
Abstract
Objective:
To examine the evidence for shared pathophysiological pathways in acute coronary syndrome and major depression and to conceptualise the dynamic interplay of biological systems and signalling pathways that link acute coronary syndrome and depression within a framework of neuro-visceral integration.
Methods:
Relevant articles were sourced via a search of published literature from MEDLINE, EMBASE and PubMed using a variety of search terms relating to biological connections between acute coronary syndrome and depression. Additional articles from bibliographies of retrieved papers were assessed and included where relevant.
Results:
Despite considerable research efforts, a clear understanding of the biological processes connecting acute coronary syndrome and depression has not been achieved. Shared abnormalities are evident across the immune, platelet/endothelial and autonomic/stress-response systems. From the available evidence, it seems unlikely that a single explanatory model could account for the complex interactions of biological pathways driving the pathophysiology of these disorders and their comorbidity.
Conclusion:
A broader conceptual framework of mind–body or neuro-visceral integration that can incorporate the existence of several causative scenarios may be more useful in directing future research and treatment approaches for acute coronary syndrome–associated depression.
Keywords
Introduction
Depression and cardiovascular disease (CVD) are prevalent and highly disabling disorders (Lopez et al., 2006). Patients who suffer an acute coronary syndrome (ACS; e.g. myocardial infarction or unstable angina) experience a threefold increased risk of developing depression (15–20% versus 6%) compared to the general population (Davidson et al., 2010; De Jonge et al., 2010; Glassman, 2007; Nemeroff and Goldschmidt-Clermont, 2012). Moreover, it is well documented that the emergence of a major depressive episode around the time of the ACS significantly increases the risk of a poor cardiac outcome, including mortality (Freedland and Carney, 2013; Parker et al., 2008). In response to these findings, a scientific statement from the American Heart Association advised that the strength of the evidence justifies formal acknowledgement of depression as a risk factor for poor medical outcomes after an ACS (Lichtman et al., 2014). The underlying pathophysiological mechanisms responsible for the bidirectional link between depression and ACS remain poorly understood (Baune et al., 2012; De Jonge et al., 2010; Freedland and Carney, 2013; Pereira et al., 2013; Stapelberg et al., 2011; Whooley, 2013) and led to this review of candidate mechanisms.
Behaviour has a clear influence. Patients with depression are more inclined to smoke, have a poor diet and be physically inactive – behaviours predisposing to cardiac events (Glassman, 2007; Joynt et al., 2003; Lane et al., 2001). After ACS, depression may lead to behaviours that increase the risk of further cardiovascular events, including a lack of self-care, reduced social interaction, underutilisation of healthcare services such as cardiac rehabilitation and poor medication adherence (Cowan et al., 2008; Lane et al., 2001). Psychosocial and behavioural factors ultimately translate into pathophysiological changes in biological systems to exert their effects on health. For example, lifestyle choices such as smoking and physical inactivity may act to increase cardiovascular pathophysiology directly but also contribute to risk indirectly through secondary disease processes associated with a metabolic syndrome or diabetes (Burg et al., 2013; Lopresti et al., 2013).
Several comprehensive reviews have considered candidate biological mechanisms, including inflammatory processes and dysregulation in autonomic and other stress-responsive circuits (De Jonge et al., 2010; Nemeroff and Goldschmidt-Clermont, 2012; Pereira et al., 2013; Thayer and Lane, 2007; Whooley, 2013), but few provide a persuasive conceptualisation of pathophysiological integration. Acknowledging the interaction of several causal mechanisms, rather than focussing upon a single contributor, may better describe the relationships between ACS and depression (Stapelberg et al., 2011). Furthermore, delineation of plausible explanatory models may require a deeper appreciation of the complexity of the disturbance(s) in key neurophysiological systems and their feedback circuits, and the noted reciprocal and perpetuating impact of one condition on the other. The heterogeneity in patient groups fitting the diagnostic labels ‘depression’ and ‘ACS’, although recognised (Baune et al., 2012; Davidson, 2012), is also rarely taken into account. This is despite the well-documented contribution of heterogeneity to disparate research findings in other conditions, such as the enigmatic chronic fatigue syndrome (Aslakson et al., 2009; Vollmer-Conna et al., 2006).
This review focusses on biological mechanisms with documented action in the periphery and in the central nervous system (CNS), and which may be activated by either psychological/behavioural stressors or a disruption of homeostasis in physiological systems. Existing genetic and acquired vulnerabilities unique to each individual are thought to shape the specific manifestations of the pathophysiology and symptomatology of ACS and depression.
For each candidate biological mechanism reviewed (immune, autonomic, platelet/endothelial and hypothalamic–pituitary–adrenal [HPA]-axis function), we will first evaluate the evidence of its pathophysiological importance to CVD/ACS and major depression separately and then highlight recent findings supporting a mediating role of these shared mechanisms in initiating or perpetuating disorder comorbidity. A framework of neuro-visceral integration is offered to conceptualise the complex and dynamic interplay of these biological systems and signalling pathways linking ACS and depression.
Methods
Search strategy
An initial literature search of MEDLINE, EMBASE and PubMed using the terms ‘depression’ or ‘depressive disorder’, and ‘acute coronary syndrome’, ‘myocardial infarction’ or ‘unstable angina’ was conducted. The terms ‘hypothalamus pituitary adrenal axis’, ‘autonomic nervous system’, ‘heart rate variability’, ‘inflammation’, ‘inflammatory mediators’, ‘C-reactive protein’, ‘cytokines’, ‘tumor necrosis factors’, ‘interleukins’, ‘dopamine’, ‘serotonin’, ‘brain derived neurotrophic factor’, ‘neurotransmitter agents’, ‘blood platelets’, ‘platelet aggregation’, ‘vascular endothelium’, ‘thrombocyte’, ‘thrombocyte aggregation’, ‘vascular endothelium’, ‘polymorphisms’, ‘genetic polymorphisms’ and ‘genes’ were combined in subsequent searches with the initial search terms.
A title abstract review was conducted and cross-referenced. Articles in English that focussed on biological pathways linking ACS and depression were included. Additionally, articles from bibliographies of retrieved papers were assessed and included where relevant. As this literature is extensive, we have, as far as possible, focussed on evidence of high quality.
Biological mechanisms mediating the link between depression and ACS
Immune activation and inflammation
CVD is a recognised covert inflammatory condition (Ross, 1999). Immune cells (e.g. macrophages) in and around an endothelial plaque signal and maintain an inflammatory response (Libby, 2012; Ross, 1999). The death of immune cells attempting to clear the plaque adds to its mass, further up-regulating inflammatory proteins and a cascade of pro-inflammatory activity (Joynt et al., 2003; Libby, 2012). Significant elevations in local (i.e. intracoronary) cytokine production profiles have recently been confirmed in patients with ACS (Cirillo et al., 2014), although the authors noted substantive individual variation. Inflammatory markers including C-reactive protein (CRP), interleukin (IL)-1, IL-6 and tumour necrosis factor (TNF)-α have been shown to predict cardiovascular events (Kaptoge et al., 2010; Shlipak et al., 2008).
The potential immunopathological effects of intense inflammatory responses are mitigated by the up-regulation of anti-inflammatory cytokines. Patients with stable angina have higher anti-inflammatory IL-10 concentrations than those with unstable angina (Smith et al., 2001). In patients with ACS, higher levels of IL-10 were shown to correlate with lower concentrations of inflammatory proteins and better health outcomes (Chalikias et al., 2005; Heeschen et al., 2003). Conversely, two large population studies suggest that higher levels of circulating IL-10 are associated with worse outcomes after ACS (Mälarstig et al., 2008; Welsh et al., 2011). Considering the dynamic interplay between cytokines with a role in inflammation, the timing of a single blood sample in relation to ACS would greatly influence relative composition of pro- and anti-inflammatory proteins in such a snapshot assessment. Thus, it has been argued that these seemingly contradictory results can be explained in terms of the complex interactions of cytokine networks (Welsh et al., 2011). Specifically, enhanced expression of an ‘anti-inflammatory’ cytokine, such as IL-10, may indicate ongoing inflammation within the tissue, thus acting as a counter-regulatory mechanism to limit further inflammation. Consistent with such a view, Mälarstig et al. (2008) concluded that circulating IL-10 should be considered a surrogate biomarker for ongoing cardiovascular inflammation and pathology, and thus is as effective as markers of systemic inflammation in risk prediction of future cardiovascular events.
A substantive literature supports an association between inflammation and depression. For example, levels of pro-inflammatory cytokines (TNF-α, IL-6 and IL-1β) were significantly higher in the blood and cerebrospinal fluid (CSF) of patients with depression compared to non-depressed controls (Dinan, 2009; Dowlati et al., 2010; Howren et al., 2009). However, considerable inter-individual variation in levels of inflammatory markers has been reported in depressed patients (Raison and Miller, 2011), and there are conflicting reports as to whether inflammatory markers revert to normal levels when depressive symptoms remit (Dinan, 2009; Miller et al., 2009).
Inflammation has been posited to link ACS/depression comorbidity. Patients with both depression and elevated inflammatory markers (e.g. CRP levels > 3 mg/L) experienced a much higher risk of ACS (Empana et al., 2005; Ladwig et al., 2005). In patients with ACS, concentrations of inflammatory markers, in particular CRP, were significantly higher in those with comorbid depression than in those without (Shimbo et al., 2006). Given the prognostic value of CRP for ACS outcome (Ramasamy, 2011), the increased morbidity and mortality risk for patients with ACS-associated depression may be mediated by an overall greater elevation in CRP levels (Shimbo et al., 2006). Other evidence has been less consistent. For example, one study showed that after adjustment for factors possibly impacting inflammation, such as smoking, physical inactivity and body mass index, the observed relationship between raised CRP and IL-6 and depression was lost (Duivis et al., 2011).
As white blood cells proliferate in response to an immunological challenge, white cell counts (WCCs) offer a simple measure of immune activation. Significantly raised WCCs have been linked to CVD with a fourfold increased risk of experiencing ACS (Brown et al., 2001; Ernst et al., 1987; Kannel et al., 1992). Additional evidence suggests that WCCs are raised as a result of distress and depression (Patterson et al., 1995; Segerstrom and Miller, 2004; Zorrilla et al., 2001). Some studies have examined the prognostic utility of WCCs for developing depression in association with ACS. Baseline WCCs predicted developing symptoms of both depression and anxiety following ACS in one study (Steptoe et al., 2013) and partially predicted cardiac mortality in patients with comorbid depression (Kop et al., 2010).
Explanations of the connection between inflammation, ACS and depression must extend beyond the direct peripheral effects of inflammation on, for example, endothelial function. Following infection or injury, immune activation and the concomitant up-regulation of pro-inflammatory proteins are typically accompanied by a set of physiological, behavioural and motivational changes, including fever, increased slow wave sleep, hyperalgesia, anorexia, anhedonia, disturbed mood and impaired concentration – termed the ‘acute sickness response’ (Hart, 1988; Vollmer-Conna, 2001). This response is triggered by the action of cytokines on CNS targets and represents a fundamental biological adaptation that shifts the organism’s priorities towards pathogen resistance, energy preservation and recovery (Poon et al., 2013). Chronic activation of inflammatory response pathways in CVD may, in vulnerable individuals, lead to the more sustained and severe pattern of behavioural and physiological changes of major depression (Gunaratne et al., 2013; Miller et al., 2013). Such complex interactions have been intensely studied, with an inflammatory contribution to neuropsychiatric conditions such as major depression being the subject of several detailed reviews (Dantzer et al., 2011; Felger and Lotrich, 2013; Liu et al., 2013; Miller et al., 2009; Raison and Miller, 2013). In essence, cytokines and their signalling pathways exert significant effects on the synthesis, release and reuptake of many neuroactive proteins including serotonin, dopamine, noradrenaline, glutamate and brain-derived neurotrophic factor (BDNF). For example, by activating the enzyme indoleamine 2,3 dioxygenase (IDO), tryptophan is converted into kynurenine instead of serotonin, a process which may affect serotonergic neurotransmission and increase the risk of depression (Dantzer et al., 2008). Kynurenine can cross the blood–brain barrier and may then be further metabolised by glial cells to generate neuroactive compounds, including quinolinic acid and kynurenic acid (Miller et al., 2013). In particular, quinolinic acid (a glutamate agonist) is well recognised for its potential to produce excitotoxicity, oxidative stress and neuronal cell death (Müller and Schwarz, 2007). Reduced levels of tryptophan and elevated kynurenine in the CSF, as well as glutamatergic hyperfunction, have been documented in depression (Capuron et al., 2003a; Müller and Schwarz, 2007; Raison et al., 2010). Additionally, the association between IDO-activity and depression was confirmed in a population-based study (Elovainio et al., 2012), although a longitudinal cohort study did not find a direct relationship between reduced tryptophan bioavailability and depression – nor establish a mediating role for tryptophan degradation in the relationship between inflammatory markers and depressive symptoms (Quak et al., 2014).
Another mechanism by which pro-inflammatory cytokines could impede monoamine function is via perturbation of tetrahydrobiopterin (BH4) activity. BH4 functions as a cofactor for several important enzyme systems involved in the biosynthesis of neurotransmitters including epinephrine, norepinephrine, dopamine and serotonin (Bendall et al., 2013). Although BH4 biosynthesis can be induced by inflammatory stimuli, pro-inflammatory cytokines also excite the activity of nitric oxide synthase (NOS) to produce nitric oxide (NO), which uses BH4 as an enzyme cofactor. Moreover, inflammatory stimuli increase oxidative stress to which BH4 is very sensitive (Bendall et al., 2013; Miller et al., 2013). Inflammation may thus increase the utilisation and ultimate degradation of BH4. BH4 is also a key regulator of endothelial NOS. Studies evaluating tissue and plasma samples from patients with CVD have revealed an inverse relationship between plasma and blood vessel BH4 levels (Antoniades et al., 2007), whereby low vascular BH4 is associated with decreased vascular function and increased vascular superoxide, and also with elevated concentrations of plasma BH4 and CRP. This example of BH4 illustrates how the same biological pathway can manifest varied localised pathologies to perpetuate CVD and/or depression.
While inflammatory proteins can impact brain functioning, acute or chronic psychological trauma and depression can in turn significantly increase inflammatory responses, decreasing neurotrophic support and the availability of serotonin and glutamate, as well as increasing apoptosis and oxidative stress (Catena-Dell’Osso et al., 2013). Up-regulated inflammatory processes may therefore be a common feature in ACS and depression, albeit for different reasons. In individuals with both conditions, elevated levels of inflammatory proteins may be perpetuated through several different pathological processes and contribute to increased morbidity and mortality.
Twin studies suggest a genetic influence on the propensity for an exaggerated inflammatory response in patients with CVD and depression (Scherrer et al., 2003; Su et al., 2009). Specific genetic polymorphisms that may contribute to vulnerability to ACS and depression have also been identified (Mulle and Vaccarino, 2013). For example, a functional polymorphism in the TNF-α gene (-308G/A) was associated with increased production of TNF-α in response to an immune challenge (Wilson et al., 1997), cardiac risk factors (Dalziel et al., 2002), metabolic syndrome (Sookoian et al., 2005) and depression (Jun et al., 2003; Lückhoff et al., 2014). Polymorphisms in the IL-6 (-174G/C and -572G/C) and CRP genes (+1444C/T) were associated with an increased inflammatory response after coronary bypass surgery (Brull et al., 2003). Additionally, the high-producing T allele of this CRP polymorphism (+1444C/T) has been associated with increased suicidal behaviour (Suchankova et al., 2013).
Mechanisms relating to platelet and endothelial function
Platelet activation and endothelial function are dysregulated in patients with CVD. Platelet activation is a finely balanced orchestration of pro-coagulant release and platelet receptor stimulation. In the event of plaque rupture, platelets aggregate to promote healing. This activation involves platelets changing shape and releasing granules adding to the ‘stickiness’ of the milieu, which can result in an occluding thrombus (Neubauer et al., 2013). Platelet activation can thus be a precipitating factor in ACS. The endothelium reacts to platelet activation by releasing NO, a potent vasodilator, which allows blood flow to be maintained (Korszun and Frenneaux, 2006; Mombouli and Vanhoutte, 1999). Damage to the endothelium and the presence of inflammatory proteins reduce NO availability, which affects blood pressure control as well as angiogenesis (Bendall et al., 2013; Le Melledo et al., 2004; Verma et al., 2002).
Several neurotransmitters with established roles in mood regulation have a dual role in platelet and endothelial function. For example, serotonin has documented peripheral action as a platelet activation agonist, initiating vasoconstriction and coagulation (Côté et al., 2004; Ziegelstein et al., 2009), and high levels of circulating serotonin may elevate thrombotic and cardiac risks (Musselman et al., 1996; Schins et al., 2003). Additionally, BDNF plays important roles in cardiac protection and recovery by mediating endothelial function and facilitating cell regeneration (Kermani and Hempstead, 2007; Okada et al., 2012). Both endothelial cells and platelets release BDNF concomitant with platelet activation (Nakahashi et al., 2000), and animal studies have demonstrated a role for BDNF in capillarisation and recovery after ischaemia (Kermani and Hempstead, 2007; Okada et al., 2012).
Centrally, BDNF is vital for neuronal longevity and neurotransmission (Lipsky and Marini, 2007). Animal models of depression have documented reduced BDNF production in the hippocampus and altered serotonergic transmission (Hashimoto, 2010). In humans, low BDNF levels have been linked to depression (Hashimoto, 2010; Lee et al., 2007; Shimizu et al., 2003), as has a polymorphism in the BDNF gene (Val66Met), which may reduce neuro-availability of BDNF (Terracciano et al., 2010). However, recent reviews conclude that the influence of BDNF on mood regulation is complex and poorly understood, with the precursor proBDNF and the mature protein mBDNF playing opposing roles in the mediation of symptoms of mood disorders (Groves, 2007; Hashimoto, 2010; Lipsky and Marini, 2007).
Serotonergic transmission has been intensely studied in depression (Karg et al., 2011). Two meta-analyses (Karg et al., 2011; Lopez-Leon et al., 2007) but not a third (Risch et al., 2009) quantified an association between polymorphisms in the serotonin transporter gene (5-HTTLPR S/L) and the risk of depression, particularly in the face of repeated life stressors. It has also been shown that post-ACS patients carrying the risk-associated S allele experienced an increased likelihood of subsequent cardiac events when they also experienced depressive symptoms (Nakatani et al., 2005).
A recent review provided a detailed description of the platelet clotting cascade and subsequent dysregulation experienced in patients with ACS and depression (Nemeroff and Goldschmidt-Clermont, 2012). These authors suggest ACS-associated depression may be an age-related phenomenon, noting the increased incidence of both ACS and new-onset depression with ageing, and the age-related decreased availability of endothelial progenitor cells (EPCs) and other cells needed for arterial repair. In support, EPCs have been demonstrated to be low in patients (average age 58 years) with either ACS or depression; however, patients with comorbid ACS and depression had the lowest EPC counts (Di Stefano et al., 2014).
Platelet and endothelial dysfunction are common to ACS and depression and exacerbated by processes associated with each disorder (Sherwood et al., 2005; Von Känel et al., 2001). Patients experiencing depression showed higher cardiac risk factors related to platelet dysfunction such as elevated fibrinogen; when re-assessed 6 months later, depression symptom remission and fibrinogen decreases were correlated (Mclean et al., 2007). Some evidence suggests a cumulative risk of platelet dysfunction resulting from comorbid ACS and depression (Kuijpers et al., 2002; Serebruany et al., 2003). However, a review (Nemeroff and Musselman, 2000) and large cohort study (Gehi et al., 2010) examining whether platelet activation functions as a mediator between CVD and depression found the evidence inconclusive.
The importance of autonomic regulation
The autonomic nervous system (ANS) is pivotal in the maintenance of body integrity, self-regulation and adaptation (Thayer et al., 2012; Tracey, 2009). Autonomic response pathways have evolved to favour sympathetic dominance to ensure adaptive survival. Experience of severe and/or persistent stress and trauma may cause prolonged reduction in prefrontal inhibitory control of stress-responsive neural circuits (Maier and Watkins, 2010), leading to an imbalance in ANS function characterised by a dominance of sympatho-excitatory circuits and loss of parasympathetic (vagal) tone. This engenders a hyper-vigilant physiological state, lacking adaptive flexibility, which has been linked to the severity and outcome of a large spectrum of diseases including ACS and depression (Hillebrand et al., 2013; Kemp et al., 2012; Pereira et al., 2013; Stapelberg et al., 2012; Thayer and Lane, 2007). In addition, the sympathetic branch of the ANS is understood to play a role in enhancing the intensity of the host response to immunogenic stimuli (Grebe et al., 2010; Kin and Sanders, 2006; Thayer et al., 2012), while the parasympathetic vagus nerve contributes to the containment of inflammatory responses (Andersson and Tracey, 2012; Tracey, 2009).
Cardiac autonomic functioning, in particular vagal modulation, can be reliably inferred from measures of beat-to-beat heart rate variability (HRV; Task Force of the European Society of Cardiology the North American Society of Pacing (Electrophysiology), 1996). One of the earliest large-scale investigations of a role for HRV in post-ACS mortality (Kleiger et al., 1987) reported a highly significant fourfold increase in mortality risk for those in the lowest quadrant of HRV over the 4-year follow-up study period. The predictive value of HRV was independent of other relevant risk factors, and its impact increased over time. Since that report, evidence has accumulated supporting a connection between low HRV and cardiovascular morbidity and mortality (Hillebrand et al., 2013; Stapelberg et al., 2012; Taylor, 2010; Thayer and Lane, 2007).
Thayer and Lane (2007) noted that all modifiable risk factors for CVD (e.g. diabetes, smoking, high cholesterol, insufficient exercise) are linked to low HRV. A reduction of HRV has been associated with poor modulation of cardiovascular responses, an increased propensity to inflammation and poorer health outcomes in ACS patients (Carney et al., 2007; Thayer and Lane, 2007). Low HRV has also been recognised as an important covariate of major depressive disorder (Kemp et al., 2012; Wang et al., 2013) and is hypothesised to underlie the link between depression and cardiovascular events (Huikuri and Stein, 2012; Stapelberg et al., 2012; Taylor, 2010). HRV parameters have been shown to negatively correlate with depression severity in patients with CVD (Stein et al., 2000) and after ACS (Carney et al., 2001), and reduced HRV persists in patients with depression following cardiac surgery (Patron et al., 2012). In patients with ACS assessed 2 months after discharge, a negative correlation between HRV and the inflammatory markers CRP and IL-6 was shown to be strongest in depressed participants (Frasure-Smith et al., 2009). While these data provide circumstantial support for an anti-inflammatory role of vagal nerve activity, no direct relationship with HRV and depression symptoms was found.
As is the case when examining the impact of inflammation on ACS and depression, the relationships between autonomic dysfunction, ACS and depression are best considered within a framework of neuro-visceral connectivity. Traditional cardiovascular research has linked altered autonomic activity to its localised effects on vascular, endothelial or cardiac function (Xhyheri et al., 2012). Similarly, reduced HRV in depression has typically been interpreted in a top-down fashion, as a reflection of ongoing psychosocial stress and associated state of hyper-vigilance (Thayer and Lane, 2007; Thayer and Sternberg, 2006). A meta-analysis documenting the failure of anti-depressant medication to enhance HRV, even when symptoms of depression are significantly improved (Kemp et al., 2010), underscores cognitive explanations of the depression–low HRV association as being in themselves insufficient. A study linking a disturbance in autonomic balance to inflammatory processes suggests that low-grade inflammation may be an additional, independent driver for centrally mediated reduction of HRV – including in those with depressive disorder (Harrison et al., 2013). An ‘internal stressor’ of ongoing inflammation may thus contribute to low HRV in both CVD and depression, while sustained low HRV, and associated reduced vagal activity, may perpetuate a physiological state unable to contain excessive inflammatory responses. Adding the neural and physiological changes associated with major depression to this mix creates a vicious feedback cycle that sustains, and possibly potentiates, autonomic imbalance and chronic inflammation, particularly in patients with comorbid ACS and depression.
HPA-axis and glucocorticoid sensitivity
Glucocorticoids function to contain the inflammatory response via a feedback loop where HPA-axis activity is induced by high levels of pro-inflammatory cytokines (particularly IL-6), and the released cortisol in turn, down-regulates further production of pro-inflammatory cytokines (Nijm and Jonasson, 2009; Walker, 2007). Administration of glucocorticoids has been associated with an increased risk of experiencing a cardiovascular event (Souverein et al., 2004; Wei et al., 2004) and the progression of atherosclerosis (Dekker et al., 2008). In patients receiving interferon (IFN)-α therapy, responders with hyper-activation of the HPA-axis were more likely to develop depression in subsequent weeks (Capuron et al., 2003b), indicating a role for HPA-axis sensitivity in immunologically triggered depression. HPA-axis dysregulation may contribute to the increased morbidity and mortality risk in patients with ACS-associated depression, although this has not been examined in any depth.
Perspectives on neuro-visceral integration as it relates to CVD/ACS-associated depression
Contemporary research has significantly advanced our understanding of the functional complexities of the major regulatory and signalling systems connecting the human mind and body. Reciprocal and dynamic influences are nowhere more apparent than in the comorbidity of medical disease and psychiatric syndromes, such as ACS and depression. Many of the biological processes previously described as important in the development and progression of either ACS or depression are now recognised as playing a role in both conditions and may be critical for understanding their linked pathogeneses.
Endothelial and vascular pathologies characteristic of ACS engage local and systemic inflammation, change autonomic reactivity and advance dysregulation of oxidative, essential enzyme and cofactor systems, including BH4 and NOS (Bendall et al., 2013). Moreover, elevation of inflammatory proteins in ACS can reduce the bioavailability and functioning of key neurotransmitter and neurotrophic systems involved in mood regulation and depression. Systemic immune activation additionally elicits a classical stress response characterised by activation of the HPA-axis and a shift in autonomic balance towards sympathetic dominance (Maier and Watkins, 2010). Reduced parasympathetic (vagal) signalling and enhanced catecholamine release from sympathetic neurons in turn perpetuate the release of pro-inflammatory proteins (Andersson and Tracey, 2012; Grebe et al., 2010; Pereira et al., 2013) and impact adversely on local cardiovascular pathology (Korszun and Frenneaux, 2006).
The premise that inflammatory processes may increase cardiovascular risk through centrally mediated mechanisms has been strengthened by a recent human neuroimaging experiment documenting that elevation of peripheral inflammatory proteins elicited distinct CNS-mediated effects on cardiovascular autonomic reactivity (Harrison et al., 2013).
Psychological stress and depression directly impact neural circuits and neurotransmitter pathways involved in the regulation of emotional and behavioural responses and activate regulatory systems that influence both local and distant mechanisms (Broadley et al., 2005; Burg et al., 2011; Maier and Watkins, 2010). Prolonged sympathetic activity can cause adverse local cardiovascular effects, with evidence from experimental studies in humans documenting a direct role for cortisol in stress-induced endothelial dysfunction and impaired baroreflex sensitivity (Broadley et al., 2005). Mental stress and depression severity have also been related to elevated endothelin-1, a potent vasoconstrictor, known to play an important role in CVD progression (Burg et al., 2011). Cardiovascular reactivity to stress has been found to be genetically influenced. A meta-analytic review of twin studies comparing heart rate and blood pressure responses to stressors demonstrated strong heritability (Wu et al., 2010). This research documents a spectrum of biological mechanisms through which depression and emotional stress directly contribute to ACS outcome.
Taken together, the accumulated evidence supports the hypothesis that disturbances in peripheral systems, notably the inflammatory response, engage similar neural networks as do psychosocial stressors and that both may ultimately affect the function of signalling pathways regulating homeostasis and body integrity, as well as mood, motivation and behaviour. As ACS constitutes a fundamental stressor of some psychological and physiological magnitude, it is important to consider the combined impact of stressors on neuro-visceral systems (Beaumont et al., 2012; Thayer et al., 2012). From the available evidence, it is clear that not all individuals with ACS have substantively raised inflammatory markers, and that those with increased inflammation do not all develop depression, so these contingencies depend on the interplay of a spectrum of factors (Figure 1). These include the severity of ACS and existing comorbidities, vulnerabilities in immune and/or behavioural genes (which can influence the intensity of the inflammatory response and endow a person with more or less resilient stress-responsive regulatory systems), as well as developmental and current life stressors, and the coping strategies (including health behaviours) adopted by each individual (Felger and Lotrich, 2013; Gunaratne et al., 2013; Raison and Miller, 2013).
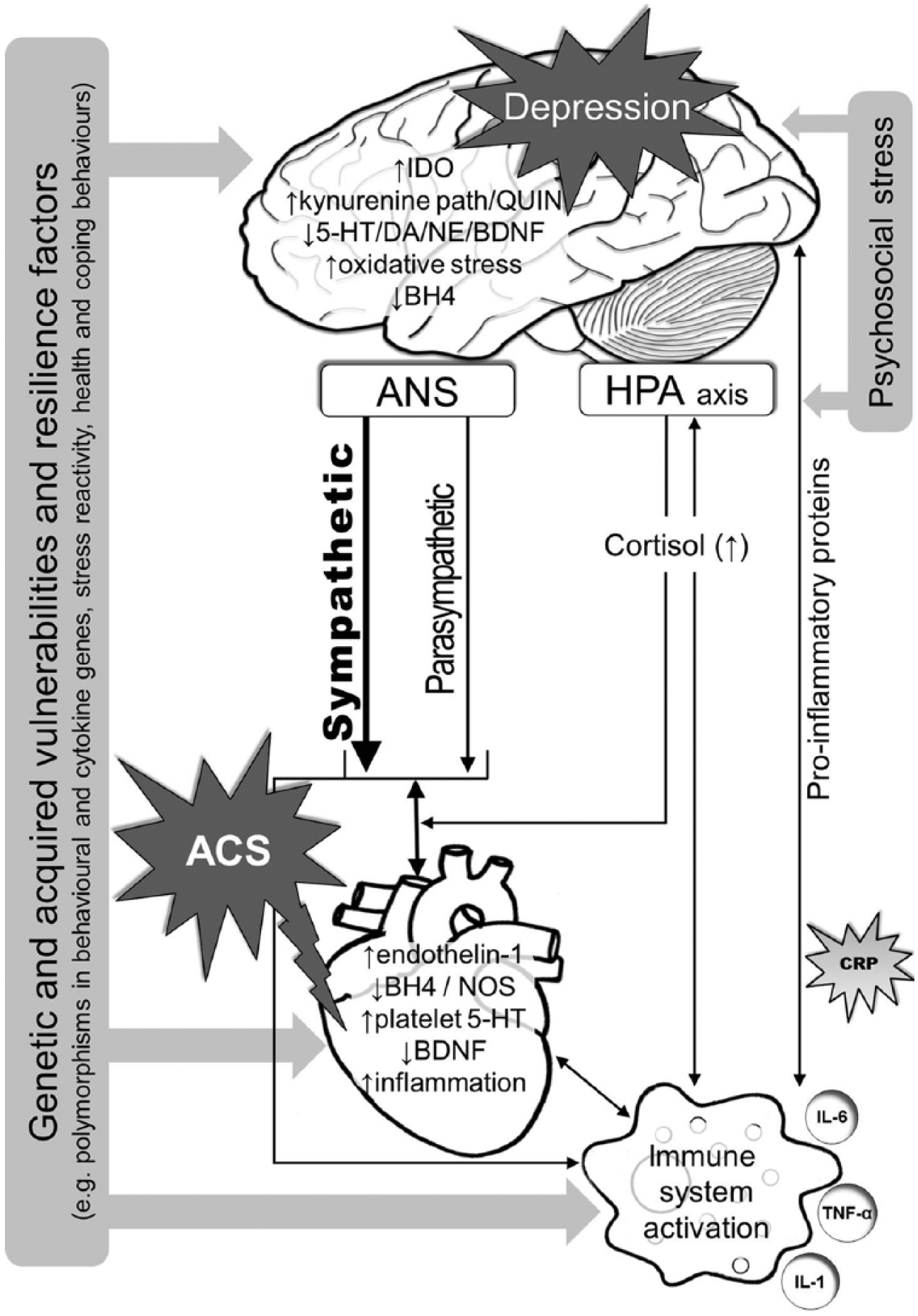
Schematic representation of the complex changes in biological signalling and response pathways potentially associated with ACS and depression comorbidity.
Translational implications
As highlighted throughout this review, ACS-associated depression is likely to arise from interactions between several bio-behavioural systems. It is therefore not surprising that identifying interventions that effectively address mortality and morbidity risk in this group has so far proved challenging. Large randomised controlled trials (RCTs) assessing the effects of anti-depressant treatment on cardiac mortality outcomes have largely proved ineffective (Carney et al., 2004; De Jonge et al., 2007; Van Melle et al., 2007). A recent meta-analysis concluded that well-conducted RCTs revealed no difference in mortality between selective serotonin reuptake inhibitors (SSRIs) and placebo in patients with ACS (Pizzi et al., 2011). However, SSRIs can only be expected to be effective if disrupted serotonergic transmission is an underlying contributor to depression.
Similarly, an initial RCT in a small number of patients with treatment-resistant depression using the anti-inflammatory agent infliximab revealed that this treatment was only beneficial for those participants whose baseline levels of CRP showed moderate elevation (i.e. CRP > 5 mg/L). By contrast, those with low baseline levels of CRP fared worse (Raison et al., 2013). Several non-pharmacological interventions designed to enhance vagal nerve activity appear to improve both HRV and depressive symptoms: for example, vagal nerve stimulation for treatment-resistant depression (Aaronson et al., 2012; Nahas et al., 2005). Patients with ACS and depression, who also have significantly reduced HRV, may find vagal nerve stimulation an effective treatment.
The findings reviewed here argue the need to individualise treatment for patients with ACS-associated depression. This could be addressed by routine testing of key biological parameters identified in ACS-associated depression (e.g. including markers of immune, HPA-axis and autonomic functioning, and metabolites of key neurotransmitters). Finally, researchers and clinicians need to be mindful that increased inflammation, autonomic dysfunction and a host of other pathophysiological changes with putative roles in ACS-associated depression can be related to poor health generally, rather than depression specifically. Improvements to diet, exercise and sleep will undoubtedly contribute to regulating these biological systems in a positive way.
Conclusion
It has become increasingly apparent that the traditional classification of ACS as a purely medical disease and depression as a mental/psychiatric condition is not helpful in guiding research efforts towards a clearer understanding of the pathophysiological mechanisms underlying each of these disorders and their frequent association. The time has come to embrace a broader integrated systems perspective of health and disease. Such a perspective will open new avenues for multidisciplinary research that, with time and discernment, will allow us to see the bigger picture. Such an approach promises greater clarity to both the assessment of risk around further cardiac events and survival following ACS and direct clinical practice towards more effective, personalised treatments for CVD/ACS and depression.
Footnotes
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Funding
This work was funded by a NHMRC Programme Grant (1037196) and by the Commonwealth Department of Health and Ageing.