
Abstract
Background
The analysis of steroids in biological matrices is challenging. One can apply immunoassay as well as gas and liquid chromatography with various types of detection, depending on the available equipment and the experience of the analyst. The question is how the methods are interchangeable between themselves. Doubts were reported having compared immunoassays and chromatography-mass spectrometry, but there are scarce data on chromatographic methods with detection types other than mass spectrometry.
Methods
Here, we present the detailed comparison of two liquid chromatographic methods for the determination of free urinary cortisol and cortisone: one with fluorescence detection (high-performance liquid chromatography [HPLC-FLD]) and the other with tandem mass spectrometry (HPLC-MS/MS). The comparison was made with 199 human urine samples. The data analysis included Passing–Bablok and Deming regression, Bland–Altman test, Wilcoxon test, mountain plot and Lin’s concordance correlation coefficient.
Results
The validation data indicated that both methods met the requirements of the European Medicines Agency. However, the statistical analysis revealed the systematic bias between the two assays. The Passing–Bablok and the Deming tests showed that the HPLC-FLD method overestimated results for cortisol and underestimated measurements for cortisone. The Bland–Altman analysis estimated the mean differences between the methods: 18.8 nmol/L for cortisol and −16.9 nmol/L for cortisone measurement.
Conclusions
Both methods’ results led to the same conclusion in observational studies, but the techniques are not interchangeable. The literature data, the observations from the clinical setting and our experience clearly indicate that the future of steroid measurements will belong to chromatography coupled with mass spectrometry.
Introduction
The analysis of steroids in biological fluids is quite challenging. On the one hand, we need the commercially available, simple methods which could be used in everyday clinical practice. On the other hand, the scientists require highly specific and sensitive methods to be applied in research focusing on the analysis of complex hormonal profiles in various biological matrices. Presently, the immunoassays (IAs) still dominate in clinical laboratories, despite their recognized disadvantages: potentially low specificity, cross-reactivity, hard-to-control matrix effects, ability to test only one compound at a time. This is because of IAs’ relatively low price, technical ease and no need for highly specialized equipment. The research laboratories focus rather on chromatographic methods, which allow separating the compounds of interest from the other biological components with resembling structure. The result is the ability to determine multiple substances in one analytical run. 1
Chromatographic methods have been widely used in the analysis of steroids for many years. Initial methods were based on paper and thin-layer systems, then the gas chromatography (GC) and the high-performance liquid chromatography (HPLC) were introduced. 2 Continuously, the application of the ultra high performance liquid chromatography enabled the better resolution and provided with advantage in terms of time-saving and increased laboratory throughput. The pivot of chromatographic bioanalytical methods is the system of detection. Less specific detectors (flame ionization detector,3–5 ultraviolet – UV,6–8 fluorescence detection – FLD 9 ) require very scrupulous sample preparation and complete chromatographic resolution of analytes to fulfil the validation criteria and ensure the reliable results. More specific ones, like mass spectrometry (MS), enable one to distinguish and simultaneously determine related compounds. Moreover, one has to realize that some methods require the process of derivatization. GC needs the transformation of steroid compounds into derivatives (most often via silylation) to increase their volatility.3–5,10–12 HPLC-FLD requires the chemical modification of steroids to obtain the fluorescent compounds.9,13
With such a wide range of analytical possibilities, the question is how comparable the different methods are. Here, we present the statistical comparison of two methods for cortisol (F) and cortisone (E) determination, both based on HPLC, but with different systems of detection: FLD and MS/MS. The analysis was performed on 199 urine samples, which provides a sufficient number of data for the convincing and reliable statistical comparison.
Materials and methods
Chemicals and reagents
Standards of F and E, prednisolone and hydrocortisone-9,11,12,12-D4 (F-D4), as well as ⩾95% formic acid, were purchased in Sigma-Aldrich (St. Louis, USA). The same company also provided quinuclidine and 9-anthroyl nitrile (9-AN). Organic solvents: acetone, acetonitrile (ACN), dichloromethane (DCM), n-hexane, methanol, all at least HPLC grade, were purchased from Merck (Darmstadt, Germany). Potassium dihydrogen phosphate (Xenon, Łódź, Poland), disodium hydrogen phosphate anhydrous and ≥85% phosphoric acid (both from Fluka Chemie, Buchs, Switzerland) were used to prepare buffers and mobile phase for the HPLC-FLD method. Ultrapure water was obtained using the Simplicity UV system (Merck-Millipore, Darmstadt, Germany), while the mobile phase for HPLC-MS/MS analysis was prepared with HPLC-MS-grade water from Merck (Darmstadt, Germany) or J.T.Baker (Deventer, The Netherlands). The details concerning the preparation of all reagents and solutions were presented previously.9,14,15
Biological samples
The urine samples were obtained from 211 women in the third trimester of pregnancy. The pregnant women were admitted to Gynecology and Obstetrics Clinical Hospital of Poznan University of Medical Sciences and recruited to participate in a research project concerning the activity of 11β-hydroxysteroid dehydrogenase 2 in hypertensive disorders of pregnancy. 15 The study was approved by the local Bioethical Committee, and the informed consent was obtained from all study participants. Each participant collected the urine for 24 h, then the total volume was noted, and the aliquot of 5 mL was taken for the analysis.
HPLC-FLD
Equipment and conditions
The HPLC-FLD method was based on the method previously developed by our research group 9 with some additional modifications. Those included the use of Kinetex XB-C18 column (100 × 4.6 mm; 5 μm) guarded by a pre-column SecurityGuard ULTRA C18 (4.6 mm; 5 μm), both from Phenomenex (Torrance, USA). The column temperature was held at 60°C. The analyses were performed on HPLC apparatus HP 1100 (Hewlett-Packard, USA) using the fluorescence detector (G1321A) with the wavelengths of excitation and emission set at 360 and 460 nm, respectively. The mobile phase consisted of ACN and 0.3 mM orthophosphoric acid (470: 530; v/v), and its flow rate was fixed at 2 mL/min. The injection volume was 50 μL.
Preparation of samples
The preparation of the biological samples comprised three steps: liquid–liquid extraction, derivatization with the use of 9-AN and the purification of the samples by the solid-phase extraction (SPE) with C18 cartridges, as described in details previously. 9 Shortly, 0.5 mL of urine was mixed with 1 mL of phosphate buffer (pH 7.5); prednisolone solution (internal standard, IS); ACN (replacing standard solutions in quality control samples) and 4 mL of DCM and shaken for 10 min. The mixture was then cooled and centrifuged. The organic layer was collected and dried under the gentle stream of nitrogen at 40°C. The dried residue was dissolved in 9-AN (150 μL) and then a mixture of triethylamine and quinuclidine (100 μL; 1:1, v/v). Next, it was stored for 30 min in water-free, dark conditions at room temperature to obtain fluorescent derivatives of F and E. The liquid solvents were again evaporated (at 30°C), and the dry residue was dissolved in ACN/water mixture (1:4, v/v) and transferred to SPE cartridges. The analytes were purified by rinsing the cartridges with water and a mixture of n-hexane and DCM, and then eluted with acetone. The eluate was evaporated to dryness at 40°C. The residue was dissolved in ACN and mobile phase, and the solution was injected into HPLC system.
HPLC-MS/MS
Equipment and conditions
The details were presented previously. 14 Briefly, the analysis was carried out in the Shimadzu system (Kyoto, Japan). It comprised the Nexera chromatograph interfaced to a triple quadrupole mass spectrometer LCMS-8030, equipped with electrospray ionization (ESI). The separation was accomplished using the Kinetex XB-C18 column (2.1 × 100 mm; 2.6 μm) from Phenomenex (Torrance, USA), and isocratic elution with the mobile phase (0.1% formic acid in H2O: 0.1% formic acid in ACN; 762:238, v/v) pumped with a flow rate of 0.30 mL/min. The desolvation line and the heat block were maintained at 250°C and 400°C, respectively. Nitrogen was used as both the nebulizing gas (2 L/min) and the drying gas (15 L/min). Argon served as the collision gas (pressure 230 kPa). The electrospray needle voltage was 4.5 kV. The MS/MS detection processed in multiple reaction monitoring mode, as presented in detail elsewhere. 14
Preparation of samples
The urine (1 mL) was mixed with 2 mL of phosphate buffer (pH 7.5), spiked with F-D4 (IS) and pure ACN (analytes’ solution in ACN was used in calibration samples), and extracted with 4 mL of DCM. The organic layer was then collected and evaporated to dryness at 40°C. The residue was dissolved in 80 μL of methanol and injected into the chromatographic column (please see Kosicka et al. 14 for details).
Statistics
Statistical analysis was carried out using the MedCalc software v17.9.7 (Ostend, Belgium). Whenever applicable, a P-value of <0.05 was considered significant. The distribution of data was always assessed with Shapiro-Wilk test. Before the analyses, cases with the results below the lower limit of quantitation (LLOQ) of any method were excluded. Such a procedure allowed to avoid a potential bias associated with different limits of quantification. Finally, comparison of the methods was performed on the results obtained from the analysis of 199 urine samples.
The Bland–Altman analysis 16 was performed to study the mean bias between the HPLC-FLD and HPLC-MS/MS measurements and to construct the limits of agreement (mean bias ± 1.96 of standard deviation; SD) with their respective confidence intervals (CIs). This technique involved constructing a graph with the absolute difference between the two paired measurements plotted against their mean value.
Subsequently, the Passing–Bablok regression 17 and the Deming regression 18 enabled estimating if there was a constant bias between the two assays. Their plots expressed the relationship between the concentrations of compounds determined by HPLC-FLD and HPLC-MS/MS method. The regression lines’ slopes and intercepts with their 95% CIs were calculated. If 95% CI for the slope contained 1 and the 95% CI for the intercept contained 0, neither proportional nor systematic differences between the two methods were recognized. The CV of the measurements, required for the Deming regression, was set at 15%, which is the highest accepted value in the European Medicines Agency (EMA) validation criteria. 19 The Passing–Bablok regression required no specific assumptions regarding the measurement errors or distribution of the samples. Therefore, it was more suitable concerning the lack of normal distribution. However, the Passing–Bablok test demands the linear relationship between the paired observations, which was confirmed with the Cusum test.
The next step was the comparison of measurements obtained by both methods using a test for paired samples. As the variables did not follow the normal distribution, the Wilcoxon test was applied.
Additionally, the agreement between the continuous variables was assessed by calculating the Lin’s concordance correlation coefficient. 20 This parameter evaluates the degree to which the paired measurements conform to a line of equality (45° line through the origin). The estimated coefficient is based on the Pearson’s correlation coefficient (a measure of precision) and a bias correction factor (a measure of accuracy). The value of the concordance correlation coefficient indicates the strength of agreement as: almost perfect (>0.99), substantial (0.95–0.99), moderate (0.90–0.95) or poor (<0.90). Additionally, the mountain plots 21 were constructed to visualize the distribution of differences better. If two assays are unbiased with respect to each other, the scatter plots should be centered over zero.
Results and discussion
Validation of methods
The most important validation parameters of the methods are presented in Table 1. Both HPLC-FLD and HPLC-MS/MS methods were validated and met the requirements contained in the EMA guidelines. 19 Briefly, the HPLC-MS/MS method is characterized by much shorter retention time of the analytes and is more sensitive than the HPLC-FLD. Using the two-fold higher volume of urine than the HPLC-FLD (1.0 vs. 0.5 mL), the HPLC-MS/MS provided a 10-fold lower value of LLOQ.
Comparison of the validation parameters.
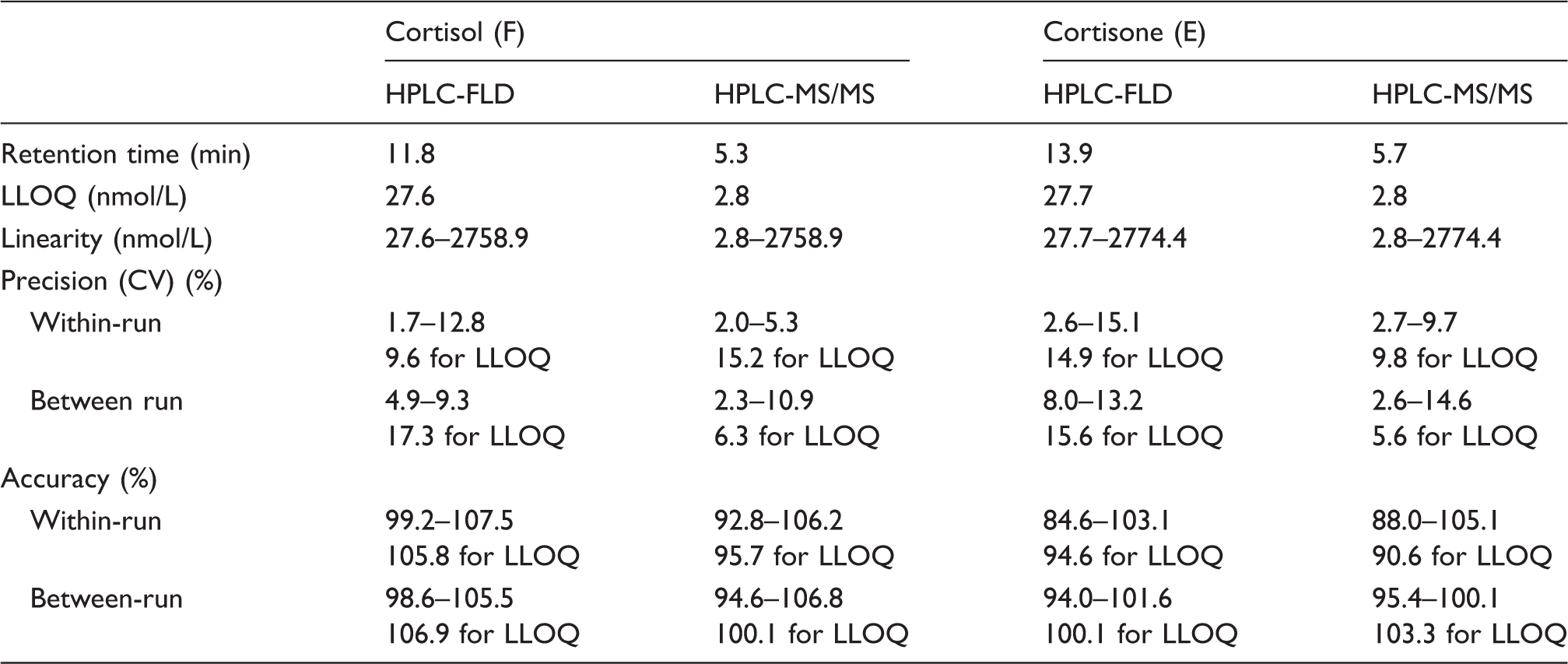
LLOQ: lower limit of quantitation; HPLC-FLD: high-performance liquid chromatography-fluorescence detection; HPLC-MS/MS: high-performance liquid chromatography-tandem mass spectrometry.
Comparison of methods
The Shapiro-Wilk test did not confirm that the differences between the measurements nor their log-transformations follow the normal distribution. Therefore, we decided to use the original data for further analyses.
The Bland–Altman analysis demonstrated a positive systematic error for F and a negative one for E (Figure 1(a) and (b)) between the HPLC-FLD measurements referenced to the HPLC-MS/MS ones, but no signs of a proportional error. The mean differences between the methods were 18.8 nmol/L and −16.9 nmol/L, respectively, and their 95% CIs did not cover the line of equality indicating significant biases between the two assays.
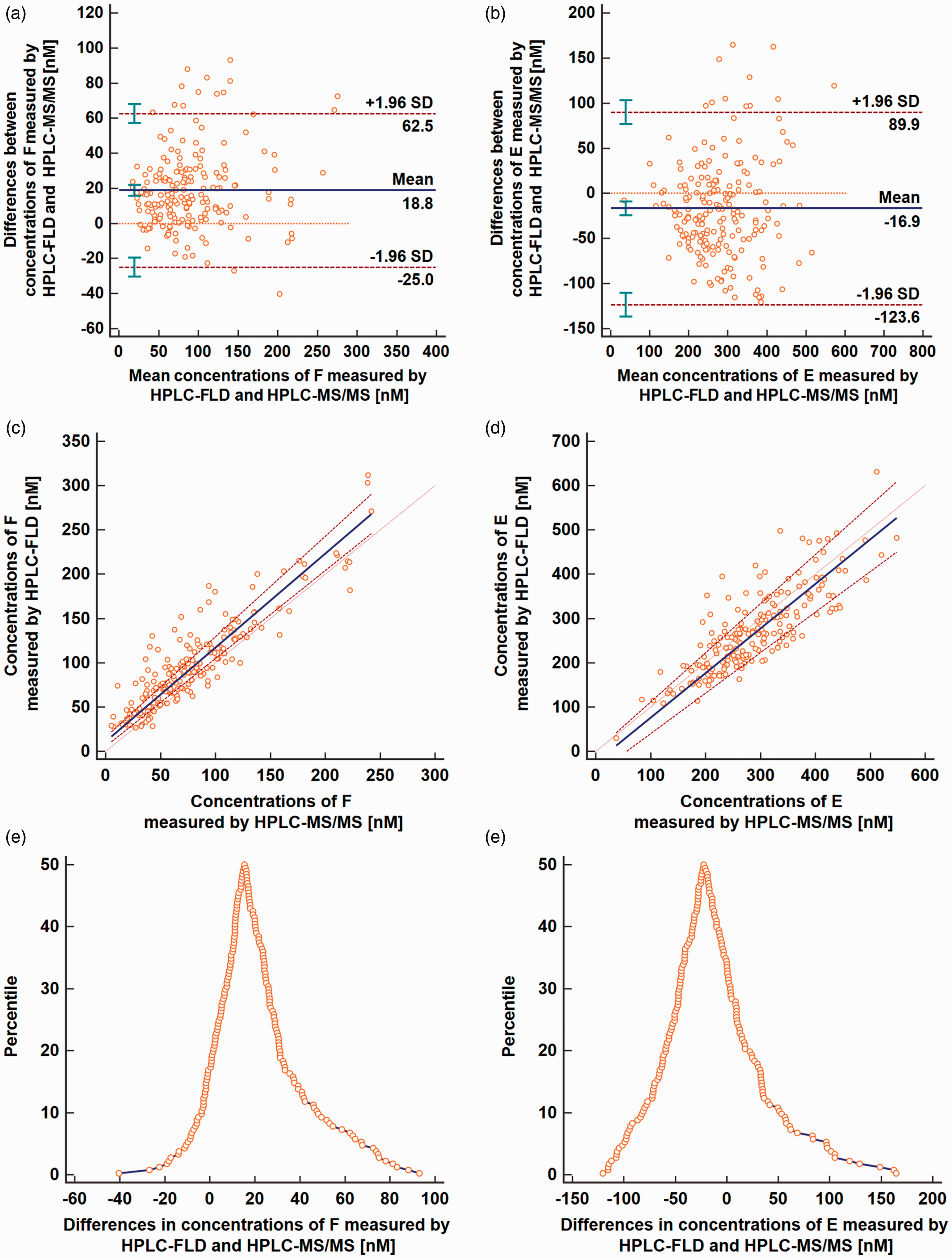
Results from the Bland–Altman analysis (a and b), the Passing–Bablok regression (c and d) and the mountain plots (e and f) for cortisol and cortisone; a and b: mean (horizontal solid bold line), LoA (horizontal dashed lines) and line of equality (horizontal solid thin line); c and d: the Passing–Bablok regression (solid bold line), its 95% CI (dashed bold lines) and identity line (dotted thin line).HPLC-FLD: high-performance liquid chromatography-fluorescence detection; HPLC-MS/MS: high-performance liquid chromatography-tandem mass spectrometry.
The Passing–Bablok and the Deming models of regression are applicable when a linear model fits the data. The Cusum test for linearity did not show significant deviation from linearity either for F (P = 0.90) or for E (P = 0.56). Both the Passing–Bablok and the Deming models showed similar results. No proportional differences were observed because the 95% CIs for the slopes contained the value 1 for all analyses (Table 2). A systematic error was found between the assays (Figure 1(c) and (d)) which confirmed the results of the Bland–Altman analysis. The HPLC-FLD method overestimated results for F (the calculated intercepts and their 95% CI from both studies were above 0) and underestimated measurements for E (the intercepts each time were lower than 0) when compared to the HPLC-MS/MS method.
Results from the analyses comparing HPLC-FLD and HPLC-MS/MS method for determination of cortisol and cortisone in human urine.
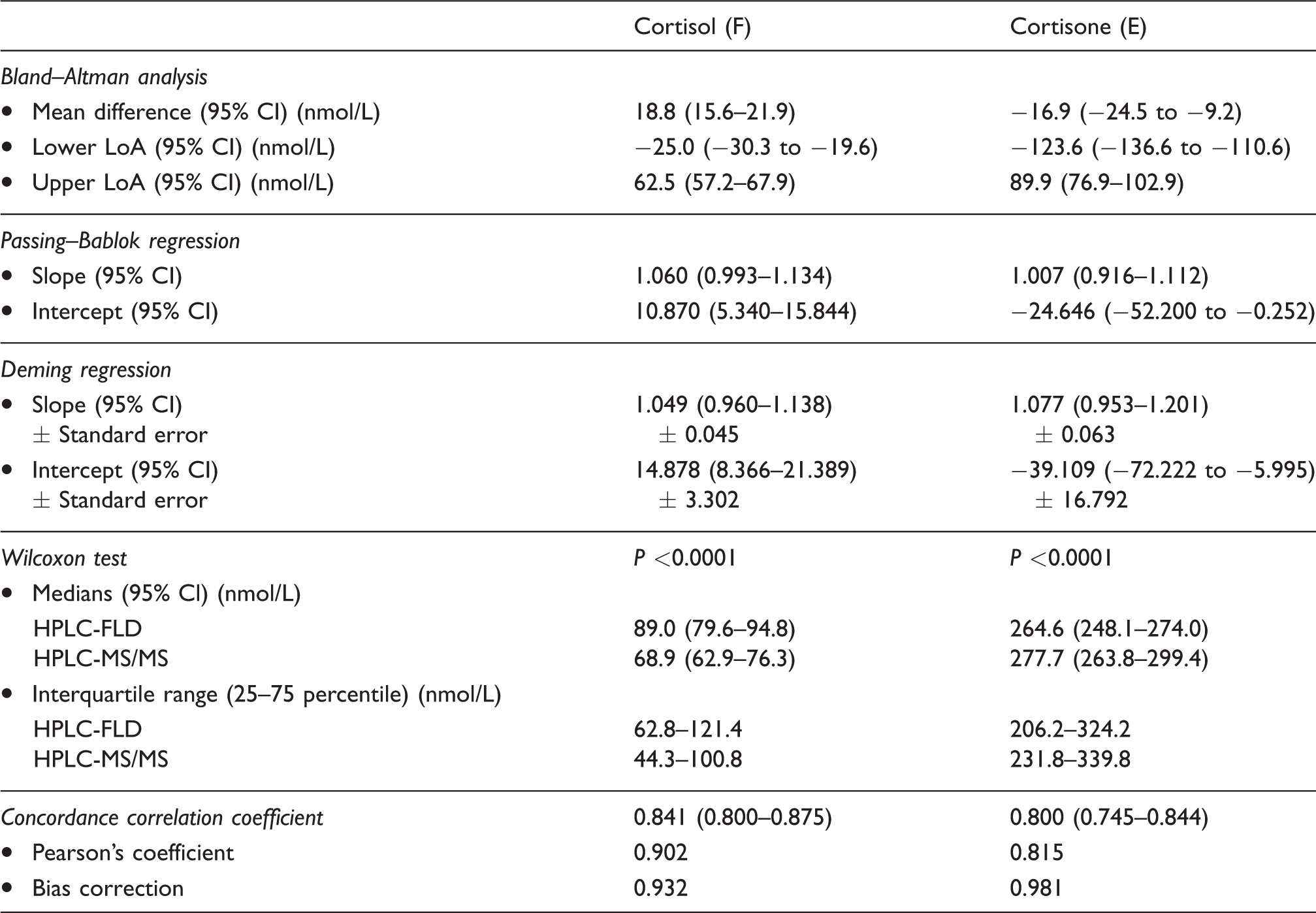
LoA: limit of agreement; CI: confidence interval; HPLC-FLD: high-performance liquid chromatography-fluorescence detection; HPLC-MS/MS: high-performance liquid chromatography-tandem mass spectrometry.
The assessment of the mountain plots (Figure 1(e) and (f)) revealed that the mean difference between HPLC-FLD and HPLC-MS/MS was about 15.4 nmol/L for F and −22.5 nmol/L for E.
The existence of a systematic error in the analyses mentioned above explains the differences revealed by the Wilcoxon test (Table 2). It showed that the concentrations of F determined by HPLC-FLD were significantly higher than those by HPLC-MS/MS (89.0 vs. 68.9 nmol/L, P < 0.0001), and the concentrations of E were significantly lower (264.6 vs. 277.7 nmol/L, P < 0.0001).
The calculated Lin’s concordance correlation coefficients were 0.8412 for F and 0.7999 for E with respective Pearson’s coefficients 0.9025 and 0.8152. These results indicate that despite powerful positive correlations between the measurements (Pearson’s coefficient >0.80), the agreement between those two assays was relatively weak (Lin’s concordance correlation coefficient <0.90). The phenomenon could be explained by the fact that the Pearson’s coefficient explains only a linear association between variables and not the actual compatibility between assays. Therefore, it is not advised as an independent tool in method comparison studies. 22
Summary
The literature data on the compatibility of bioanalytical methods are equivocal.23–28 Usually, a trend toward higher F concentrations in biological matrices was observed in IAs as compared to MS/MS assays,27,28 especially in patients undergoing metyrapone treatment. 26 This phenomenon is explained by the cross-reactivity of antibodies against F with structurally related compounds. Moreover, when IAs methods were involved, the F assay’s bias was reported to be affected by patient gender, and different biological matrix components, which resulted from various clinical conditions of the patients. 29 Such interferences are more natural to avoid in HPLC and GC methods owing to extraction, the chromatographic resolution itself and a system of detection. On the other hand, an excellent agreement in urinary F measurements by GC-MS and HPLC-MS/MS was previously reported. 28 Such high compatibility of the chromatographic methods is hardly achievable between MS and other types of detection. The presented results indicate that the compared HPLC-FLD and HPLC-MS/MS methods for determination of F and E in urine are not interchangeable. They both fulfilled the criteria for analytical methods established by EMA, 19 but the systematic biases exist between the assays. They resulted in overestimating concentrations for F and underestimating values for E in HPLC-FLD as compared with HPLC-MS/MS. The possible hydrolysis of the steroid conjugates in the analytical process needs to be addressed. It has to be emphasized though that in the analysis of glucocorticoid balance in pregnant women with hypertension, we came to the same conclusions when applying HPLC-FLD 15 and when repeating and extending the analyses using the HPLC-MS/MS. 30 The value of urinary free cortisol (UFF) should be lower than the urinary free cortisone (UFE) in healthy humans. The UFF/UFE ratio usually is close to 0.5 and should not be higher than 0.8.31,32 Higher values indicate dysfunction of the 11β-hydroxysteroid dehydrogenase 2 which converts cortisol to cortisone in the kidneys and in placenta. 33 Lower values were observed in our project involving women in the third trimester of pregnancy. In the cohort, the metabolism of cortisol was increased and the levels of UFF were from 3 (in normotensive controls) to 5 (hypertensive patients) times higher than those of UFE. 30 Therefore, our experience shows that both HPLC-FLD 9 and HPLC-MS/MS 14 methods are suitable for the observational study of glucocorticoid balance in hypertensive pregnant women. However, the assays cannot be compared between themselves because they are not in total agreement. There are doubtless advantages of the HPLC-MS/MS methods (specificity, lower LLOQ, ease of samples preparation), which make it the method of choice for our further research. In conclusion, all of the above makes it quite obvious that the MS measurements will defeat the opponents and be the future method of choice in the steroid analysis.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research was supported by the National Science Center (Narodowe Centrum Nauki) in Poland (grant number 2012/05/B/NZ7/02532).
Ethical approval
The study was approved by the Bioethical Committee (approval nos 954/11 and 991/15) at Poznan University of Medical Sciences.
Guarantor
KK, AS, FKG.
Contributorship
AS and KK have equally contributed to the paper. KK conceived the study, performed the analyses, analysed the data and wrote the first draft of the manuscript; AS performed the analyses, analysed the data and wrote the first draft of the manuscript; ASG was involved in protocol development, patient recruitment and data analysis; MK was involved in protocol development, patient recruitment and data analysis; GB was involved in protocol development and supervised the clinical aspects of the study; FK was involved in protocol development and supervised the analytical aspects of the study. All authors reviewed and edited the manuscript and approved the final version of the manuscript.