
Abstract
Objective:
Whilst dopaminergic dysfunction remains a necessary component involved in the pathogenesis of schizophrenia, our current pharmacological armoury of dopamine antagonists does little to control the negative symptoms of schizophrenia. This suggests other pathological processes must be implicated. This paper aims to elaborate on such theories.
Methods:
Data for this review were sourced from the electronic database PUBMED, and was not limited by language or date of publication.
Results:
It has been suggested that multiple ‘hits’ may be required to unveil the clinical syndrome in susceptible individuals. Such hits potentially first occur in utero, leading to neuronal disruption, epigenetic changes and the establishment of an abnormal inflammatory response. The development of schizophrenia may therefore potentially be viewed as a neuroprogressive response to these early stressors, driven on by changes in tryptophan catabolite (TRYCAT) metabolism, reactive oxygen species handling and N-methyl
Conclusions:
Outside of the dopaminergic model, the potential pathogenesis of schizophrenia has yet to be fully elucidated, but common themes include neuropil shrinkage, the development of abnormal neuronal circuitry, and a chronic inflammatory state which further disrupts neuronal function. Whilst some early non-dopaminergic treatments show promise, none have yet to be fully studied in appropriately structured randomized controlled trials and they remain little more than potential attractive targets.
Keywords
Introduction
Leading theories of schizophrenia pathogenesis postulate that multiple ‘hits’ may be required in susceptible individuals in order to unveil the clinical syndrome. The first such hits potentially occur in utero, and may involve disruption of developing neuronal architecture and epigenetic changes as well as the establishment of an abnormal inflammatory response. Such downstream effects may then result in increased expression of inflammatory markers, tryptophan catabolites (TRYCATs) and other damaging reactive oxygen species (ROS), which in combination with other secondary hits such as methylation abnormalities and N-methyl
Results and discussion
Neuroprogression in schizophrenia: structural brain changes and clinical correlates
The development of schizophrenia is proposed to involve the alignment of multiple potential sources of pathology as a series of hits within susceptible individuals which culminates in the clinical syndrome (Figure 1).
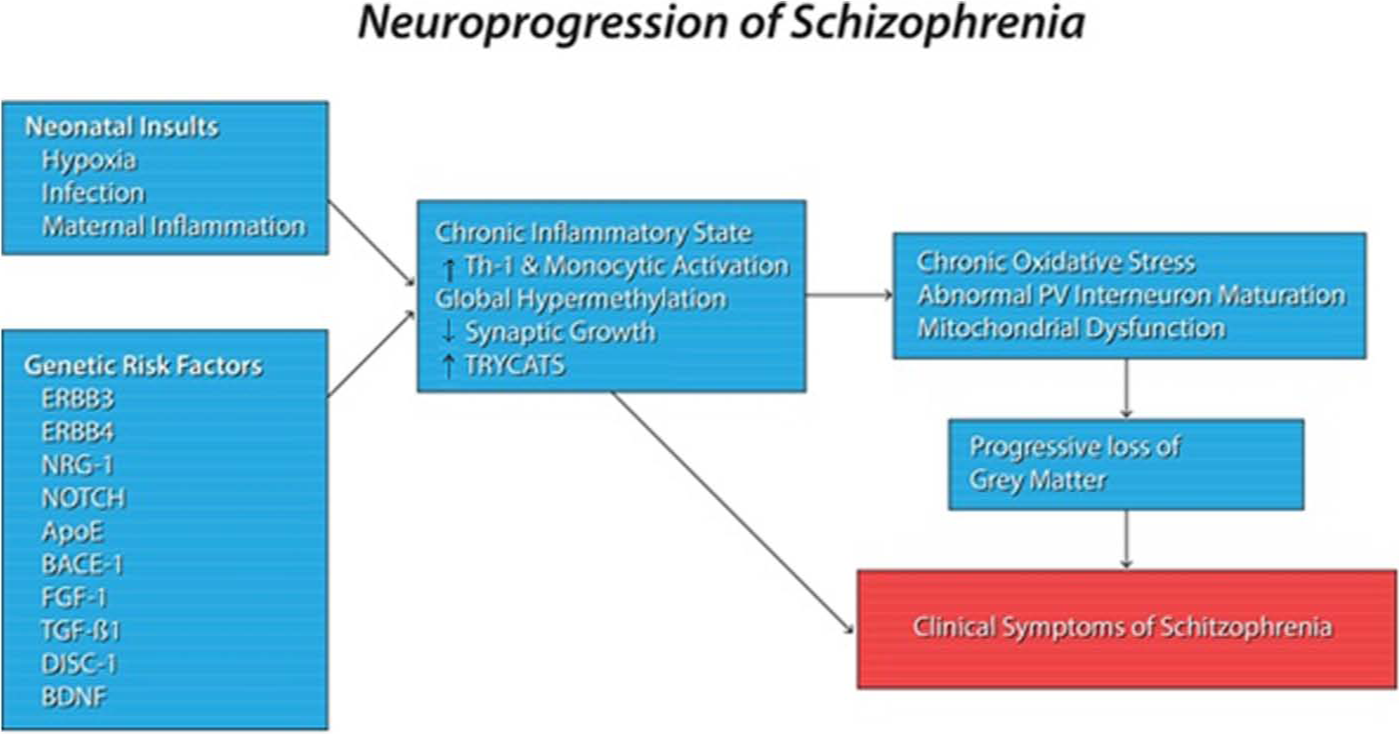
Factors that may contribute to the pathogenesis of schizophrenia. The development of schizophrenia is proposed to involve a series of hits on genetically susceptible individuals. Such hits potentially lead to the abnormal maturation of neuronal circuits, allowing expression of the clinical syndrome. Genetic risk factors for schizophrenia, potentially coupled with a first hit during embryogenesis, may set about a process of abnormal synaptic maturation in susceptible individuals. The combination of a chronic inflammatory state involving an increased Th-1 and monocytic response, secondarily to altered gene expression secondary to a neonatal insult, a state of global hypermethylation, increased levels of TRYCATs, and polymorphisms of genes suggested to be responsible for neuronal dendrite growth and function may lead to shrinkage of the neuropil with subsequent abnormalities of neural connectivity. Such aberrant maturation may be combined with neuronal dysfunction from chronic oxidative stress, mitochondrial abnormalities, and NMDA receptor hypofunction to induce a progressive loss of gray matter, which has been suggested to culminate in the clinical symptoms of schizophrenia.
One of the most striking examples of the potential neuroprogression seen in schizophrenia relates to changes in cortical white and gray matter seen within the disease. Schizophrenia may be associated with progressive enlargement of brain ventricles (van Haren et al., 2012) mediated in part through abnormalities in both the cortical gray matter and underlying reductions in white matter surface area (Colibazzi et al., 2013). Structural brain abnormalities typically present early in the disease course, and appear to be apparent even within the prodrome (Ellison-Wright et al., 2008; Glausier and Lewis, 2013; Iwashiro et al., 2012; Jung et al., 2012; Lim et al., 1996). Although by no means conclusive, structural deficits noted within studies include: increased lateral ventricular volumes (Lam et al., 2012; Vita and de Peri, 2007); decreased frontal lobes and thalamic volumes (Andreasen et al., 2011); gray matter loss within the anterior cingulate, superior temporal gyrus and hippocampus (Eack et al., 2010); and decreased overall cerebral size (Cobia et al., 2012). The most dramatic changes in both white and gray matter have been suggested to occur within the frontal lobes (Ho et al., 2003) followed by temporal, frontal and parietal white matter loss (Olabi et al., 2011). Voxel-based morphometry (VBM) has demonstrated that decreases in gray matter density in the left amygdala are more pronounced in older than in younger patients (Hulshoff Pol et al., 2001). Some studies, although not all, have suggested that decrements of gray matter within the left hippocampus, left superior frontal gyrus, left superior temporal gyrus, bilaterally in the supramarginal gyri, thalami, superior temporal lobes, occipitotemporal lobes, precunei, and posterior cingulate and insular gyri may play a role in pathogenesis (Lewis, 2012; van Haren et al., 2007). Part of the inconsistency comes from research using voxel-based approaches that are centered on the assumption that white matter abnormalities amongst individual patients occur in the same general spatial locations, which may not be true for all individuals suffering from schizophrenia (White et al., 2013).
Such changes within gray and white matter may correlate with symptoms and progression (Goff, 2013), although some authors have questioned the observed imaging findings, suggesting that sampling bias or a as yet unknown effect of chronic antipsychotic treatment may be contributing to the progressive decline seen (Zipursky et al., 2013). Patients with smaller anterior cingulate volumes may display greater difficulties with executive functioning and cognition, whereas insular alterations have been suggested to cause more somatic hallucinations (Shergill et al., 2001). In contrast, frontal and temporal lobe changes produce deficits in language, attention and working memory (Perlstein et al., 2001). Decreases in white matter within the anterior cingulate (Yao et al., 2013), the right anterior internal capsule, right anterior commissure, and bilaterally within the genu and truncus of the corpus callosum suggest aberrancy of inter-hemispheric connections (Hulshoff Pol et al., 2004), whereas cortico-thalamic decrements may disrupt both glutaminergic and dopaminergic neuronal circuitry (Carlsson et al., 2001). Post-mortem analysis of the brains of individuals suffering from schizophrenia has suggested the observed decrements in white and gray matter are not associated with prominent gliosis and neuronal loss (Selemon et al., 1995). Instead, the loss of gray matter in schizophrenia is hypothesized to be secondary to neuropil shrinkage, with loss of dendritic spine density, decreased arborization and loss of synaptic connections with neighboring neurons the central driving factors (Figure 2). This pruning leads to disruption of neuronal circuitry and may be driven by aberrant neuroplasticity genes such as those associated with myelination and oligodendria, including receptor tyrosine kinase erb-B3 (ERBB3), receptor tyrosine kinase erb-B4 (ERBB4) and neuregulin-1 (NRG-1) (Agartz et al., 2006), disrupted in schizophrenia-1 (DISC-1) (Bray et al., 2010; Dresner et al., 2011), and brain-derived neurotrophic factor (BDNF) proposed as risk factors for the development of schizophrenia (Agartz et al., 2006; Neddens et al., 2011; Szeszko et al., 2005).
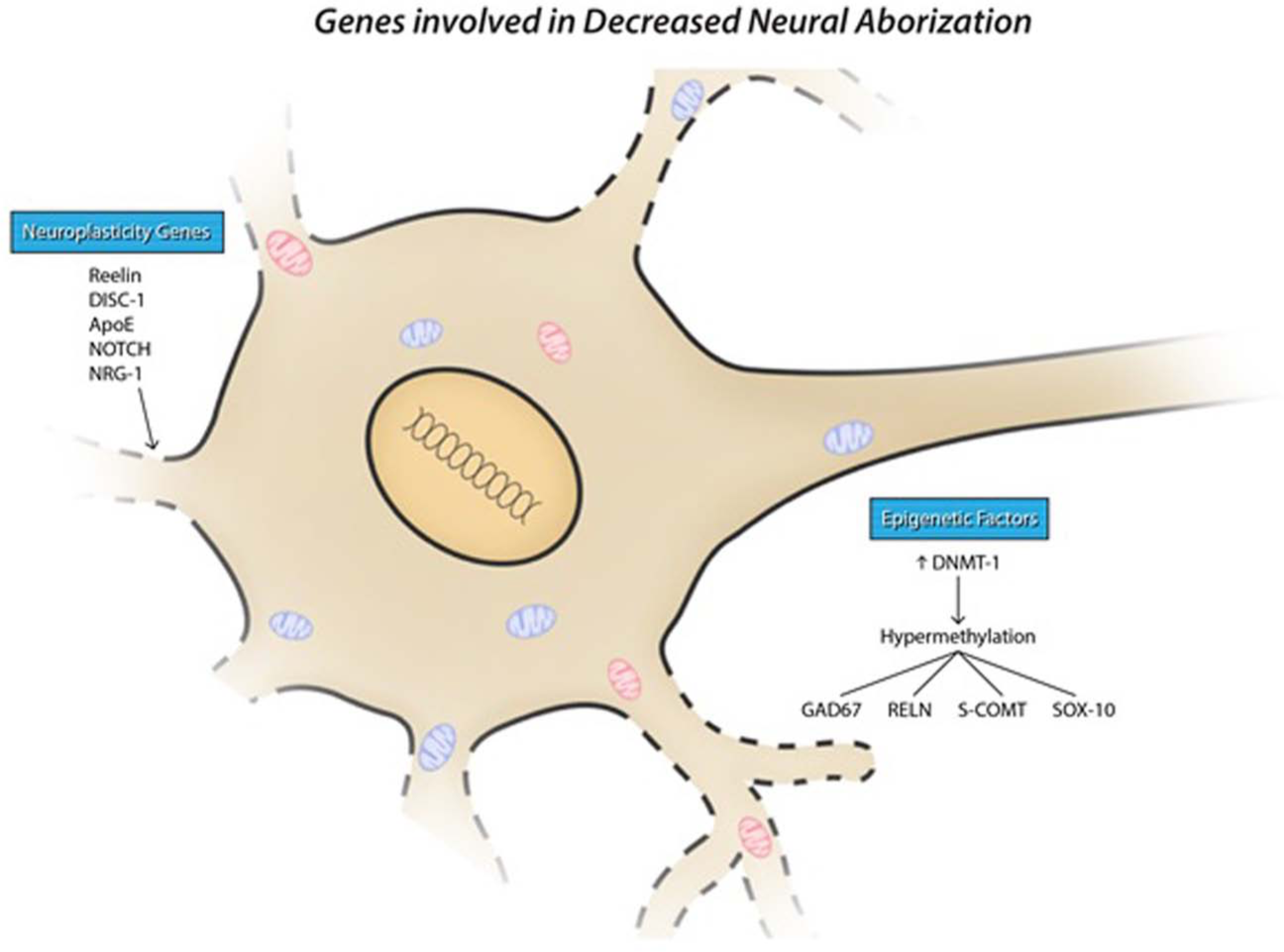
Proposed mechanisms of neuropil shrinkage in schizophrenia. One of the proposed mechanisms of neuroprogression in schizophrenia is through a loss of synaptic connections and shrinkage of the neuropil. Genes such as RELN, DISC-1, ApoE, NOTCH and NRG-1 have been shown to be involved in the growth and development of connections between neurons during embryogenesis and development, and mutations of these are observable risk factors for the development of schizophrenia. The chronic inflammatory state seen in schizophrenia has similarly been suggested to contribute to the progressive changes seen within the disease. Hypermethylation of genes such as RELN may also in part be responsible for neuroprogression through down-regulation of their protein products.
Typical antipsychotics such as haloperidol have been associated with reversal or slowed progression of putamen volume loss, whereas atypical antipsychotics have been suggested to exert more of an effect on the thalami (Dazzan et al., 2005), caudate nucleus and anterior cingulate (Carlsson et al., 2001; Chakos et al., 1994). This neuronal pruning and associated volume loss has been proposed as one of the core pathological mechanisms which underlies the neuroprogressive changes seen in schizophrenia, and indeed appears to begin soon after an early embryological insult.
Biochemical basis of neuroprogression in schizophrenia
Embryonic insults and schizophrenia development
The neuroprogressive nature of schizophrenia is proposed to begin as early as in utero in susceptible individuals. Indeed, many of the proposed early risk factors for schizophrenia relate to embryonic insults. Seasonality of birth, paternal age, obstetric complications leading to fetal hypoxia (Cotter and Pariante, 2002; Winter et al., 2008) and vitamin D deficiency (Anderson et al., 2013) have all been suggested to contribute to the early ‘hits’ for the pathogenesis of schizophrenia. It has also been recently proposed in a study of the Norwegian populace that the degree of fetal growth restriction in utero is linearly related to increased risk (Eide et al., 2013). In addition, conditions which stimulate a maternal and therefore fetal inflammatory response are implicated, infectious or otherwise. The inflammatory response drives embryonic changes in gene expression which are most pronounced during the second trimester (Dammann and Leviton, 1997; Leonard et al., 2012; Miller et al., 2013a). The developing central nervous system (CNS) is exquisitely sensitive to the effects of pro-inflammatory cytokines (Tohmi et al., 2007). Altered cytokine production has been demonstrated in multiple maternal-fetal compartments, including the placenta, amniotic fluid, fetal liver and brain (Boksa, 2010). Maternal exposure to increased levels of tumor necrosis factor alpha (TNF-α) and interleukin-8 (IL-8) may be associated with an increased risk of schizophrenia in the offspring (Boksa, 2010; Brown et al., 2004). Exposure to IL-8 within the second/third trimesters or pregnancy has been related to increases in cerebrospinal fluid (CSF) volume (Ellman et al., 2010).
Increases in pro-inflammatory cytokines including IL-1β, IL-6, TNF-α, interferon alpha (IFN-α) and IFN-β have been suggested to be associated with structural brain deficits in animal models (Soumiya et al., 2011). Such models show disruption of cortical laminar formation, particularly layer V which is associated with dysregulation of gamma-aminobutyric acid (GABA) interneurons (Charych et al., 2009). Animal models utilizing poly(I:C) produce maturation-dependent hyperactive function of the subcortical dopaminergic system with subsequent cognitive deficits (Ozawa et al., 2006). It is postulated that risk factors such as birth asphyxia or gestational diabetes mellitus are also mediated by alterations in inflammatory control mechanisms (Cannon et al., 2002). Hypoxia is thought to induce susceptibility genes both directly through the hypoxic stressor itself and indirectly through the induction of an inflammatory response and is believed to underlie part of the observed association with obstetric complications (Nicodemus et al., 2008). The hypoferremic state which occurs secondary to inflammation may also impact detrimentally on the developing fetal brain (Beard et al., 2006; Kwik-Uribe et al., 2000). Such embryological insults and the chronic inflammation they subsequently induce have been suggested to begin the neuroprogressive process which ultimately culminates in the clinical symptoms of schizophrenia.
Inflammation, Th-1 and Th-2-like responses
After a series of early hits in utero, which may serve to induce a series of inflammatory changes as one of the first hits required, schizophrenia may potentially be characterized by a chronic inflammatory state with increased levels of pro-inflammatory cytokines and altered levels of Th-1-like and Th-2-like cytokines (Anderson and Maes, 2013; Miller et al., 2011). Hypothesized signs of inflammation and monocytic activation include increased levels of pro-inflammatory cytokines (e.g. TNF-α, IL-1 and IL-6), acute phase proteins (APPs) and complement factors (Maes et al., 1994, 1995b, 1997b; Mayilyan et al., 2006; Miller et al., 2011; Naudin et al., 1996; Shintani et al., 1991; Wong et al., 1996). Patients with schizophrenia can show reduced levels of endogenous anti-cytokines or anti-proinflammatory cytokines, including clara cell secretory protein 16 (CC16)(Maes et al., 1996).
It has previously been theorized that schizophrenia is accompanied by a Th-2-like response (Schwarz et al., 2001). Two recent meta-analyses (Miller et al., 2011; Potvin et al., 2008) were unable to confirm this hypothesis. The latter provided evidence that schizophrenia is accompanied by a chronic mild inflammatory state and a Th-1-like response, evidenced by increased levels of IFN-γ, IL-12 and sIL-2Rs (soluble interleukin-2 receptors). The same meta-analysis found that IL-1 and IL-6 are state markers whereas increased IL-12, IFN-γ and sIL-2R are trait markers of the disorder. High levels of pro-inflammatory and Th-1-like cytokines (e.g. IL-1β, IL-6 and IL-2) are also present in the cerebrospinal fluid (CSF) of patients with schizophrenia (Garver et al., 2003; McAllister et al., 1995). Drexhage et al. (2011) demonstrated signs of a mixed T-cell response, suggesting simultaneous activation of both Th-1 and Th-2 responses (Anderson and Maes, 2013). Increased CSF levels of IL-1 have been found to be a predictor of acute psychotic relapse in patients recently withdrawn from haloperidol (McAllister et al., 1995), suggesting that immune-inflammatory dysfunction plays a role in the neurobiology of relapse (Emsley et al., 2013). The association between cytokine abnormalities and acute exacerbations of schizophrenia seems to be independent of antipsychotic medications (Miller et al., 2011). Some cytokines (e.g. IL-1β, IL-6 and TGF-β) as well as abnormal blood lymphocyte parameters (CD4/CD8) (Miller et al., 2013b) have been suggested to represent state markers for acute exacerbations of psychosis.
Increased IL-1, through activation of the hypothalamic-pituitary-adrenal (HPA) axis, may contribute to the exaggerated stress responses observed in schizophrenia (Sapolsky, 2003). Polymorphisms of the IL-1 gene complex may be linked to both schizophrenia development and ventricular enlargement, which has also been suggested of IL-8 (Ellman et al., 2010). IL-10 promoter variants, producing higher levels of IL-10, have been proposed to be more common in schizophrenia (Bocchio Chiavetto et al., 2002). Other authors have suggested that other up-regulated genes in schizophrenia not normally seen in other chronic inflammatory states include gamma-inducible human guanylate binding protein-1 (GBP-1), serpin peptidase inhibitor, clade A member 3 (SERPINA3) and interferon induced transmembrane proteins-2 and 3 (IFITM2, IFITM3) (Saetre et al., 2007).
Antipsychotics appear to have no uniform effects on the inflammatory and Th-1 and Th-2-type responses in patients suffering from schizophrenia. Antipsychotic agents may suppress inflammatory processes in schizophrenia, such as the acute phase response (Maes et al., 1997a). IL-6 and sIL-6R levels may also normalize with effective antipsychotic therapy (Maes et al., 1997a). Antipsychotic agents may increase the initially lowered levels of CC16 and enhance the production of the sIL-1R antagonist, both resulting in anti-inflammatory effects (Maes et al., 1996; Song et al., 2000). There are divergent effects of antipsychotics on Th-1 characteristics. Some authors have shown that lower levels of markers indicative of Th-1 immune activation including ICAM-1 and IFN-γ normalize with treatment (Schwarz et al., 2000). The levels of soluble CD8 (sCD8) and the leukemia inhibitory factor receptor (LIF-R), however, are increased during treatment with antipsychotic agents, indicating increased T-cell activation and immunosuppressive effects, respectively (Maes et al., 2002). Clozapine has profound immune effects, including increases in IL-6, CC6, soluble interleukin-6 receptor antagonist (sIL-RA), sIL-2R, sIL-1RA and LIF-R, indicating that clozapine has both anti- and pro-inflammatory effects (Maes et al., 1994, 1995a).
Immune cells themselves may also serve as surrogate state or trait markers, with the overall CD4/8 ratio being significantly increased in antipsychotic naïve patients and normalizing with antipsychotic treatment, whereas CD56 or CD3 appear to have more use as markers of the trait (Miller et al., 2013b).
Neurodevelopmental animal models of schizophrenia, such as post-natal social isolation rearing (SIR) (Moller et al., 2013) and prenatal poly(I:C) challenge (Meyer and Feldon, 2012), have also demonstrated the presence of a pro-inflammatory state, including disordered TRYCAT metabolism (Moller et al., 2012) and an altered pro- versus anti-inflammatory cytokine ratio (Moller et al., 2013).
Chronic systemic inflammation in schizophrenia may explain why patients are more prone to develop metabolic abnormalities such as insulin resistance. Although contributory effects of antipsychotic agents cannot be ignored, metabolic disturbances also appear in antipsychotic naïve patients (Na et al., 2012; Thakore, 2004). The metabolic syndrome itself is a state of chronic inflammation, with increased levels of C-reactive protein (CRP), IL-1, IL-6, TNF-α and leptin (Carrizo et al., 2008). The chronic inflammatory state seen within individuals suffering from schizophrenia has been proposed to be partly responsible for the higher levels of oxidative stress seen within such individuals, which in turn may provide one of the driving forces behind the neuroprogressive nature of schizophrenia.
Oxidative stress
Schizophrenia appears to be associated with a state of chronic oxidative and nitrosative stress (O&NS), which may originally be induced during embryogenesis, continually driven by associated abnormalities including a state of chronic inflammation and mitochondrial abnormalities, and may potentially be a major factor behind the observed neuropil shrinkage. The brain is particularly vulnerable to O&NS-mediated damage due to high concentrations of lipids in membranes of CNS structures, high concentrations of susceptible proteins, and poor endogenous anti-oxidant defense systems (Cui et al., 2004). O&NS may contribute to the neuroprogression of schizophrenia (Moller et al., 2011; Schiavone et al., 2012). Levels of glutathione, a cornerstone of the human anti-oxidant defense system, have been suggested to be reduced by up to 40% within the caudate nucleus (Bitanihirwe and Woo, 2011), CSF, medial prefrontal cortex and striatum in studies of both in vivo imaging and post-mortem analysis in patients with schizophrenia (Do et al., 2000). Variance in the glutamate-cysteine ligase regulatory subunit (GCLM) gene, leading to impaired production of glutathione, is a proposed risk factor for schizophrenia (Tosic et al., 2006). Markers of lipid peroxidation (e.g. malonyldialdehyde (Morera et al., 2009) and isoprostanes (Dietrich-Muszalska and Olas, 2009)), DNA damage and protein carbonylation (Catts et al., 2012; Pedrini et al., 2012) produced in states of increased oxidative stress have been shown to be elevated in the plasma of individuals suffering from schizophrenia. Patients with lower levels of cell membrane polyunsaturated fatty acids (PUFAs) may show more negative symptoms and a higher incidence of the metabolic syndrome, and have been suggested to have a poorer reaction to antipsychotic therapy (Bentsen et al., 2012). In particular, lower levels of nervonic acid may predispose to conversion from the at-risk state to overt illness (Amminger et al., 2012). Expression of superoxide dismutase (SOD), involved in the control of superoxide radicals, is also elevated in schizophrenia (Melamed et al., 1998; Singh et al., 2008; Zhang et al., 2007).
Damage to arachidonic acid (AA) through lipid peroxidation has been implicated in impairment of action potential generation in hippocampal pyramidal cells, decreased synthesis of prostaglandins and impaired GABA receptor functioning, and may lead to decreased GABA, but increased dopamine uptake within synapses (Pellmar, 1986; Rafalowska et al., 1989; Schwartz et al., 1988). Some proteomic research investigating alterations in protein regulation in the prefrontal cortex demonstrate that nearly half of the altered protein expression is associated with genes pertaining to oxidative stress or mitochondrial function (Prabakaran et al., 2004). Similar proteomic analysis of erythrocytes has demonstrated significant up-regulation of selenium binding protein-1 (SBP-1) and glutathione-S-transferase (GSTA3), and down-regulation of thioredoxin peroxidase (PRDX5), all proposed to be involved in the regulation of ROS (Prabakaran et al., 2007).
Dopamine, through its metabolites dopamine-semiquinone or dopamine-quinone, has been shown to be a significant source of ROS within the CNS (Grima et al., 2003). Oxidative stress causes post-translational modification of the dopamine transporter and this inhibits the uptake of dopamine (Kim and Andreazza, 2012). NMDA receptor activation results in the release of glutathione (Carter, 2006) and increased downstream production of nitric oxide (NO) (Levkovitz et al., 2007) is thought to be a means of counteracting the toxic effects of ROS and oxidative dopamine metabolites. Indeed, NO and its metabolites have been implicated in the neuroprogression of schizophrenia (Bernstein et al., 2011), while typical and atypical antipsychotics have diverse effects on NO synthase activity (Nel and Harvey, 2003). It has been suggested that dopamine receptor supersensitivity, glutamate dysfunction and the release of O&NS may underlie post-relapse emergence of treatment non-responsiveness (Emsley et al., 2013).
Mitochondria, whose morphology and function have been suggested by multiple authors to be abnormal in schizophrenia, are a potent source of free radicals (de Oliveira et al., 2011). Studies using a SIR neurodevelopmental animal model have demonstrated that altered behavior, immune dysfunction, and regional brain changes in dopamine may be associated with abnormalities in cortico-striatal mitochondrial function (Moller et al., 2013). A mitochondrial DNA sequence variation suggested to be associated with schizophrenia affects nicotinamide adenine dinucleotide phosphate (NADPH)-ubiquinone reductase (complex I) leading to increased production of superoxide (Marchbanks et al., 2003). O&NS have been shown to normalize with the use of atypical antipsychotics, with down-regulation of pro-inflammatory and up-regulation of anti-inflammatory gene transcripts (Chittiprol et al., 2010). O&NS appear to be a central component of much of the pathogenesis of schizophrenia. Increased oxidative stress appears to begin in utero and is henceforth propagated by the subsequent chronic inflammatory state as well as other potential mechanisms of schizophrenia pathogenesis such as mitochondrial dysfunction. In addition, proposed normal pathways which serve to protect the CNS from oxidative stress such as glutathione or NMDA receptor activity appear to be dysregulated in schizophrenia, potentially worsening oxidative stress.
NMDA receptor hypofunction
As aforementioned, NMDA receptor activation with the subsequent release of glutathione and downstream NO action is thought to be one of the protective mechanisms against ROS damage within the CNS. Individuals suffering from schizophrenia may have hypofunction of NMDA receptors which may exaggerate the neuro-toxic effects of ROS. NMDA receptor antagonism induces the rapid release of glutamate with hypothesized subsequent excito-toxic effects on neurons secondary to the excessive influx of calcium (Kegeles et al., 2012; Krebs et al., 2006). Genetic variations in the calcium channel gene CACNA1C are demonstrated in schizophrenia, which impacts on glutamate transmission (Bray et al., 2010; Moosmang et al., 2005; Schizophrenia Psychiatric Genome-Wide Association Study Collaborators, 2011). Genetic ablation of NMDA receptors within mouse models induces schizophrenia-like behavioral disturbances (Belforte et al., 2010). Hypofunction of NMDA receptors has been suggested to lead to the inhibition of inhibitory GABA interneurons which serve to control the rate of firing of cortical pyramidal cells (Carlsson, 2006) leading to a state of concurrent GABA deficit and glutamate excess (Figure 3) (Lisman et al., 2008). Interneurons which express parvalbumin (PV) are amongst the most studied in relation to NMDA receptor dysfunction. PV interneurons have shown decreased expression mRNA for the NR2A subunit of the NMDA receptor and decreased expression of glutamate decarboxylase (GAD)-67 (Lewis et al., 2011; Woo et al., 2008, 2010) within the frontal cortex of individuals suffering from schizophrenia (Lewis et al., 2004), although it is currently unclear whether such alterations in GAD-67 result in the demonstrated reductions seen in PV interneurons (Curley et al., 2013). Similarly, some authors have linked polymorphisms of the GAD-1 gene with schizophrenia (Gavin and Akbarian, 2012). SIR has been shown to be associated with altered NMDA receptor density (Toua et al., 2010) and oxidative stress, observed as an increased expression of NADPH oxidase 2 (NOX2) (Schiavone et al., 2009). NOX2 is an innate immune enzyme responsible for superoxide production that has been suggested to control glutamate release in the prefrontal cortex (Schiavone et al., 2012; Sorce et al., 2010) and may compromise cortical PV interneurons (Powell et al., 2012; Schiavone et al., 2009).
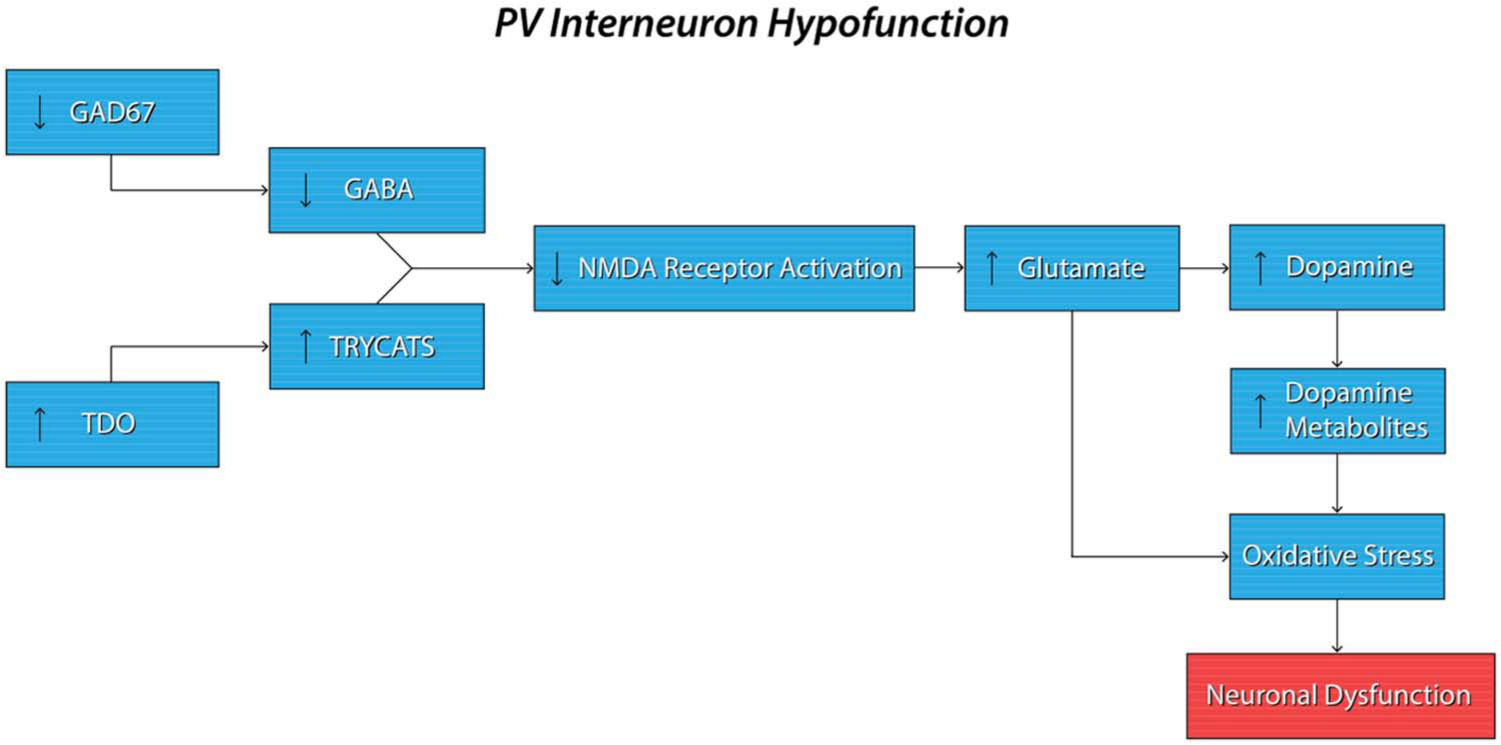
Proposed mechanisms of action of NMDA receptor dysfunction in schizophrenia. NMDA receptor dysfunction is thought to be a critical component of both the hyperglutaminergic and hyperdopaminergic state seen in schizophrenia. A decrease in endogenous GABA production through dysfunction of the GAD67 gene locus (possibly secondarily to hypermethylation of its promoter probands), as well as an increase in endogenous NMDA receptor antagonists TRYCATs, has been suggested to lead to a reduction in their function. NMDA dysfunction may subsequently cause an increase in the release of the excito-toxic neurotransmitter glutamate. An increase in the release of glutamate may be partly responsible for the hyperdopaminergic state seen in individuals suffering from schizophrenia. Both glutamate and dopamine, mainly through its reactive metabolites, have been shown to increase oxidative stress within neuronal cells and lead to their subsequent dysfunction.
Several proposed risk-factor genes including DISC-1, NRG-1, ERBB4 and Reelin have been shown to be selectively altered within PV interneurons (Hikida et al., 2007; Matrisciano et al., 2013). Dysregulation of these genes may result in abnormalities of synaptic structure and function (Javitt et al., 2012). Reelin is abundantly expressed within GABA interneurons in neuronal layers I and II of the cortex, within the putamen, hippocampus, and caudate and has been suggested to interact with numerous proteins including BDNF, the integrin receptor, the apolipoprotein E2 (ApoE2) receptor and the very-low density lipoprotein (VLDL) receptor to regulate dendrite spine outgrowth and structure (Dong et al., 2005; Guidotti et al., 2011). DISC-1 has been shown to be involved in the regulation of neuronal outgrowth, migration and proliferation (Young-Pearse et al., 2010). The NOTCH family of proteins may regulate dendritic structure and map to regions associated with schizophrenia in linkage analysis (Passos Gregorio et al., 2006). NRG-1 may regulate NMDA expression within glutaminergic synaptic vesicles, and acting through ERBB4 inhibits NMDA receptor currents after receptor activation (Talbot et al., 2004). Both PV interneurons and the circuits they regulate mature extensively during adolescence, corresponding to the temporal appearance of schizophrenia-like symptoms (Sullivan and O’Donnell, 2012). PV interneurons are the last subset of interneurons to mature and receive the highest amount of glutaminergic synapses from thalamic afferent neurons (Gulyas et al., 1999).
Glutamate dysfunction originates from various sources. Mitochondrial dysfunction, altered pro-inflammatory cytokines and altered conversion of tryptophan to quinolinic acid (an NMDA agonist) versus kynurenic acid (KA) (a negative NMDA receptor modulator) have been proposed to underlie the altered glutamate activity and disturbances in O&NA (Moller et al., 2013). The hyperglutaminergic state may lead to dysregulation of PV interneurons with subsequent atrophy of the hippocampus and subiculum (Schobel et al., 2013). KA, one of the TRYCATs, has been suggested to be increased in individuals suffering from schizophrenia (Parsons et al., 1997). Increased levels of 3-hydroxykynurenine, another TRYCAT, may be seen in antipsychotic-naïve patients (Myint et al., 2011), particularly within the prefrontal cortex (Schwarcz et al., 2001). Tryptophan-2,3-dioxygenase (TDO), one of the two enzymes capable of converting tryptophan to TRYCATs, has been shown to be up-regulated within schizophrenia (Lee and Meltzer, 2001). This may be secondarily to increased glucocorticoid levels (Anderson and Maes, 2013). This same pathway can also be induced by activation of indeloeamine-2-3-dioxygenase (IDO), which in turn may be induced by IFN-γ and pro-inflammatory cytokines, including TNF-α and IL-1. This may lead to further activation of the TRYCAT pathway (Anderson and Maes, 2013). Increases in TRYCATs may promote hypofunction of NMDA receptors and amplification of the hyperdopaminergic and hyperglutaminergic state (Banerjee et al., 2012).
KA, through inhibition of α7 nicotinic acetylcholine receptors, may also reduce GABAergic post-synaptic signaling. Activation of IDO has been shown to induce the catabolism of tryptophan and increase the production of excitotoxic and neurotoxic TRYCATs, including 3-hydroxykynurenine and quinolinic acid. The latter has been associated with neuroprogressive processes (Anderson and Maes, 2013).
The association between altered tryptophan and TRYCAT metabolism and neurodevelopmental stressors has been demonstrated in animals using the SIR model (Moller et al., 2012), as well as its possible causal relationship with altered behavior and frontal cortical dopamine release (Moller et al., 2013). Transcription studies have demonstrated alterations in proteins involved in the binding and transmission of glutamate within the thalamus, including those which interact with the NMDA receptor (Bruneau et al., 2005). Proteins for the degradation and transport of breakdown products of glutamate have also been suggested to be up-regulated within the thalamus, including glutamine synthase (GS), vesicular glutamate transporter (VGLUT), phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), excitatory amino-acid transporters (EAAT) 1 and 2 (Bruneau et al., 2005), glutamine-synthase-like protein (GSLP) and glutamate dehydrogenase (GDH) (Burbaeva et al., 2006). Glutamine, the breakdown product of glutamate, has been shown to be increased within the thalamus in both antipsychotic naïve and chronically medicated patients (Theberge et al., 2003).
Hyperstimulation of glutaminergic circuits within the nucleus accumbens may produce a hyperdopaminergic state (Muller, 2008). Similar to PV interneurons, the maturation of glutaminergic circuitry has been proposed to be completed around late adolescence (Woo et al., 1997), correlating with the temporal neuroprogession of schizophrenia. Much of the NMDA receptor dysfunction seen in schizophrenia has been proposed to be associated with differences in proband methylation, which may occur as one of the early hits needed to induce pathogenesis of the disease.
Epigenetic factors
There is emerging evidence that schizophrenia is associated with abnormalities of proband methylation and altered gene expression in addition to polymorphisms of proposed risk factor genes, a hypothesis which is supported by differences in methylation patterns observed between monozygotic twins discordant for schizophrenia (Petronis, 2004). DNA methyl-transferase1 (DNMT1), the main DNMT protein implicated in schizophrenia, has been shown to be actively involved in cellular replication and general cell maintenance (Ruzicka et al., 2007). Normally the expression of DNMT1 is down-regulated after terminal differentiation has occurred. In individuals suffering from schizophrenia, DNMT1 has been suggested to be expressed in unexpectedly high levels within GABA-interneurons (Ruzicka et al., 2007). Studies have suggested differences in the expression of DNMT1 within GABA interneurons of different layers of the cortex; layers I, II and IV have demonstrated the greatest up-regulation of DNMT1 (Ruzicka et al., 2007). The increased expression of DNMT1 may result in hypermethylation with the subsequent down-regulation of GAD-67 and reelin (RELN) (Ruzicka et al., 2007), which may contribute to the GABA interneuron hypofunction seen within schizophrenia probands (Benes et al., 2007; Breese et al., 2000; Woo et al., 2008). Genome-wide acetylation patterns within peripheral mononuclear cells have demonstrated an overall decrease in the ‘open’ chromatin mark of acetylated histone 3, with an increase in the ‘closed’ chromatin mark, histone 3 dimethyl K9 (H3K9me2) in schizophrenia probands (Gavin and Sharma, 2009). Other genes that have variously shown increased methylation and down-regulation within studies include: soluble catechol O-methyltransferase (S-COMT) (Melas et al., 2012) and numerous oligodendrocyte genes including the sex-determining region of the Y (SRY)-related 3-hydroxy-3-methyl-glutaryl (HMG)-box (SOX)-10, myelin oligodendrocyte glycoprotein, cyclic-nucleotide phosphodiesterase, oligodendrocyte lineage transcription factor 2, and myelin-associated oligodendrocyte basic protein (Iwamoto and Kato, 2009). In affected twin studies, hypomethylation of alpha-N-acetylgalactosaminide alpha-2,6-sialyltransferase 1 (ST6GALNAC1), involved in the regulation of cell-to-cell interactions, has been found by some groups (Dempster et al., 2011). GAD-1 polymorphisms may also be associated with the development of schizophrenia (Volk et al., 2012), and some polymorphisms show association with increased amounts of the ‘closed’ chromatin mark (Huang et al., 2007). Polymorphisms of the serotonin receptor type 2 (HTR2A) have shown association with promoter hypermethylation in schizophrenia probands. The administration of atypical antipsychotics which target this receptor has been suggested to result in normalization of HTR2A receptor expression (Abdolmaleky et al., 2011). Clozapine may increase expression of the ‘open’ acetylation mark within the GAD-67 promoter, an effect which may not be emulated with genetic ablation of the D2 or D3 receptor (Huang et al., 2007). The administration of methionine (the precursor molecule to S-adenosyl methionine (SAM), the substrate used by DNMT1 to methylate CpG islands) to patients with schizophrenia has been shown to exacerbate symptoms and increases the methylation status of GAD-67 and RELN (Guidotti et al., 2011) within the prefrontal cortex and hippocampus (Tremolizzo et al., 2005). Sodium valproate has been shown to decrease methylation of GAD-67 and RELN within the brains of individuals suffering from schizophrenia (Tremolizzo et al., 2002). This ability of valproate has been proposed to be unrelated to its anticonvulsant action, and instead may be due to its ability to directly interfere with the action of histone deacetylase (HDACs), thus inducing promoter demethylation (Detich et al., 2003). Valproate may help stabilize the ‘open’ chromatin state and induce the action of DNA de-methylase (Detich et al., 2003). Such changes in promoter methylation provide an alternative and potentially attractive therapeutic target alongside the receptor blockade necessary for the action of known anti-psychotics.
Mitochondrial dysfunction
Mitochondria, both as a potent source of ROS which may drive some of the neuroprogressive changes seen in schizophrenia, as well as primary dysfunction of the organelle proper, may be partly responsible for the neuroprogression seen within schizophrenia. Dramatic changes in both the number and structure of mitochondria have been observed in the post-mortem analysis of individuals suffering from schizophrenia, notably within oligodendrocytes in the pre-frontal cortex (Clay et al., 2011; Regenold et al., 2009). Proposed mitochondrial abnormalities include alterations in gene expression, dysfunction of the oxidative phosphorylation system leading to increased production of ROS (Figure 4), and hypoplasia of mitochondria, particularly within the substantia nigra pars compacta (Ben-Shachar, 2002; Karry et al., 2004). Mitochondria have been shown to be a major source of ROS. Mitochondrial diseases can be associated with secondary neurotransmitter disturbance (Garcia-Cazorla et al., 2008), which may hold relevance for dopaminergic dysfunction noted in schizophrenia. An increase in striatal dopamine and ATP are associated with increased self-directed behaviors and reduced prepulse inhibition in SIR rats (Moller et al., 2013). Conversely, altered frontal lobe mitochondrial ATP is correlated with negative symptoms and neuropsychological deficits (Yacubian et al., 2002), congruent with an SIR-induced reduction in frontal cortical ATP, dopamine and social interactive behaviors (Moller et al., 2013). Mitochondrial damage may be partially due to increased levels of oxidized dopamine metabolites, suggested by the high concentrations of monoamine oxidase found on mitochondrial surfaces (Ben-Shachar, 2002). Cavelier et al. have suggested the activity of complex IV is decreased between 43% and 63% in the frontal cortices of individuals suffering from schizophrenia (Cavelier et al., 1995). Reductions of complex I activity have also been observed, but this may be confounded by the toxic effects of anti-psychotics on mitochondria (Prince et al., 1997). Polymorphisms of mitochondrial proline dehydrogenase (PRODH), encoding a protein that converts proline to glutamate within mitochondria, may also be a risk factor for schizophrenia (Kempf et al., 2008). The G72/G30 gene locus, encoding the protein DAOA (Filiou et al., 2012), is implicated in both dendritic branching and splitting of mitochondria (Kvajo et al., 2008). Polymorphisms of the glutamate-cysteine ligase catalytic (GCLC) subunit gene with seven to nine GAG repeats may also be associated with schizophrenia (Gysin et al., 2007). Glutathione has been suggested to protect receptors which are particularly sensitive to the detrimental effects of ROS, which include the NMDA receptor (Choi and Lipton, 2000). The use of N-acetyl cysteine (NAC) may ameliorate the oxidative insult caused by the hyperdopaminergic state induced by the use of amphetamines within the frontal cortex of rats through augmentation of glutathione levels (Dean et al., 2011). NAC has also been found to reverse induced striatal oxidative stress in rats in vivo in a dose-dependent manner (Harvey et al., 2008), which may serve as a potential novel therapeutic source in looking to slow the neuroprogression of schizophrenia.
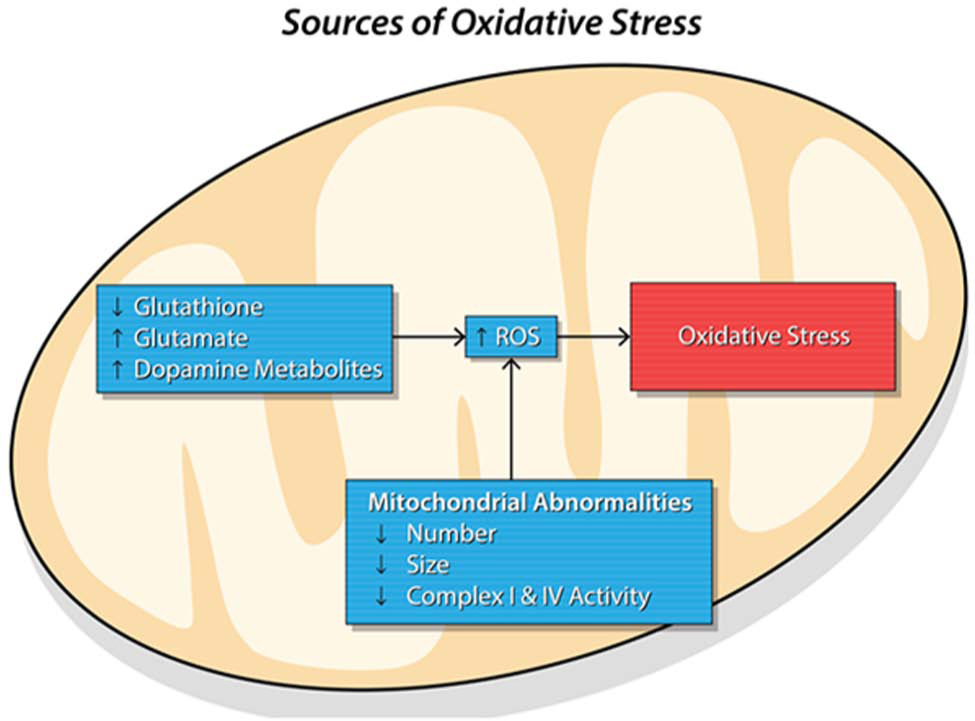
Proposed sources of oxidative stress in schizophrenia. Oxidative stress and the subsequent neuronal dysfunction it produces has been proposed as one of the mechanisms behind the neuroprogression seen within schizophrenia. Sources of oxidative stress are protean. Glutathione, one of the main anti-oxidants within the body, has often been suggested to be decreased within the CNS of individuals suffering from schizophrenia. There has also been demonstration of up-regulation of anti-oxidant genes, including SOD. Excito-toxic neurotransmitters glutamate and the metabolites of dopamine may also be increased and a source of oxidative stress within schizophrenia. Mitochondria have emerged as a prominent source of ROS. Mitochondrial abnormalities proposed within schizophrenia which may contribute to the production of ROS include a decrease in the number and size of mitochondria, abnormal morphology, and dysfunction of complex I and IV.
Immune-inflammatory and redox mechanisms as novel targets in anti-psychotic action
Although D2 receptor blockade remains a necessary and sufficient component for antipsychotic action (Howes and Kapur, 2009; Kapur and Mamo, 2003), many of the currently used antipsychotics display a wide range of novel therapeutic actions which go beyond simple receptor blockade (Figure 5). Current drug treatments, acting primarily at D2/3 receptors, are generally effective against positive symptoms; however, negative and cognitive symptoms continue to remain relatively refractory (Howes et al., 2012). Apart from its other pharmacological properties, clozapine has been suggested to modulate proinflammatory cytokine and ROS release (Hu et al., 2012; Maes et al., 1994; Moller et al., 2013; Song et al., 2000; Sugino et al., 2009), reverses oxidative stress (Moller et al., 2011), corrects aberrant TRYCAT metabolism (Moller et al., 2012), and, additionally, has proposed effects on NOX2 (Hu et al., 2012) and redox-impaired PV interneurons (Cabungcal et al., 2013). These antioxidant actions, in conjunction with positively changing the neuroprotective-neurodegenerative ratio in TRYCAT metabolism (Moller et al., 2012; Myint et al., 2011), may ameliorate the enlarged ventricles and decreased dendritic spine density evident in schizophrenia. Many if not all of these actions have been suggested to be linked to effects on dopamine (Emsley et al., 2013; Moller et al., 2013).
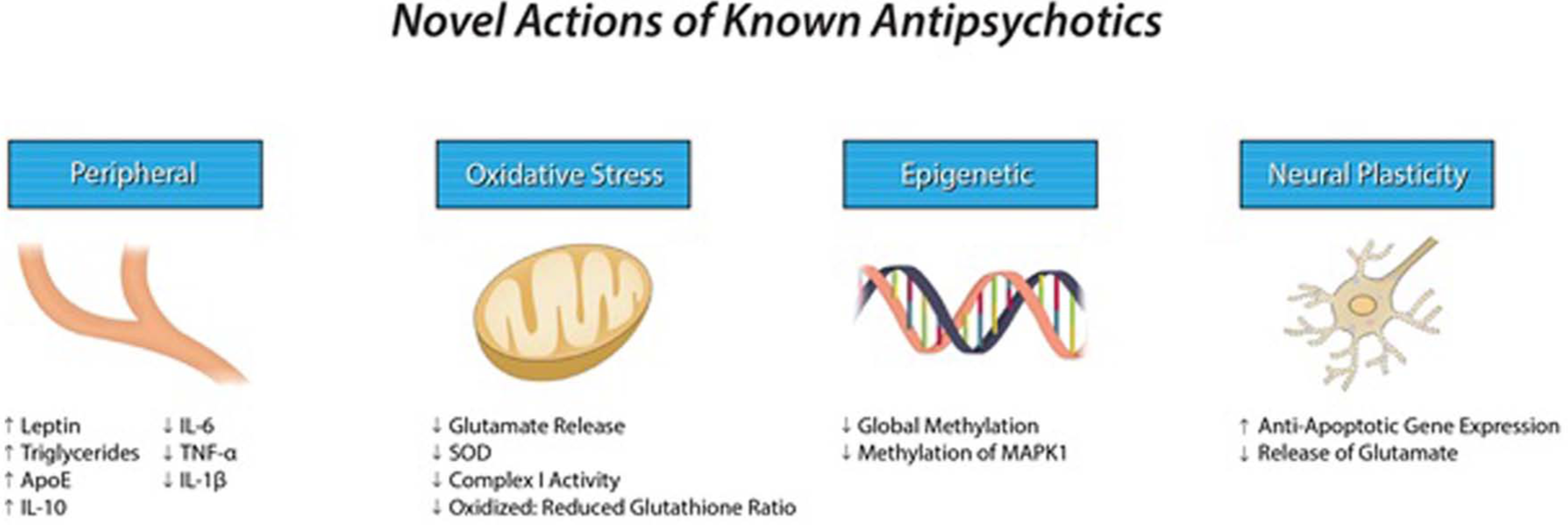
Suggested novel actions of known antipsychotics. Whilst the function of antipsychotics currently employed against schizophrenia is related to their ability to antagonize D2 and 5-HT2 receptors, they may also have a number of other actions which may slow neuroprogression in schizophrenia. Whilst the various antipsychotics show no uniform effects on the immune response, some studies have suggested they decrease levels of the pro-inflammatory cytokines IL-6, TNF-α, and IL-1β, whilst also increasing levels of the anti-inflammatory IL-10. Antipsychotics may reduce oxidative stress through reducing the release of the excito-toxic neurotransmitter glutamate. They may also have anti-inflammatory effects by increasing levels of reduced glutathione and reducing the expression of SOD. Antipsychotics have been suggested to increase the expression of anti-apoptotic genes within neurons as well as decreasing the burden of global hypermethylation seen in schizophrenia.
The early introduction of clozapine, olanzapine or risperidone has been shown in some, but not all, studies to slow or even arrest the progressive decrease in gray matter (van Haren et al., 2007) and subsequent psychotic episodes (Mittal et al., 2008). Atypical antipsychotics may increase leptin levels (Baptista and Beaulieu, 2002) and activate lipogenic genes mediated through increased expression of the sterol regulatory element-binding protein (SREBP-1c) transcription factor (Guillou et al., 2008). Antipsychotic medications may also increase the activation of ApoE (Vik-Mo et al., 2009).
Atypical antipsychotics, as opposed to typical (haloperidol is often the primary comparator), appear to decrease the chronic inflammatory state. Levels of IL-1β, IL-6 and TGF-β may all normalize with anti-psychotic treatment (Miller et al., 2011). Clozapine, risperidone and olanzapine have been proposed to reduce the expression of pro-inflammatory cytokines IL-6 and TNF-α whilst simultaneously increasing levels of the anti-inflammatory IL-10 in animal models (Sugino et al., 2009).
Haloperidol, chlorpromazine and fluphenazine are much more potent inhibitors of complex I than is clozapine (Barrientos et al., 1998; Maurer and Moller, 1997). Clozapine has been associated with a decrease in the expression of SOD, a decrease in peripheral levels of malonyldialdehyde and a decrease in the ratio of oxidized to reduced glutathione (Moller et al., 2011). Clozapine may also act through the NMDA glycine receptor in addition to its antagonism of D2 and 5-HT2A, which may explain some of its novel actions (Schwieler et al., 2004).
Patients chronically treated with haloperidol have been suggested to have higher levels of global DNA methylation as opposed to those treated with aripriprazole, clozapine, risperidone or olanzapine (Melas et al., 2012). Clozapine may demethylate GABAergic promoter genes such as MAPK1, separate to any direct effect on DNMT1 (Browning et al., 2005). Antipsychotics may be able to decrease the release of glutamate from cortical nerve terminals when such nerves are stimulated; however, it has also been suggested they do not inhibit the basal release of glutamate (Goto et al., 2012; Yang and Wang, 2008). Understanding the novel actions of known antipsychotics may help inform the future development of adjunct therapeutic agents, which may help counter the neuroprogressive nature of the disease.
Novel treatments
N-acetyl cysteine
There has been increasing interest in the use of a number of adjunct therapeutic approaches in the treatment of schizophrenia given the emerging evidence of its neuroprogressive nature. NAC has been shown to modulate glutamate activity (Bauzo et al., 2012), in part through potentiation of the cysteine/glutamate antiporter which increases glutamate reuptake, as well as being a substrate for the production of glutathione (Miyake et al., 2011). NAC has also been shown to decrease ROS production (Harvey et al., 2008; Kerksick and Willoughby, 2005), and may also correct mitochondrial dysfunction with subsequent improvements in cognition (Sandhir et al., 2012). It may also bolster the bio-behavioral effects of clozapine as suggested in an animal model of schizophrenia (Moller et al., 2013). NAC has been investigated as a novel adjunctive treatment for schizophrenia (Ferrari et al., 1995; Mayer and Noble, 1994).
Carmeli et al. administered NAC in a randomized, double-blind, crossover protocol for 60 days, followed by placebo for another 60 days, and studied its effects on multivariate phase synchronization (MPS) within 128-channel resting state EEGs. They found that NAC significantly increased MPS within the right temporal, left parieto-temporal, and bilateral prefrontal regions, where glutathione deficiency had been shown to produce detrimental effects on beta/gamma oscillations (Carmeli et al., 2012). Berk et al. looked at the effects of 2 g per day of oral NAC versus placebo as an adjunct to the treatment of chronic individuals suffering from schizophrenia over a 24-week period. They noted an improvement in the clinical global impression (CGI) (–0.22 (–0.41, –0.03), p=0.025) scores, CGI-severity (–0.26 (–0.44, –0.08), p=0.004) and positive and negative symptoms scale (PANSS) total scores (–5.97 (–10.44, –1.51), p=0.009), an effect size consistent with modest benefits (CGI-S effect size 0.43 (0.08, 0.79), PANSS total effect size 0.57 (0.22, 0.93)) (Berk et al., 2008, 2011b). Lavoie et al. looked at 2 g of NAC against placebo for 60 days in a randomized, double-blind, crossover trial. By recording 128-channel auditory-evoked potentials (AEPs) and mismatch negativity (MMN) during a frequency oddball discrimination task they showed that treatment with NAC significantly improved MMN generation (p=0.025) against placebo (Lavoie et al., 2008). Whilst NAC has shown promise in initial trials, it is still largely unknown what role it might have to play in preventing neuroprogression.
Polyunsaturated fatty acid supplementation
Supplementation of essential fatty acids has been investigated as an adjunct treatment for schizophrenia. Sivrioglu et al. (2007) administered 1000 mg of omega-3 fatty acids (eicosapentaenoic acid (EPA) and docosahexanoic acid (DHA)), combined with 1000 mg of vitamin C and 400 IU of vitamin E, to 17 chronic individuals suffering from schizophrenia treated with haloperidol over a 4-month period. The patients demonstrated improvements within multiple testing domains, including the brief psychiatric rating scale (BPRS) (40% mean reduction), scale for the assessment of negative symptoms (SANS) (mean reduction 52.2%) and the Simpson Angus Scale (SAS) (p<0.005) (Sivrioglu et al., 2007). Amminger et al., in a randomized, double-blind, placebo-controlled trial in 81 patients, studied the effects of supplementation of 1.2 g/day omega-3 PUFA over 40 weeks. They found the difference in cumulative risk between the two groups for progression to psychosis was 22.6%, as well as significance differences in positive symptoms (p=0.01), negative symptoms (p=0.02) and overall symptomatology (p=0.01) (Amminger et al., 2010). A recent Cochrane review on PUFA supplementation, which included eight eligible studies, concluded that the results of such trials remain inconclusive at present, and further larger and well-conducted studies were needed to answer the question of whether PUFA supplementation is effective in the treatment of individuals suffering from schizophrenia (Joy et al., 2006).
Non-steroidal anti-inflammatory drugs
Muller et al., in a double-blind placebo-controlled trial looked at the use of 400 mg of celecoxib against placebo as an adjunct therapy in patients treated with amisulpride (range 200–1000 mg) over a 6-week period. They noted significantly improved scores with celicoxib in PANSS negative (p=0.03), PANSS global (p=0.05) and PANSS total (p=0.02), and in CGI ratings (p≤0.001) (Muller et al., 2010). Muller et al. again studied the effects of 400 mg of celecoxib against placebo in a double-blind trail in patients on risperidone over a period of 5 weeks. There were improvements in PANSS total scores (p=0.05), with the celecoxib group additionally showing improvement in all subscale scores (Muller et al., 2002). Akhondzadeh et al., in a double-blind, placebo-controlled trial of 60 patients, studied the effects of 400 mg adjuvant celecoxib in patients chronically treated with risperidone over an 8-week period. They noted a significant change in PANSS total scores at the conclusion of the 8 weeks (p<0.001), as well as improvements in PANSS subscales (Akhondzadeh et al., 2007). In a randomized, controlled trial (RCT), significantly greater reductions in PANSS total and positive subscale scores were seen with aspirin than placebo, and trends were seen on other subscales (Laan et al., 2010). Lastly, Laan et al. explored the adjunctive use of aspirin and showed reduction in total and positive PANSS scores (Laan et al., 2010). The overall objective benefit of the use of non-steroidal anti-inflammatory drugs (NSAIDs), however, has yet to be fully elucidated.
Minocycline
Minocycline modulates glutamate-induced excitotoxicity, and also displays anti-inflammatory, antioxidant, and neuroprotective properties. As these mechanisms dovetail with the pathophysiological pathways to neuroprogression in schizophrenia, minocycline has attracted interest as a potential therapy. It may have clinical utility based on case reports (Dean et al., 2012; Kelly et al., 2011) as well as RCT data. In a RCT in 144 people treated within the first 5 years of illness, improvements in negative symptoms were seen in minocycline-treated individuals (Chaudhry et al., 2012). Much like the other novel treatments explored, there is a need for more research on each with better study design before any firm decisions about their potential use as an adjunctive treatment can be made.
Conclusion
Many seemingly diverse theories on the pathogenesis of neuroprogression in schizophrenia appear to be aligning on similar underlying mechanisms (Wood et al., 2009). Given that many of these theories postulate that schizophrenia is a progressive illness with evolving changes over time involving a state of chronic inflammation of immune-inflammatory pathways, it is prudent to explore new treatment strategies which may ameliorate such changes (Dodd et al., 2013). However, none of the novel therapies explored have yet been shown to fully quell the chronic inflammatory state, nor reverse the observed structural and neurochemical deficits noted within individuals suffering from schizophrenia. Hence, further research into such attractive alternative targets is warranted, and a greater understanding of the pathogenesis of schizophrenia will allow us to guide our therapeutic targets in the future.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Declaration of interest
MB has received grant support from the National Institutes of Health (NIH), Simons Autism Foundation, Cancer Council of Victoria, CRC for Mental Health, Stanley Medical Research Foundation, MBF, NHMRC, beyondblue, Geelong Medical Research Foundation, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Organon, Novartis, Mayne Pharma and Servier. MB has been a speaker for Astra Zeneca, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen Cilag, Lundbeck, Merck, Pfizer, Sanofi Synthelabo, Servier, Solvay and Wyeth, and served as a consultant to Astra Zeneca, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen Cilag, Lundbeck and Servier. BH has participated in speakers/advisory boards and received honoraria from Organon, Pfizer and Servier, and has received research funding from Lundbeck. JD, SM and MM declare no conflicts of interest.