
Abstract
Objective:
The glutathione (GSH) pathway is the main antioxidant system to protect against oxidative stress in the human brain. In this study, we tested whether molecular components of the GSH antioxidant system are changed in dorsolateral prefrontal cortex tissue from people with schizophrenia compared to controls.
Method:
The levels of total glutathione and reduced GSH were determined by fluorometric assay via quantifying thiols in extracts from frontal cortex of 68 people. Immunoblotting was used to measure levels of enzymes responsible for maintaining GSH, the glutamyl-cysteine ligase (GCL) catalytic subunit (GCLC) and the GSH peroxidase (GPx)-like protein (n = 74). Quantitative reverse transcription polymerase chain reaction (RT-PCR) was used to measure GCLC messenger RNA (mRNA) expression.
Results:
Both total glutathione (t(66) = 2.467, p = 0.016) and reduced GSH (t(66) = 3.001, p = 0.004) levels were significantly less in people with schizophrenia than in controls. However, there were no significant differences in either GCLC-like protein (t(72) = −1.077, p = 0.285) or GCLC mRNA expression (t(71) = −0.376, p = 0.708) between people with schizophrenia and control subjects. There was also no significant difference of GPx-like protein levels between schizophrenia and controls (t(72) = −0.060, p = 0.952). Moreover, no significant correlations of putative confounding factors with GSH changes were detected.
Discussion:
These results suggest that people with schizophrenia have impaired GSH antioxidant capacity, alongside normal levels of key regulatory proteins.
Introduction
In brains, increased reactive oxygen species (ROS; such as H2O2, O2− and OH−) and antioxidant system deficits can lead to oxidative stress, which may lead to neuron damage/death and cause tissue damage (Betteridge, 2000; Klein and Ackerman, 2003); oxidative stress is a common condition in ageing and occurs with neurodegeneration (Barnham et al., 2004; Mariani et al., 2005). There is increasing evidence of oxidative stress being involved in schizophrenia from both studies of peripheral blood and studies of brain tissue (Flatow et al., 2013; Gawryluk et al., 2011; Mahadik and Mukherjee, 1996; Mukherjee et al., 1996). For instance, Flatow et al. (2013) performed a meta-analysis to examine the evidence for oxidative stress from peripheral measures at different clinical stages of schizophrenia. This meta-analysis showed that oxidative stress was identifiable in schizophrenia at all stages of illness and occurred regardless of lifestyle factors or antipsychotic treatment of the patients (Flatow et al., 2013). Importantly, the results from this meta-analysis suggested the possibility that it is possible that oxidative stress is one potential cause of schizophrenia or that oxidative stress may form an important core component of the biological basis for schizophrenia.
In human brain, glutathione (total glutathione) is a key component of one of the most important antioxidant systems to protect against metabolic stress (Bains and Shaw, 1997; Dringen, 2000; Dringen et al., 2000). Glutathione is a tripeptide (composed of three amino acids: cysteine, glutamate and glycine) that buffers ROS in cells. The synthesis of glutathione is supported by two enzymes: glutamate cysteine ligase (GCL), the rate-limiting enzyme; and GSH synthetase (Figure 1). There are two different states of glutathione in brain: reduced glutathione (referred to here as GSH), where the molecule can neutralize ROS; and oxidized glutathione (referred to here as GSSG), where it cannot. GSH detoxifies ROS via glutathione peroxidases (GPx). After this reaction, every 2 moles of GSH are oxidized to 1 mole of GSSG; then GSSG is recycled back to GSH by GSH reductase (GR) (Figure 1). In healthy cells, more than 90% of total glutathione is in the reduced GSH form (Halprin and Ohkawara, 1967), which has a very narrow homeostatic range. So keeping total glutathione and reduced glutathione (GSH) at sufficient levels as well as maintaining a healthy balance of GSH to GSSG, which reflects the redox state of a cell, are necessary for preventing the human brain from experiencing oxidative damage.
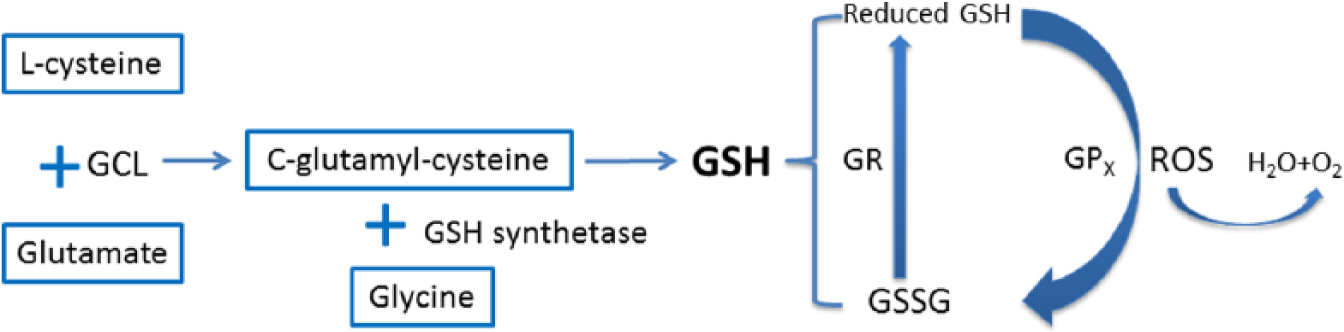
Glutathione synthesis and recycling pathway. Synthesis of glutathione is supported by two enzymes: glutamate cysteine ligase (GCL) and GSH synthetase. First,
Previous studies have found evidence of GSH deficits in schizophrenia suggesting less ability to buffer oxidative events in the brain. Do et al. (2000) reported that based on examination of cerebrospinal fluid samples, unmediated people with schizophrenia (n = 26) had a significant reduction of total glutathione (27%, p < 0.05) as compared to matched unaffected controls (n = 14). Using the non-invasive proton magnetic resonance spectroscopy, they also reported that there was less total glutathione (reduced by 52%, p = 0.0012) in medial prefrontal cortex of people with schizophrenia than in matched controls (Do et al., 2000). These results suggest that the brains of people with schizophrenia are under oxidative stress, and the prefrontal cortex may be one area experiencing oxidative stress. However, these findings are not from direct measures in brain tissue. Postmortem tissue studies are needed to support the evidence, and to confirm the magnitude of oxidative stress in the cortex of schizophrenia, and could also determine the putative molecular changes that coincide with GSH deficits by testing the levels of proteins important for synthesis and breakdown of GSH.
An abnormal GSH system in schizophrenia has previously been reported from studies of human postmortem brains. Yao et al. (2006) investigated the GSH system in caudate nucleus (from 12 schizophrenia cases and 26 matched controls). Using high-performance liquid chromatography with coulometric multi-electrode array system (HPLC-CMEAS) to measure GSH, they reported reduced GSH in postmortem brain tissue from schizophrenia cases. However, this study only included 12 schizophrenia cases, and it did not determine GSH levels in cortex. We aimed to determine the GSH levels in the dorsolateral prefrontal cortex (DLPFC), a brain area with known neuropathology and neurophysiological abnormalities in schizophrenia (Catts et al., 2014; Hashimoto et al., 2008; Weickert et al., 2003; Weinberger, 1987). Evidence of GSH deficits in prefrontal cortex has been found in animal models mimicking some aspects of schizophrenia. When phencyclidine (a N-methyl-
It is possible that the levels or the activity of enzymes that regulate GSH and GSSG levels are changed in the brain of people with schizophrenia, which could lead to altered glutathione or imbalanced GSH to GSSG levels. We hypothesized that there would be a decrease in total glutathione in people with schizophrenia and that the pathway of GSH synthesis regulating total glutathione levels, and the GSH recycling pathway, which can affect GSH levels, would be altered. In support of this, Gysin et al. (2007) found deficits of GSH synthesis in schizophrenia using cultured skin fibroblast cells from people with schizophrenia and matched controls. The cells were treated with dimethyl sulfoxide to induce oxidative stress, and the schizophrenia samples had significantly reduced GCL activity (26%, p = 0.002) compared to control samples. Moreover, a significant reduction in protein levels of a subunit of GCL, ‘GCLC’ (29%, p < 0.001), was correlated with the GCL activity reduction. The results of this study suggest that less GCLC may be a key factor contributing to less GSH. However, other studies have not replicated this result (Gawryluk et al., 2011). Gawryluk et al. (2011) found that GCLC protein levels were not significantly different between individuals with schizophrenia (n = 14) and controls (n = 12) by Western blotting in postmortem prefrontal cortex. They also found no changes in the key GSH recycling enzyme GR levels, but reduced GPx levels in schizophrenia. This suggests there may be no deficit in GSH synthesis enzymes, but that an impaired GSH system may be caused by reduced levels of GPx in schizophrenia.
In this study, we examined the GSH antioxidant system in postmortem DLPFC tissue from people with schizophrenia and matched healthy controls. The quantities of GSH and the major enzymes in this system (GCLC and GPx-like protein) were measured. We also tested the GCLC messenger RNA (mRNA) expression, as this is the rate-limiting enzyme in the GSH synthesis pathway and strongly regulated by neuronal activity (Baxter et al., 2015; Gawryluk et al., 2011). It is hypothesized that decreased total glutathione and reduced GSH levels and lower GCLC-like or GPx-like protein levels would be found in schizophrenia. There are several confounding factors (antipsychotics, illicit drugs and smoking) that might induce oxidative stress in schizophrenia. Thus, in this study, we investigated whether these confounding factors may contribute to oxidative stress through correlational analysis with GSH changes in schizophrenia.
Materials and methods
Dorsolateral prefrontal cortex cohort description
Tissue from the DLPFC of schizophrenia patients and matched control subjects was obtained from the New South Wales Brain Tissue Resource Centre. All frozen tissue was sampled from the middle third (rostro-caudally) of the middle frontal gyrus anterior to the premotor cortex, corresponding to the BA46. Samples were trimmed to include primarily grey matter. All research was approved and conducted under the guidelines of the Human Research Ethics Committee at the University of New South Wales (HREC 12345). This cohort included 37 people with schizophrenia and 37 individuals matched controls (Weickert et al., 2010). The schizophrenia and controls were matched according to age, brain pH, RNA integrity number (RIN) and postmortem interval (PMI); there were no statistically significant differences between them (Table 1). In this study, immunoblotting and quantitative polymerase chain reaction (qPCR) experiments were performed based on this cohort. However, for the GSH assay, the 37/37 cohort needed to be narrowed down because some tissue quantities were not sufficient. Based on the tissue remaining, a new 34/34 cohort was designed for the GSH assay (Supplementary Table 1).
New South Wales Brain Tissue Resource Centre dorsolateral prefrontal cortex cohort.
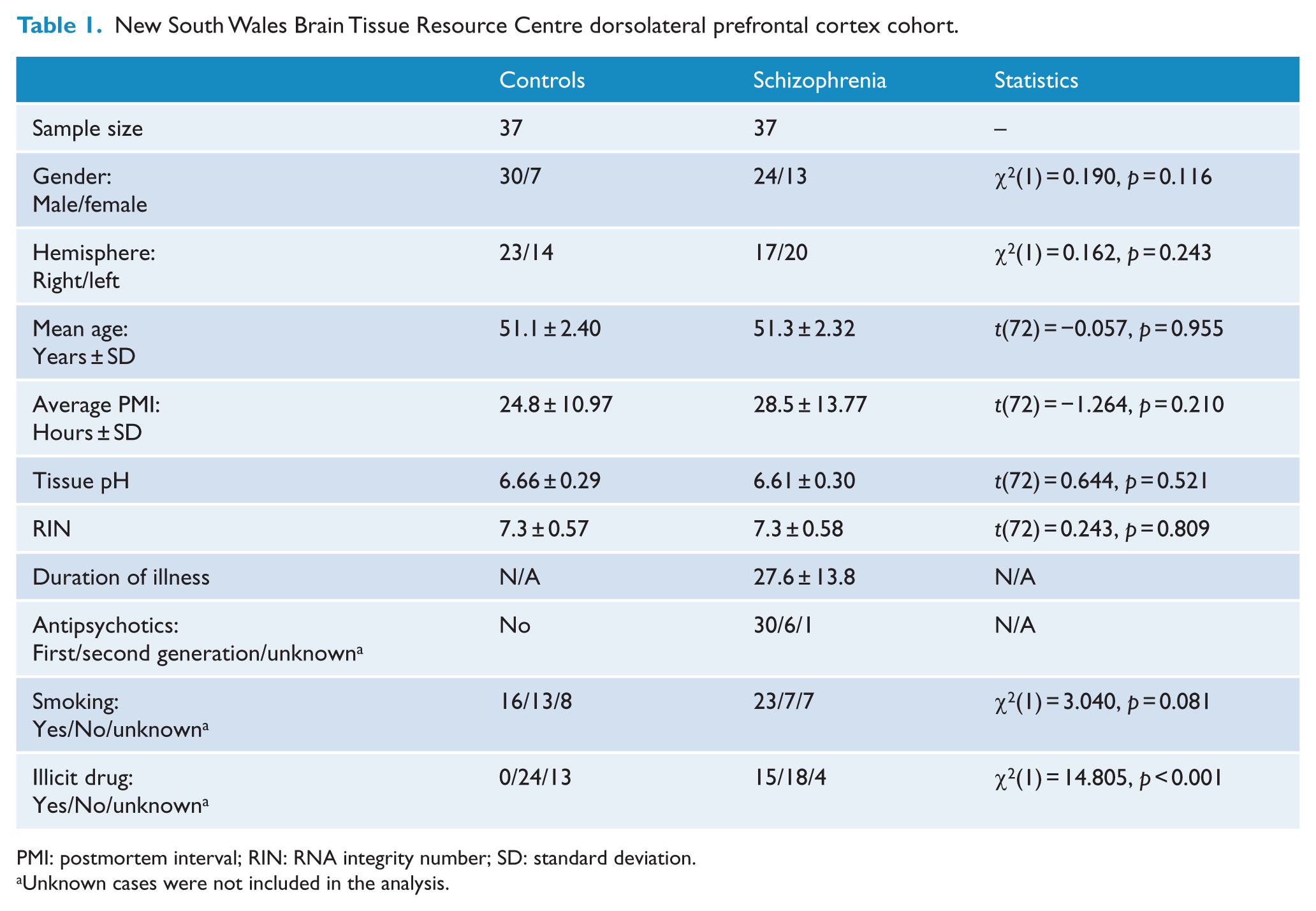
PMI: postmortem interval; RIN: RNA integrity number; SD: standard deviation.
Unknown cases were not included in the analysis.
Glutathione assay
The GSH Assay Kit (Abnova, KA2520) was used to measure both total glutathione and reduced GSH levels. For each individual case in the 34/34 cohort, 20 mg cortical grey matter tissue was weighed out. According to the GSH Assay Kit protocol, 200 μL ice-cold phosphate-buffered saline (PBS) homogenizing buffer was added to the tissue to make a 100 mg/mL solution. Tissue was homogenized with a plastic pestle in microfuge tubes for 1–2 minutes on ice and then centrifuged at 500g (Eppendorf, 5417R) for 10 minutes at 4°C. The supernatant was recovered and an aliquot immediately utilized for the GSH assay; the 100 mg/mL (tissue) solution was diluted to 5 mg/mL for GSH measurement. Bradford Reagent (Sigma-Aldrich, Cat#B6916) was used to determine the concentration of total protein in the tissue extracts, and we did not find a significant difference in protein concentration between diagnostic groups suggesting that there was not a systematic diagnostic difference in our protein extraction efficiency. The components of the GSH assay kit can be found in Supplementary Table 2. This kit uses a proprietary non-fluorescent dye that becomes strongly fluorescent upon reacting with thiols on GSH. The assay was performed in black 96-well plates. Fifty microliters of GSH standard (10, 5, 2.5, 1.25, 0.625, 0.3125, 0.1563, 0.0781 and 0 µM) was added to the appropriate wells, 50 μL assay buffer was used as assay blank control and 50 μL of each diluted sample solution was added into the corresponding well. Then, 50 μL of GSH Assay Mixture was added into the wells of GSH standards, blank control and test samples to make the total assay volume of 100 μL/well. For total glutathione assay, 50 μL of total glutathione Assay Mixture was used, which reduces all GSSG to GSH, and thus in this assay the quantity measured constitutes total glutathione. The reduced GSH and total glutathione assays were done on separate plates, with all measures done in duplicate on the same plate, and then the experiment was repeated again. The reaction was incubated (protected from light) at room temperature for 30 minutes and then the fluorescence was read by a micro-plate reader (BMG LABTECH, FLUOstar OPTIMA) at Ex/Em = 490/520 nm. Each sample on the assay plate was normalized to four identical internal control samples to allow calculation of the average across duplicate plates. The intra-assay variability in the internal controls ranged from 2.77% to 3.76%.
Immunoblotting
For immunoblotting, the amount of protein loaded and relevant antibodies concentrations were optimized in pilot tests. To measure GCLC protein expression, western blotting was performed as previously described (Fillman et al., 2013; Wong et al., 2009). First, 40 mg of frozen tissue from the DLPFC was homogenized in 400 mL of homogenization buffer and then protein solutions were diluted to 1 μg/μL using Laemmli sample buffer (BIO-RAD, Cat#161-0730) with 5% 2-mercaptoethanol. Acrylamide gels (8%) were run at 120 V for 60 minutes for protein separation. After separation, proteins were transferred to a nitrocellulose membrane (BIO-RAD, Cat#162-0115) and then the membrane was blocked for 1 hour with non-fat skim milk (Blotting-Grade Blocker, BIO-RAD, Cat#170-6404). The monoclonal GCLC antibody (1:5000, Abcam, Cat#ab55435, immunogen corresponding to amino acid 528-638 of human GCLC) was added to the membrane for incubation overnight at 4°C. The following day, the membrane was washed by tris-buffered saline and tween (TBST). A horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000, Millipore, Cat#AP192) was added to the membrane for 1 hour at room temperature. After one further round of washing in TBST, sensitive enhanced chemiluminescent (ECL) (Millipore, Cat#WBKLS0500) was applied to the membrane for 1 minute at room temperature in the dark to allow visual detection of the immuno-positive bands using the Chemidoc (BIO-RAD, Universal Hood II). The expected 73-kDa band was quantified by optical density (OD) using Image Lab software (Version 5.0, BIO-RAD). Each gel was normalized to β-actin expression and internal controls, which did not differ between diagnostic conditions. The experiment was performed twice and data averaged.
Western blotting was also performed for GPx-like proteins. However, according to the images of the GPx western blotting, multiple unexpected bands were detected. Based on those bands’ locations, they likely represent protein dimers, trimers and tetramers of GPx (Supplementary Figure 1). Thus, the results of western blotting of GPx-like proteins cannot readily be used in analysis, and instead dot blotting was utilized to test total GPx-like protein levels. The polyclonal anti-GPx (1: 3000, Abcam, Cat#ab21966, immunogen corresponding to C terminal amino acids 175-194 of Human Glutathione Peroxidase, UniProt ID: P07203) primary antibody and the HRP-conjugated secondary antibody (1:2000, Millipore, Cat#AP147) were used in the dot blotting. The dot blotting was performed as previously described (Beltaifa et al., 2005). Generally, the process of dot blotting is similar to the western blotting; only the dot blotting does not include gel electrophoresis. Without separating protein by size, all protein of each sample was blotted and blocked on a single blot and so β-actin was not used to normalise. The average of all controls OD was used as the internal control value for each membrane. The experiment was performed twice and data averaged.
Quantitative PCR for GCLC
Preparation of complementary DNA (1.14 ng/well) and qPCR experiments to measure GCLC mRNA were performed as described previously on the DLPFC cohort (Weickert et al., 2010). The GCLC (Hs00155249_m1) probe was used in the qPCR, with ACTB (Hs99999903_m1), GAPDH (Hs99999905_m1), TBP (Hs00427621_m1) and UBC (Hs00824723_m1) used as normalizing housekeeping genes, as those genes did not change expression with diagnosis (all p > 0.343) (Weickert et al., 2010). Cycling conditions, controls and reaction amounts were the same as previous studies (Weickert et al., 2009, 2010). All reactions were performed in triplicate.
Statistical analysis
Data analysis was performed using SPSS (IBM, SPSS Statistics 22). Outliers for each diagnostic group were identified by Grubbs test using GraphPad software. Only one outlier (in GCLC mRNA measure, control group) was identified and removed from analysis. Normal distributions of data were tested by the Kolmogorov–Smirnov and the Shapiro–Wilk tests. Only the western blot GCLC-like protein data were not normally distributed within diagnostic groups; therefore, the data were log10 transformed to obtain normal distributions. Pearson or Spearman’s correlation tests were performed as appropriate. Student’s independent t-test and analysis of covariance (ANCOVA) were employed to analyse data between groups. Significance was set at p < 0.05.
Results
Total glutathione and reduced GSH assay
By independent t-test, we found that people with schizophrenia have less (~12%) total glutathione (t(66) = 2.467, p = 0.016) and less (~16%) reduced GSH (t(66) = 3.001, p = 0.004) than controls (Figure 2). There was no significant correlation of either total glutathione or reduced GSH with confounding or clinical factors such as age, pH, PMI, duration of illness and antipsychotic dose (all r < 0.2, p > 0.05).
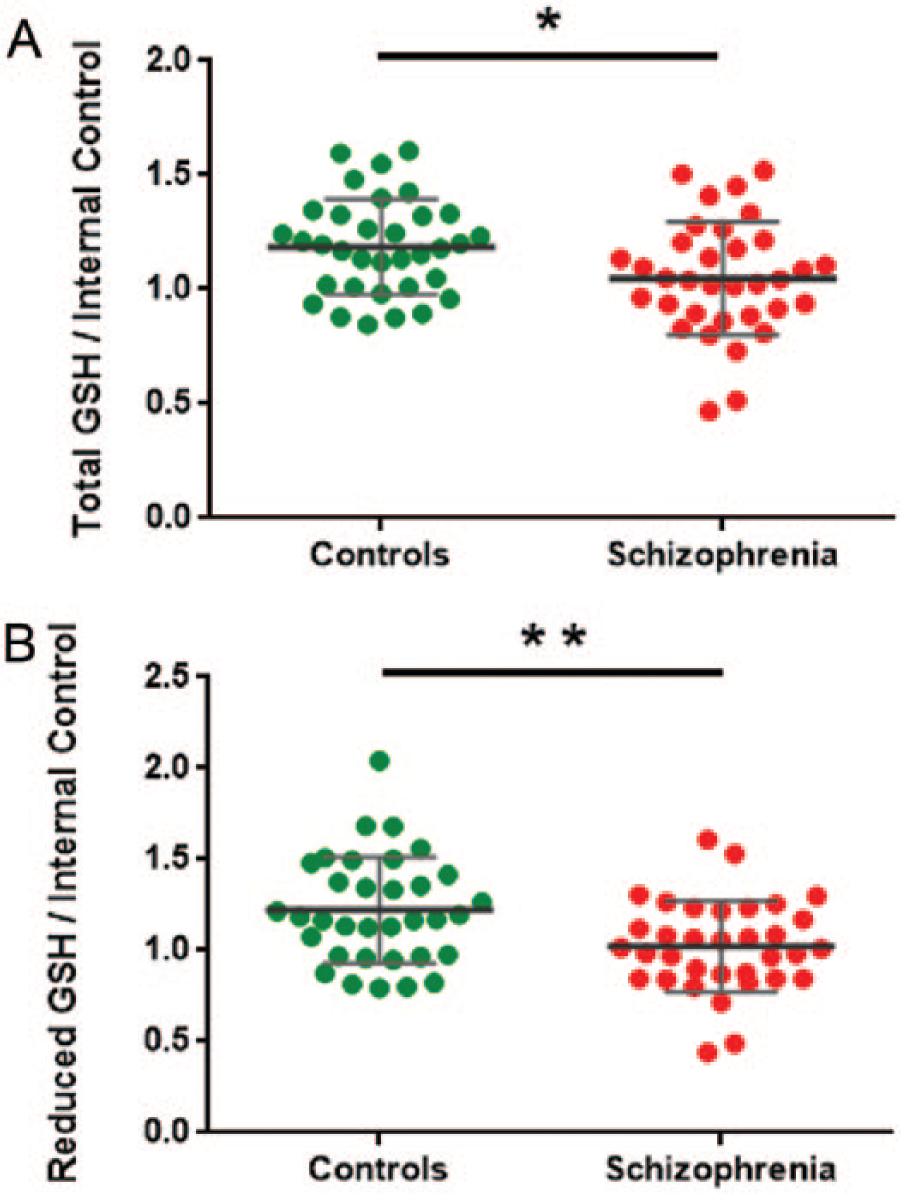
(A) Total glutathione and (B) reduced GSH (GSH) levels measured by GSH assay kit analyses in schizophrenia (red) and control (green) individuals. All glutathione data were normalized to the mean of internal controls; error bars denote standard deviation of the mean. *p < 0.05; **p < 0.01.
GCLC western blot and qPCR
GCLC-like protein levels were significantly correlated with PMI in people with schizophrenia (r = 0.423, p < 0.01). However, there was no significant correlation between GCLC-like proteins and other potentially confounding factors, such as duration of illness, antipsychotic dose, age and pH (all r < 0.15, p > 0.05). Moreover, GCLC mRNA levels were significantly correlated with pH in schizophrenia (r = −0.332, p = 0.045), while there was no significant correlations of GCLC mRNA with other potentially confounding factors (all r < 0.11, p > 0.05).
Based on an independent t-test analysis, we did not detect a significant difference between the level of GCLC-like protein in people with schizophrenia compared to controls (t(72) = −1.007, p = 0.285). There was also no significant diagnostic difference for the GCLC-like protein between schizophrenia and controls when we co-varied for PMI (F(1,71) = 0.530, p = 0.469). Moreover, GCLC mRNA expression did not differ between people with schizophrenia and controls (t(71) = −0.376, p = 0.708), even after co-varying for pH (F(1,70) = 0.020, p = 0.889) (Figure 3).
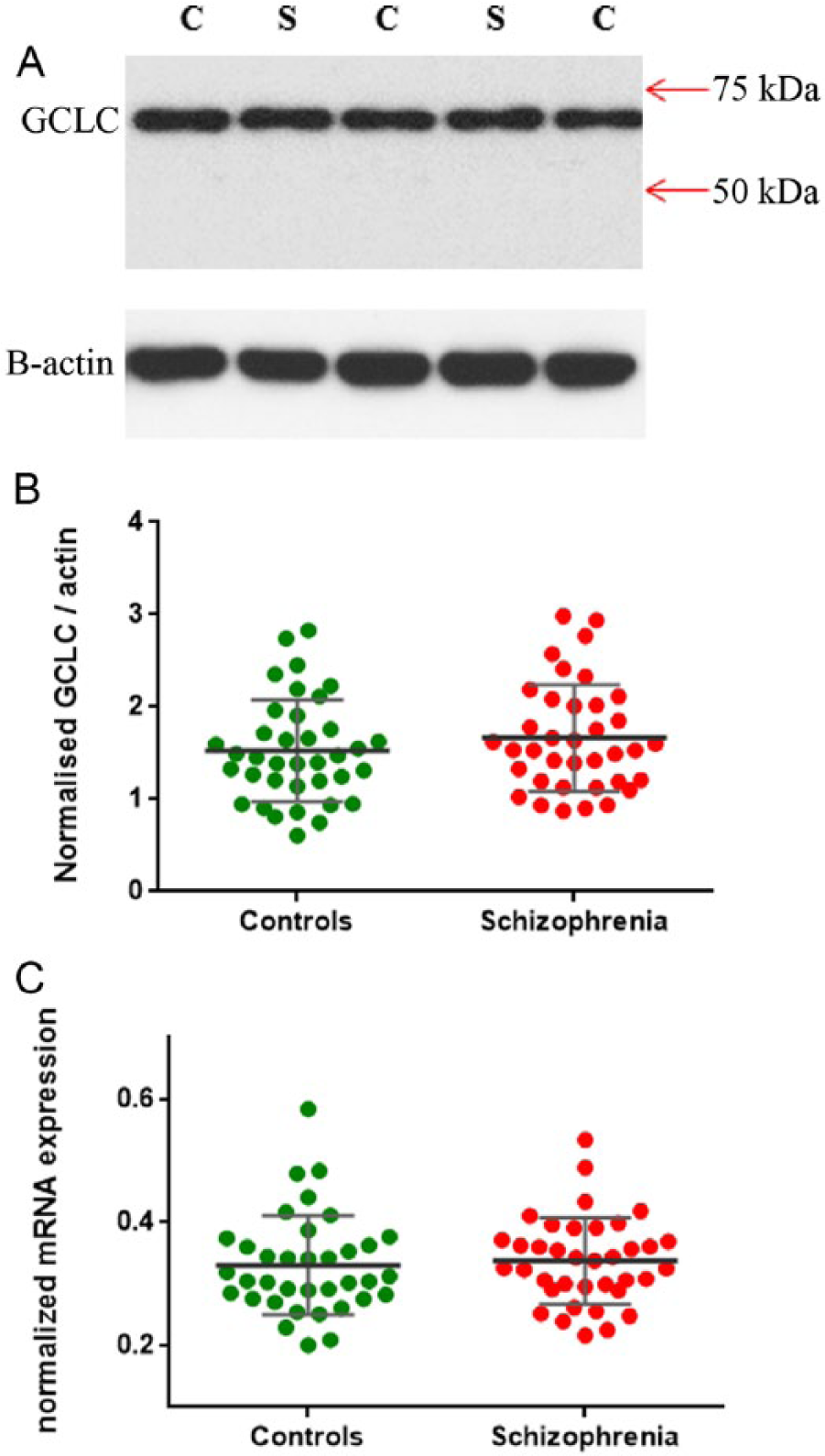
(A) Western blot image of glutamate cysteine ligase catalytic subunit (GCLC). GCLC bands were observed at the expected 73 kDa and the β-actin bands indicate equal loading across samples. C: controls; S: schizophrenia. There were no significant differences in GCLC protein (B) and mRNA expression (C) levels between schizophrenia (red) and controls (green). GCLC protein data were normalized to β-actin and internal controls; GCLC mRNA data were normalized to the mean of housekeeping genes expression; error bars denote standard deviation of the mean.
GPx-like protein dot blot
We did not detect a significant difference of GPx-like protein levels between people with schizophrenia compared to controls (t(72) = −0.060, p = 0.952) (Figure 4). No significant correlations between GPx-like protein levels and potentially confounding factors, such as duration of illness, antipsychotics, age, pH and PMI, were found (all r < 0.16, p > 0.05).
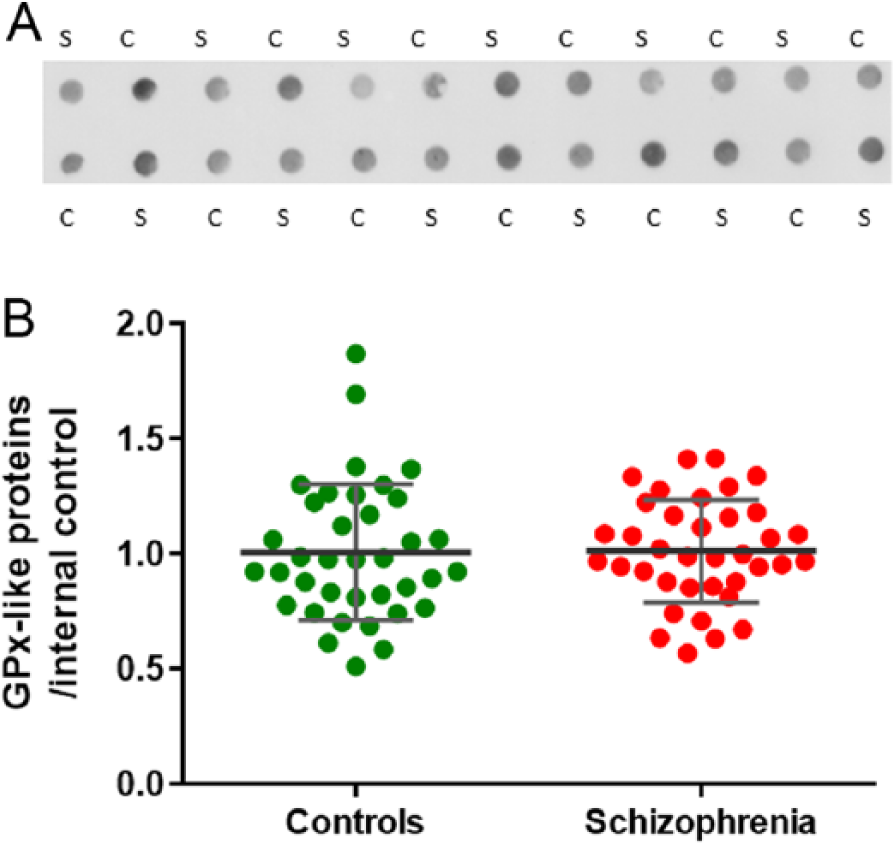
(A) Dot blot image of glutathione peroxidase (GPx)-like protein. C: controls; S: schizophrenia. There was no difference in GPx-like protein (B) between schizophrenia (red) and controls (green). GPx-like protein data were normalized to internal control; error bars denote standard deviation of the mean.
Effect of categorical demographic or potentially confounding illness factors on glutathione pathway measures
There were no significant differences between males and females overall in the measured GCLC-like protein (t(72) = 0.171, p = 0.476), GCLC mRNA (t(71) = 0.821, p = 0.415), GPx-like protein (t(72) = 0.100, p = 0.921), reduced GSH (t(66) = −0.092, p = 0.927) and total glutathione (t(66) = 0.430, p = 0.669). There were no significant differences between left and right hemisphere in GCLC-like protein (t(72) = −0.792, p = 0.431), GCLC mRNA (t(71) = 1.189, p = 0.238), GPx-like protein (t(72) = 0.812, p = 0.420), reduced GSH (t(66) = 0.820, p = 0.415) and total glutathione (t(66) = 0.474, p = 0.637). Similarly, there were no significant interaction effects of sex (all F < 0.958, p > 0.331) or hemisphere (all F < 1.524, p > 0.221) when these molecular measures were considered in a two-way analysis of variance (ANOVA) with diagnosis as the second independent variable.
There were no significant differences between individuals with schizophrenia who died by suicide or by other means in GCLC-like protein (t(35) = 0.013, p = 0.990), GCLC mRNA (t(35) = 0.350, p = 0.729), GPx-like protein (t(35) = −1.291, p = 0.205), reduced GSH (t(33) = 0.988, p = 0.330) and total glutathione (t(33) = 1.015, p = 0.318). There was no significant difference of total glutathione (t(31) = 0.640, p = 0.527) and reduced GSH (t(31) = 0.541, p = 0.592) levels from people who took first- or second-generation antipsychotics as their primary medication. In people with schizophrenia, a history of illicit drug use was not statistically related to total glutathione (t(28) = 0.644, p = 0.525) or to reduced GSH (t(28) = 0.883, p = 0.385) levels. Moreover, a history of tobacco smoking was not statistically related to total glutathione (t(25) = 0.297, p = 0.769) or to reduced GSH (t(25) = 0.962, p = 0.345) levels.
Discussion
In this study, we found that people with schizophrenia have less total glutathione and less reduced GSH. A reduction of total glutathione would be expected to result in reduced capacity of the brain antioxidant system. We also measured GCLC protein and mRNA expression levels in DLPFC tissue because GCLC regulates GCL activity, the rate-limiting enzyme in the GSH synthesis pathway. However, we did not find the expected deficits of GCLC at either the protein or mRNA levels in schizophrenia. Moreover, there was no significant difference between diagnostic groups in GPx-like protein levels, which is an important enzyme in the GSH recycling pathway. All these results suggest that brains of people with schizophrenia have increased oxidative stress, but that the cause of GSH deficits in schizophrenia does not appear to be a direct result of reduced GSH-related protein levels. Furthermore, confounding factors (type of antipsychotics, illicit drugs and smoking) may not be major contributors to oxidative stress in schizophrenia in our cohort.
Our findings demonstrating GSH deficits in the brains of people with schizophrenia are consistent with previous postmortem studies (Gawryluk et al., 2011; Yao et al., 2006). The advantage of our study is that the postmortem cohort included a greater number of cases, resulting in a more powerful analysis. However, our study did not confirm enzyme deficits in the GSH synthesis or GSH recycling pathway. There are many possible reasons to explain this result. On one hand, although previous postmortem studies suggested there was reduced GCLC and GPx levels in people with schizophrenia (Gawryluk et al., 2011; Yao et al., 2006), they included a small number of cases in their studies. So, as the sample size was increased, this may reduce sampling bias and may better reflect the population of people with schizophrenia. Also, there has been increasing concern about the reproducibility of data obtained using antibodies (Bordeaux et al., 2010). It is possible that variation between antibody specificity or even between batches of antibodies contribute to varied results across multiple studies of the same disorder. On the other hand, the reasons for GSH deficits in people with schizophrenia may be heterogeneous leading to different results in different cohorts. Therefore, some brain GSH/GSSG imbalances may not be related to abnormal levels of glutathione synthesis or recycling pathway enzymes but could be related to changes in other factors not measured in our study, such as changes in enzyme activity and/or levels of essential enzyme co-factors. We did not measure enzyme activity or co-factor levels in our study as this would require special preparation of tissue homogenates, but this should be considered in future experiments. Additionally, some inflammatory cytokines may be involved in oxidative stress pathways in schizophrenia. Increased ROS can activate transcription factors that eventually lead to the release of inflammatory cytokines, which can deplete GSH, and cause further oxidative stress (Biswas and Rahman, 2009; Morris et al., 2014). As our laboratory has found elevated inflammatory cytokines in a subset of people with schizophrenia (Fillman et al., 2013, 2014, 2015), it is possible that inflammation contributes to GSH deficits in some people with schizophrenia. Furthermore, as certain foods (such as milk and eggs) contain GSH, diet could affect GSH levels. People who have consumed food with a high level of GSH for long time may have more antioxidants. However, diet cannot be included as a factor in this study because the recorded pre-mortem information does not have a description of diet.
Both animal studies and clinical research have determined that antipsychotics may be relevant to oxidative stress in schizophrenia (Kropp et al., 2005; Martins et al., 2008; Reinke et al., 2004). Kropp et al. (2005) found that antipsychotic drugs could increase lipid peroxidation (using malondialdehyde in human plasma as the marker), suggesting that antipsychotics may induce oxidative stress. Moreover, first- and second-generation antipsychotics might have differential effects on oxidative stress. Using thiobarbituric acid reactive substances and protein carbonylation as a marker, animal studies have shown that the first-generation antipsychotics can induce oxidative stress, while the second-generation antipsychotics may not contribute to an increase in ROS (Martins et al., 2008; Reinke et al., 2004) and could even reduce oxidative stress (Dakhale et al., 2005). However, these studies did not investigate the effect of antipsychotics on GSH levels. In our study, we did not find a statistically significant effect of different classes of antipsychotics on GSH levels in people with schizophrenia. It is possible that antipsychotics (especially the first-generation antipsychotics) intake may induce oxidative stress, but that this effect does not occur by disturbing the GSH system.
There are many other factors that can contribute to oxidative stress, such as ageing, brain pH and history of illicit drug use and smoking. Similar to the analysis of effect of antipsychotics in this study, we did not find significant associations between GSH deficits and these factors. Flatow et al. (2013) used meta-analysis to summarize the many oxidative stress markers and found that the levels of markers in different schizophrenia-relevant studies are quite variable, perhaps suggesting different stages of illness may involve oxidative stress via different molecules or pathways.
To summarize, our results demonstrating cortical GSH deficits suggest that people with schizophrenia have impaired antioxidant capacity in brain in the chronic illness stage even up to the time of death. Moreover, although our study did not show any significant effect of potential confounders on GSH level, it cannot be ruled out that those factors still contribute to oxidative stress. Having found GSH deficits in people with schizophrenia, we next need to determine how GSH deficits (oxidative stress) contribute to schizophrenia. Hardingham and Do (2016) hypothesized that there is a linkage between N-methyl-
Footnotes
Acknowledgements
We thank Danny Boerrigter for technical assistance. We express our gratitude to the individuals who donated postmortem tissues used in this study and their families. Tissues were received from the New South Wales Brain Tissue Resource Centre at the University of Sydney which is supported by the Schizophrenia Research Institute and National Institute of Alcohol Abuse and Alcoholism (NIH [NIAAA] R28AA012725). Y.Z. and V.S.C. are joint first authors.
Declaration of Conflicting Interests
Y.Z. and V.S.C. report no competing interests. C.S.W. is on an advisory board for Lundbeck, Australia.
Funding
This work was supported by Schizophrenia Research Institute (utilizing infrastructure funding from the NSW Ministry of Health and the Macquarie Group Foundation), the University of New South Wales and Neuroscience Research Australia. C.S.W. is a recipient of a National Health and Medical Research Council (Australia) Principle Senior Research Fellowship (APP1117079).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.