
Abstract
Hyperkalaemia is a common biochemical finding that can allude to preanalytical or truly pathological causes. Here, we present a case of a 41-year-old female patient who has regularly presented with incidences of isolated hyperkalaemia since 2012, with otherwise normal renal function and no other associated symptoms. Investigations into the patient’s family history revealed similar biochemical findings in her brother and eldest son. Familial causes of hyperkalaemia were investigated and an eventual diagnosis of pseudo-hypoaldosteronism type 2C was established. This is a rare congenital renal tubular disorder – also known as Gordon syndrome – that can cause a characteristic triad of symptoms that include hyperkalaemia, metabolic acidosis and hypertension. The presence and severity of each of these symptoms is dependent upon the disease-causing mutation that occurs in WNK4, WNK1, CUL3 or KLHL3 genes. These mutations alter the regulation of sodium/chloride co-transporter (NCC) expression on the luminal membrane of the principal cells of the distal convoluted tubule, disrupting normal homeostatic regulation of electrolyte reabsorption and excretion. The resolution for treating this condition is the administration of a thiazide diuretic, which directly counteracts the effects of NCC co-transporter overexpression and consequently aims to resolve the symptoms that arise as a result of this aberrant signalling. The case described here uniquely presents an extremely rare pathogenic variant in the conserved acidic motif of WNK1 resulting in a clear electrolyte phenotype with no hypertension.
Introduction
Hyperkalaemia is a common biochemical finding that can be caused by a wide range of clinical conditions and can be ultimately life-threatening. 1 Pseudo-hypoaldosteronism type 2 – also known as Gordon syndrome – is an extremely rare autosomal dominant condition that causes an isolated hyperkalaemia with otherwise normal renal function, in addition to hypertension, hyperchloraemia and metabolic acidosis. 2
This condition is known to manifest as a result of mutations to one of the four genes responsible for the regulation of normal sodium and chloride reabsorption in the distal convoluted tubule and collecting duct in the nephron via the thiazide-sensitive sodium/chloride co-transporter (NCC). These mutations occur in one of the following genes: WNK4, WNK1, CUL3 and KLHL3, with symptom severity being found to be mutation-dependent. 3 The proteins encoded by these genes are primarily responsible for the regulation of NCC expression and subsequent function on the luminal membrane of the principal cells of the distal convoluted tubule. WNK4 is a direct inhibitor of NCC expression, whereas WNK1 acts as a negative regulator of WNK4. 4 The mutations in these proteins that are responsible for the pathogenesis of pseudo-hypoaldosteronism type 2 equate to a loss of WNK4 function or gain of WNK1 function that consequently results in increased NCC membrane expression and action. The remaining two proteins that also contribute to this regulatory process include CUL3 and KLHL3 regulatory proteins that regulate WNK4 and WNK1 activity through ubiquitin-mediated degradation. Mutations in these proteins are ultimately known to cause loss of enzymatic function that results in an accumulation of intracellular WNK4 and WNK1, thereby promoting dysfunctional regulation of NCC membrane expression. 3
Deregulation of NCC expression results in an increased concentration of sodium and chloride entry into the principal cells of the distal convoluted tubule that directly affects potassium excretion through two primary mechanisms. The first being the disruption of the electrochemical gradient between the luminal membrane and the lumen itself. The increase of intracellular influx of negatively charged chloride reduces the electronegative gradient between the lumen and the intracellular space resulting in lower levels of electrochemically-driven potassium excretion. 5 The second mechanism is the decreased expression of surface ROMK potassium channels through abnormal WNK4/WNK1 signalling that causes a reduction in the capacity of the distal portion of the nephron to excrete potassium.6,7 Both of these mechanisms subsequently lead to increased intracellular concentrations of potassium in the distal convoluted tubule that are reabsorbed back into circulation through the potassium/chloride co-transporter (KCC) on the basolateral membrane.
Individuals with Gordon syndrome present with salt-sensitive hypertension which responds well to thiazides. Increased activity of NCC leads to enhanced sodium retention and volume expansion resulting in low or low normal plasma renin activity. Plasma aldosterone level can be inadequately low or even normal due to the opposite influences of low renin and high potassium levels.3,8,9
Case report
Here, we report a case of an initially unexplained hyperkalaemia in a 41-year-old female patient. The patient first presented in 2012 following routine biochemistry investigations prior to administration of NSAID treatment for chondritis. Normal serum creatinine, urea and sodium coupled with normal liver function were observed alongside an isolated hyperkalaemia of 6.8 mmol/L (reference range 3.5–5.3 mmol/L). No discernible symptoms or risk factors associated with hyperkalaemia were noted upon presentation, and common causes of hyperkalaemia were ruled out in this patient, including diabetes mellitus, medications and acute tissue damage (Table 1). However, it was evident that sample separation was initially performed over 6 h after sample collection which appeared to explain this incident of hyperkalaemia through in vitro mechanisms of potassium release from red blood cells postvenepuncture. The patient has since presented with isolated hyperkalaemia on a number of subsequent occasions, with no definitive preanalytical or pathogenic cause being identified (Figure 1). This triggered appropriate investigations to elucidate the cause of this.
An overview of laboratory results obtained from biochemistry investigations.
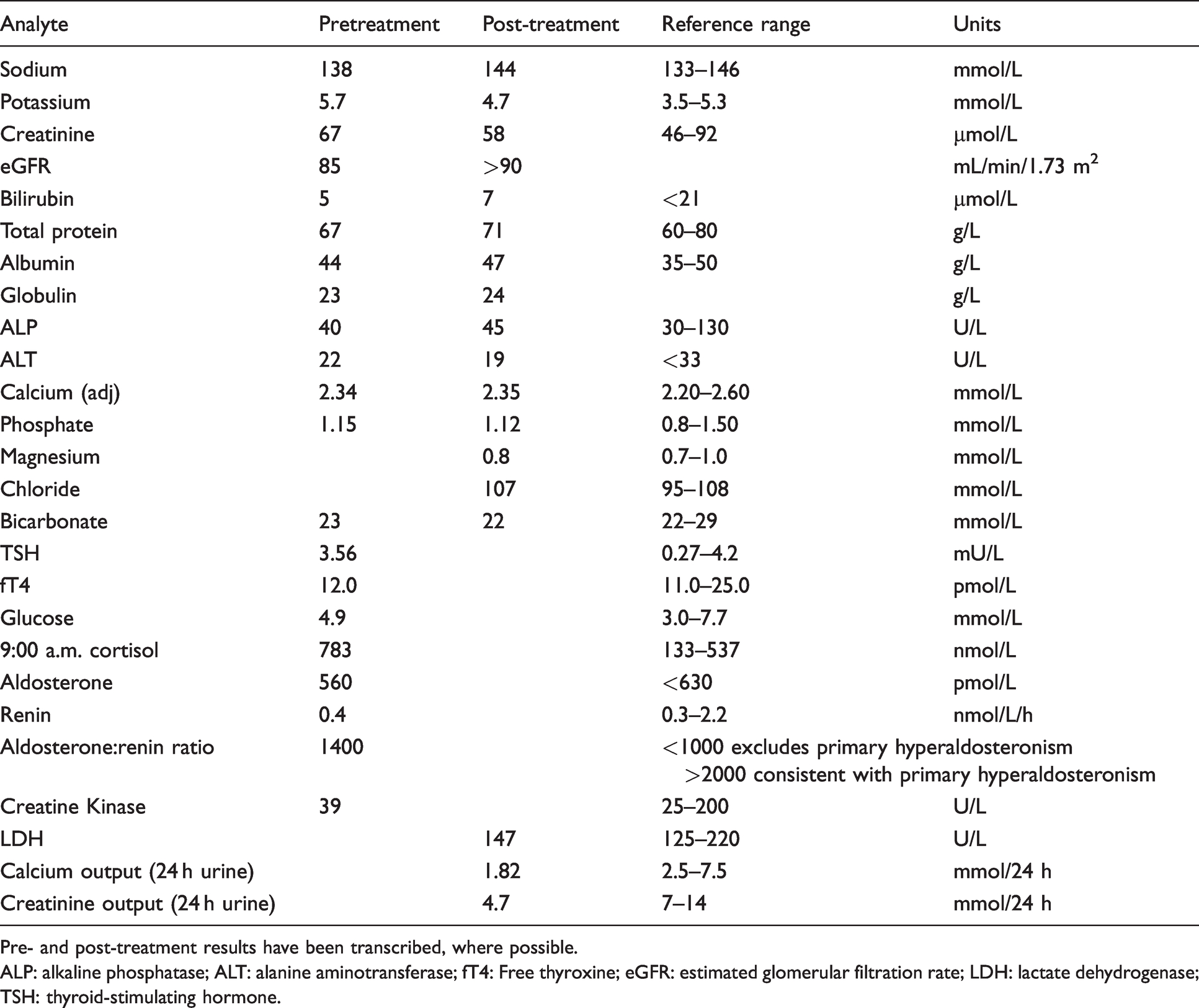
Pre- and post-treatment results have been transcribed, where possible.
ALP: alkaline phosphatase; ALT: alanine aminotransferase; fT4: Free thyroxine; eGFR: estimated glomerular filtration rate; LDH: lactate dehydrogenase; TSH: thyroid-stimulating hormone.
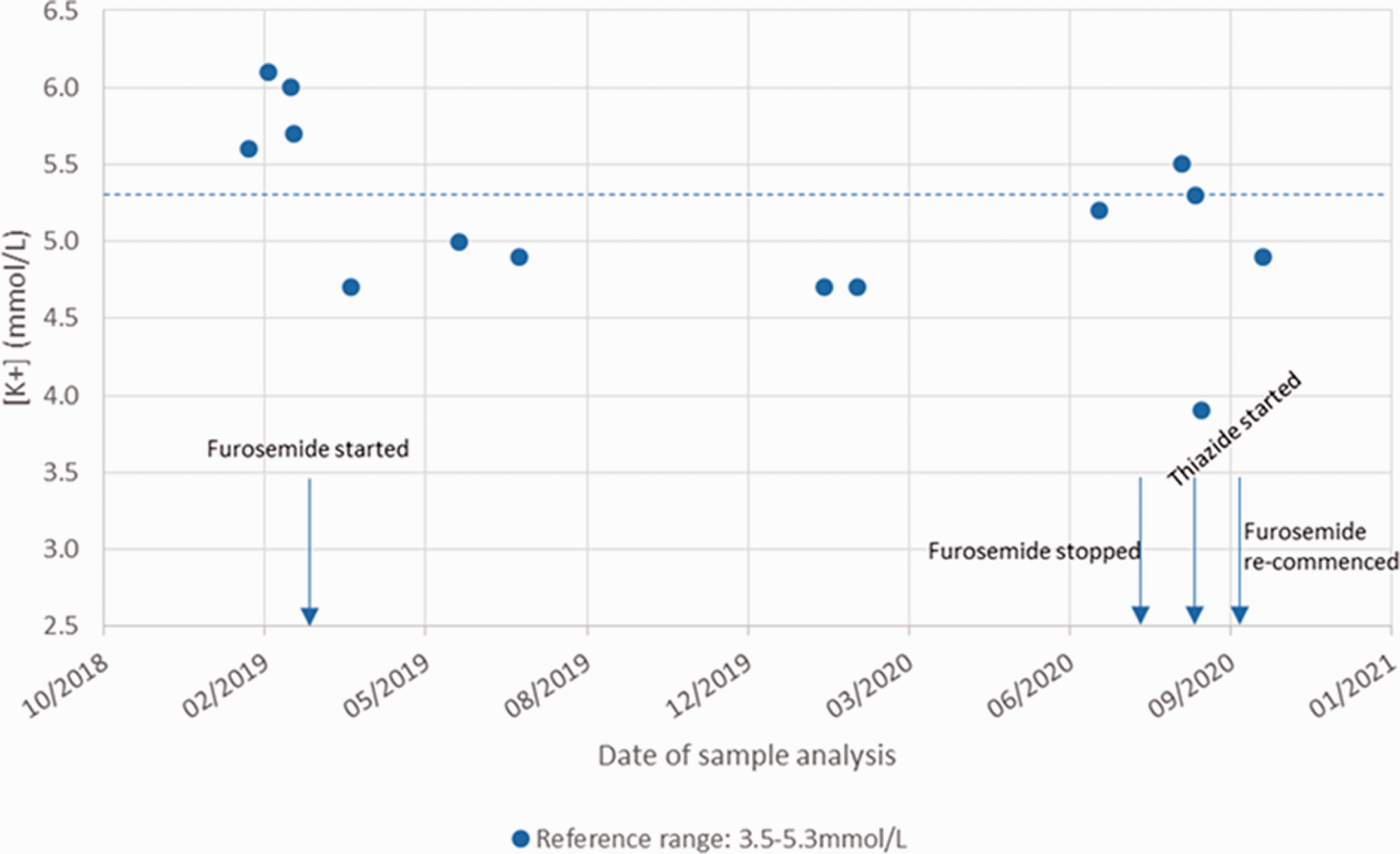
The timeline of investigations into our patient’s potassium results is illustrated. The approximate dates of diuretic therapy are also highlighted by blue arrows. The dotted horizontal blue line also indicates the upper limit of the reference range used in these investigations.
Adrenal insufficiency was ruled out on the basis of an adequate 9:00 a.m. cortisol concentration. However, renin concentration was at the low end of normal with inadequately normal aldosterone level. The patient was also found to have unremarkable liver, bone and thyroid function tests, and acute tissue damage, haemolysis and diabetes were also excluded following biochemical assessment (Table 1). A renal ultrasound also revealed normal, non-obstructed kidney and bladder physiology. Haematology investigations revealed an entirely normal full blood count and an erythropoietin concentration of 7 mU/mL, thus effectively ruling out an increased red blood cell turnover as a potential underlying cause of hyperkalaemia. The patient was also found to have a slightly elevated heart rate (85 bpm) and was normotensive (109/68 mmHg).
Interestingly, it was noted that a number of the patient’s relatives had also presented with incidences of hyperkalaemia, including her brother, eldest son and maternal aunt. No associated symptoms or diagnoses had been established in any of these cases. This therefore triggered further investigations into potential familial causes of isolated hyperkalaemia. Initially, the patient was investigated for a diagnosis of familial pseudo-hyperkalaemia. Familial pseudo-hyperkalaemia is an inherited defect in the ABCB6 gene that encodes the ATPase binding cassette protein. 10 This increases the temperature sensitivity of the mechanisms responsible for mediating intracellular potassium concentration in red blood cells. This causes an increased leakage of potassium as a result of defective ATPase activity and is solely restricted to in vitro environments and is therefore not reflective of in vivo hyperkalaemic status. 11 However, both biochemical and genetic investigations did not reveal an underlying diagnosis of familial pseudo-hyperkalaemia in our patient.
Exclusion of the aforementioned causes of hyperkalaemia coupled with a family history of persistent hyperkalaemia then prompted clinical suspicion of a pseudo-hypoaldosteronism type II diagnosis. As a result, genetic testing was initiated, which revealed a mutation in the highly conserved acidic motif of WKN1 (c.1903G>A p.(Asp635Asn)). Mutations in this motif prevents ubiquitination and hence proteasomal degradation of kidney-specific isoforms of WNK1 leading to its abundance in the distal nephron. Consequently, this results in an increase in the luminal membrane expression of NCC in the distal convoluted tubule. This provided a suitable diagnosis for this patient and sufficiently identified the cause of her isolated incidences of hyperkalaemia. Extended genetic testing has been offered to the immediate members of the patient’s family who have also presented with hyperkalaemia. However, to our knowledge, this has not yet been extended to asymptomatic first-degree relatives of the patients’ family.
Interestingly, this particular pathogenic variant is extremely rare and appears to have only been previously reported in the literature once before in a 2-year-old male patient who was also normotensive and hyperkalaemic; this patient did however demonstrate a mild acidosis.12 Subjects carrying missense mutations in the WNK1 conserved acidic motif have a clear electrolyte phenotype, but with no hypertension, especially in comparison with those who had similar WNK4 mutations.
Gordon syndrome patients are typically known to be highly sensitized to therapeutic intervention with thiazide diuretics. Thiazide diuretics are known to act through competitive inhibition of the NCC on the luminal membrane of the principal cells of the distal convoluted tubule. These treatments can therefore be used to directly counteract the effects of increased NCC expression that is associated with Gordon syndrome. 13 The patient was treated with bendroflumethiazide which was initially successful in correcting her hyperkalaemia. However, it was not well tolerated, and the patient was swapped to furosemide. The patient’s immediate family members, namely her brother and eldest son, who both presented with hyperkalaemia were able to tolerate thiazide treatment which successfully reduced their potassium concentrations to within the reference range. While furosemide is defined as a loop diuretic and does not have direct inhibitory effects on NCC activity, it does contribute to increasing potassium excretion and lowering plasma concentrations through competitive inhibition of the NKCC2 transporter found in the ascending loop of Henle. This increases the distal delivery of potassium and results in secondary mineralocorticoid excess which contributes to increasing renal potassium excretion. 14
Discussion
Gordon syndrome is an incredibly rare condition with an unknown prevalence and only around 180 individuals being reported in the literature. 13 The clinical presentation of this disorder can vary depending on the disease-causing mutation present in patients but is generally considered a diagnosis of exclusion. There are four pathogenic mutations in WNK1, WNK4, KLHL3 and CUL3 genes that can result in the clinical manifestation of Gordon syndrome. These mutations are listed in order of the severity of symptoms they elicit. While hypertension, metabolic acidosis and hyperkalaemia are considered the primary features of this condition, patients may not present with all three of these symptoms concurrently. There are also additional features that may include hypercalciuria, hypocalcaemia, decreased bone mineral density and renal calcium stones, although these are known to be associated more intrinsically with WNK4 mutations. 3
Hélène Louis-Dit-Picard et al. have recently described a small cohort of patients with missense variants all located in the WKN1 acidic motif, which is highly conserved across members of the WNK family and pivotal for their ubiquitination. While this particular pathogenic point mutation in the WNK1 acidic motif (WNK1 c.1903G>A p.(Asp635Asn)) has been previously reported in the literature before, to our knowledge this is the first detailed case report written on such a patient. 12 Although the function of the acidic motif has never been fully elucidated, it remains interesting that there is a discrepancy in phenotype depending on the WNK1 or WNK4 gene affected with similar genetic variants at the acidic motif of the kinase. Patients with heterozygote mutations in the acidic motif of WNK1 tend to be normotensive while those with similar mutations in WNK4 are hypertensive. The full understanding of the mechanisms underlying the absence of hypertension remains to be fully elucidated. The WKN1 gene gives rise to two main isoforms: a long catalytically active isoform and a short kidney-specific isoform only expressed in the distal nephron. Mutations in the WKN1 acidic motif selectively abolishes ubiquitination and degradation of the kidney-specific WKN1 isoform suggesting that this is the mechanism deriving altered potassium metabolism and aberrant activation of NCC. The full understanding of the mechanisms underlying the absence of hypertension in patients with WKN1 acid motif mutations remains to be identified.
Incidences of isolated hyperkalaemia coupled with no associated pathological symptoms can be indicative of the numerous sources of pseudo-hyperkalaemia. However, when investigations such as these do not provide a satisfactory cause of such a finding, it can often be difficult to ascertain an exact diagnosis for patients. Spot urine potassium measurement from hyperkalaemic individuals can be useful primarily to aid in diagnosis. In Gordon syndrome it is expected that urine potassium concentrations would be abnormally low, due to the reduced capacity of renal excretion of potassium. Unfortunately, this test was not performed for our patient.
This case report highlights a rare genetic cause of isolated hyperkalaemia that manifests in the form of Gordon syndrome, that when identified can be corrected with the use of thiazide diuretic treatment. However, due to the rarity of this condition Gordon syndrome can easily be overlooked as the precipitating cause of a patient’s hyperkalaemia, and there is seemingly a lack of appropriate clinical guidance available to sufficiently address how to approach the investigation and treatment of this disease. For example, the most recent clinical practice guidelines for the treatment of hyperkalaemia in adults, published by the Renal Association, did not include Gordon syndrome as one of the possible causes of chronic hypokalaemia. 15 The case presented in this report highlights that while Gordon syndrome is a very rare disorder, the symptoms it causes can present with varied severity, which can be particularly alarming in the context of acute and unexplained hyperkalaemia. The lack of awareness of such a syndrome as a result of its rarity can therefore prolong the time to diagnosis, use inappropriate treatment for patients and take up considerable resources through the investigation of unrelated pathological causes. It is hoped that this report can serve as a useful source of information to increase awareness around this condition and to improve the way in which rare causes of hyperkalaemia are investigated and diagnosed.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not required. Written informed patient consent has been obtained for publication of this case report.
Guarantor
SZ.
Contributorship
TL prepared the first draft of the text. All authors reviewed and edited the article and approved the final version.