereas the α3a and α4 (∼180 kD) chains are considered as truncated chains (Figure 6). Interactions of cells with laminin α-chains are critical for cell-matrix interactions. At least seven distinct integrin heterodimers (α1β1, α2β1, α3β1, α6β1, α7β1, α9β1, α6β4, and αvβ3) as well as dystroglycan, HSPGs, and HNK-1/L2 bind to sites on laminin α-chains (Rao and Kefalides 1990; Gee et al. 1993; Hall et al. 1993; Sung et al. 1993; Mercurio 1995; Mecham and Hinek 1996; Colognato and Yerchenco 1996).
Laminin trimer subunit composition
The laminin α-chain genes are all expressed in both mouse embryos and adults, but in distinct spatiotemporal patterns. All chains are localized to the ECM and, with a few exceptions, more specifically to BMs. All BMs studied to date contain at least one α-chain, and many BMs have more than one α-chain subfamily member. Contrary to early reports, the α1 laminin chain has the most restricted distribution, whereas the α5 laminin chain is the most widely distributed α-chain in adult vertebrates (Durbeej et al. 1996; Miner et al. 1997). Patterns of α-chain expression are established embryonically, but some individual BMs switch α-chains as development proceeds; this sort of switching has been detailed for kidney development (Durbeej et al. 1996; Miner et al. 1997; Miner 1998).
Here we focus on the most recently described members of the α-chain subfamily, α4 and α5. These α-chains are more widely expressed than the α1-, α2-, and α3-chains. Miner et al. (1998) detailed α5 expression in murine development; it was present in most embryonic and extraembryonic BMs at early stages but became restricted to a distinct subset of BMs as development proceeded. The α5-chain localized to practically all γ1-positive BMs at E8.5, including BMs underlying the neural folds and the surface ectoderm as well as BMs associated with gut epithelium. The α5-chain was also a prominent component of BMs around somites. In later stages of development, α5 was expressed in a subset of BMs and remained abundant in the surface ectodermal BM throughout embryogenesis.
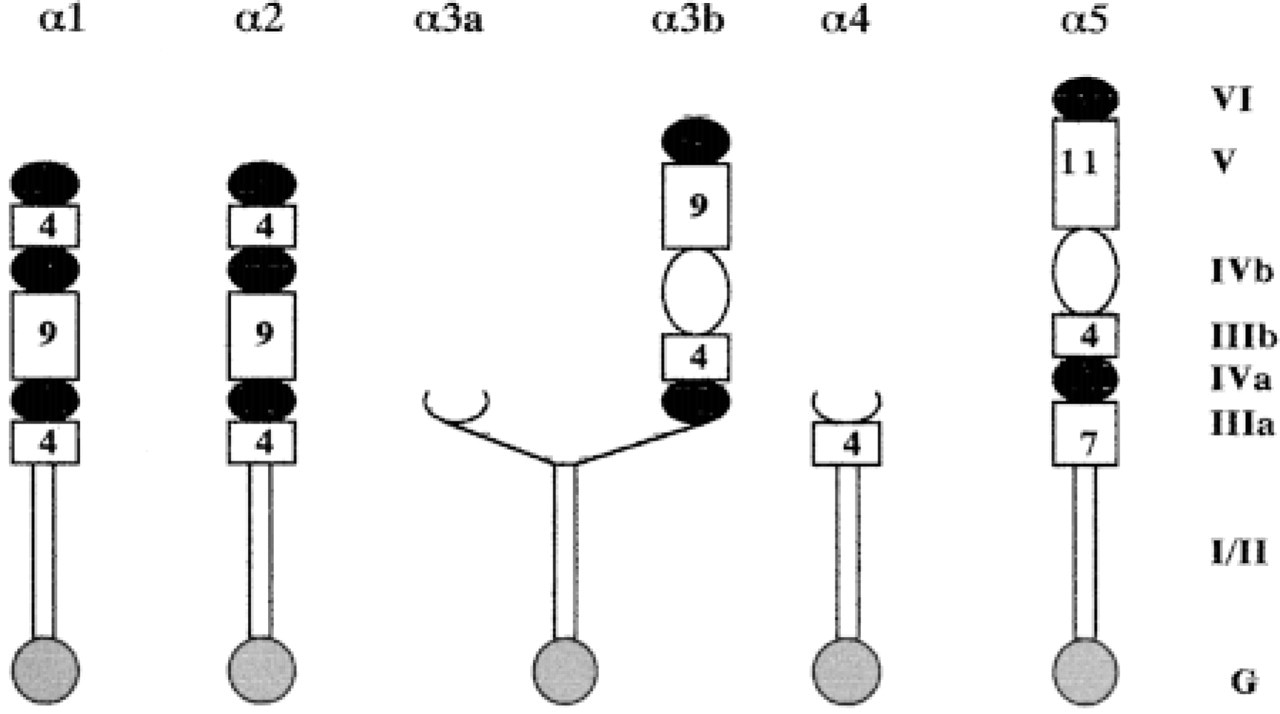
The laminin α-chain subfamily. The original numbering of domains is based on accepted nomenclature (Engvall and Wewer 1996; Sasaki et al. 1998). Carboxy-terminal globular domain, G; α-helical domains (I and II); cysteine-rich domains containing EGF-like repeats (IIIa, IIIb, and V); globular regions (IVa, IVb, and VI). The number of EGF repeats is shown in the boxes.
Ribonuclease protection analysis of various E17.5 and adult mouse tissues revealed that low to moderate levels of α4 and abundant levels of α5 RNA were present in all tissues tested, with lowest expression in the liver for both. In situ hybridization performed on E15.5 day embryos revealed that laminin α4 was expressed strongly in mesenchymal tissues of the head, dorsal root ganglia, and intestine, and was observed diffusely in skeletal and cardiac muscle. Laminin α5 was strongly expressed in skin, lung, olfactory epithelium, the superficial layers of the tongue and palate, salivary gland, intestine, and the most superficial cells of the liver (Miner et al. 1997).
Analysis of protein expression and localization was carried out using polyclonal antibodies against the α4 and α5 laminin chains. In adult murine kidney, α4, although absent from all renal, epithelial, and arterial BMs, was found in many capillaries of the medulla. A more widespread expression was found for the α5-chain, for which virtually all BMs, including those of glomeruli, arteries and all tubules were positive. α4 expression in heart was restricted to capillaries and was found at low levels in many myocyte BMs (Virtanen et al. 1996). In the heart, α5 localized to the arterioles and capillaries and was found at low levels in many myocyte BMs. The α5-chain co-localized with α3 in most alveolar BMs of the lung, whereas α4 was expressed in a subset of alveolar BMs (Sorokin et al. 1997). In the tissues studied, the α4- and α5-chains co-localized with the either the β1- or the β2-chain and the γ1-chain. Expression patterns of isoforms containing α4 and α5, as well as other laminin isoforms, in the peripheral nervous system and the spinal cord are reviewed by Lentz et al. (1997). Overall, α5 has a widespread distribution in adult tissues and might replace α1 either in endothelium or in epithelium, which lack α1.
Because of its widespread distribution, it was not surprising that mice null for the α5 laminin chain result in fetal lethality (Miner et al. 1998). Many phenotypes were described: failure of neural tube closure, exencephaly, failure of digit separation, syndactyly, and dysmorphogenesis of placenta. All defects were apparent after E9 and no homozygotes survived beyond E17. Development of many tissues and organs was perturbed by the absence of the α5-chain, including limb, neural tube, placenta, lung, heart, intestine, and kidney. More specifically, the kidneys were either small or absent and had defective glomerulogenesis (Miner and Li 2000). Defects in lung lobe septation and bronchiolar branching were observed, and the left ventriole of the heart was reduced in size. Lethality was most likely caused by defective placentation in embryos; α5 may be important in placental endothelial cell migration and blood vessel branching, trophoblast adhesion to BM, and BM formation. In addition to the roles suggested by the α5 null mice, α5 is an early molecular marker for sexual differentiation and is regulated by the testis-determining factors (Frojdman et al. 1999). All this underlines the importance of BMs in development because in the absence of the α5-chain, defective matrix assembly was observed. It will be interesting to see whether the deletion of the α4-chain gene will have a milder phenotype based on its restricted distribution.
Laminin chains are not always localized to the BM. This is evidenced by the recent description of the γ3-chain (Iivanainen et al. 1999; Koch et al. 1999). Iivanainen et al. (1999) described the γ3-chain as being generally expressed in endothelial cells of arterioles and capillaries as well as interstitial cells of certain tissues. Alternatively, Koch et al. (1999) revealed a broad tissue distribution in skin, heart, lung, and the reproductive tracts. In skin, the γ3-chain was localized within the BM of the dermal-epidermal junction at points of nerve penetration. The chain was also prominent along the apical surface of ciliated epithelial cells of lung, oviduct, epididymis, ductus deferens, and seminiferous tubules. This distribution of γ3-containing laminins on the apical surfaces of a variety of epithelial tissues is novel and suggests the possibility that apical laminins are important in morphogenesis and structural stability of the cilitated processes of these cells (Koch et al. 1999). Western blotting analysis showed that γ3 is complexed to α2 and β1 laminin chains, and therefore represents the twelfth laminin isoform described (Iivanainen et al. 1999).
Summary
Basement membranes are far more complex than conventional electron microscopy originally led us to believe. The proteins discussed above illustrate the heterogeneous complexity of BM structure and function. Tissue specific BMs, in terms of composition, are now revealed, as are further roles for their components. Further complexity is likely to arise as the human genome project is completed and further BM proteins are discovered. These discoveries are only a prelude to the vast amount of work that lies ahead. Interactions among other BM proteins will have to be elucidated, as well as potential cell surface receptors. Post-translational modifications are many and varied. It is not clear why there are multiple HSPGs in BMs. Pehaps there are core protein-specific glycanation events that yield HS chains with specific fine structure and binding attributes. Multiple collagens can be glycanated, but the reasons are not clear. Normal functions of these proteins in vivo may offer insight into many inherited and acquired BM-associated diseases and, perhaps, future prospects for gene therapy.
Footnotes
Acknowledgements
Supported in part by National Institutues of Health grant RO1 AR36457 to Dr J.R. Couchman.
We thank Dr G.J. Cole (Ohio State University) for the gift of the agrin antibody.