
Abstract
A 21-year-old female had recurrent presentations to the emergency department with myalgia, vomiting, abdominal pain and subsequently developed generalized seizures. She was volume depleted with a plasma sodium of 125 mmol/L (reference interval: 135–145) and she had fluctuating hypertension. Acute porphyria was suspected and confirmed with raised urine porphobilinogen/creatinine ratio of 12:4 μmol/mmoL (reference interval < 1:5) and she was treated with intravenous haem arginate. Urinary porphyrin/creatinine ratio was 673 nmol/mmoL (reference interval <35) and faecal porphyrins 2430 μmol/kg dry weight (reference interval: <200) were markedly elevated, with raised faecal CIII:CI ratio, consistent with acute coproporphyria. Diagnosis was confirmed by the demonstration of a novel missense variant in the coproporphyrinogen oxidase gene c.863T > G (p.Leu288Trp) predicted to be deleterious and which segregated with three other affected family members. Although CT head was normal, magnetic resonance imaging scan revealed symmetrical signal abnormalities and swelling in the parietal and occipital lobes consistent with posterior reversible encephalopathy. Over several days, her seizures ceased and sodium and blood pressure normalized. The aetiology of the acute porphyric attack was likely multifactorial with contributions from a recent viral illness and caloric deprivation. No drug precipitant was identified. We postulate that untreated hypertension played a key role in the development of posterior reversible encephalopathy. Early clinical suspicion and urine porphobilinogen testing are the key components in preventing morbidity and mortality in acute porphyrias.
Introduction
Porphyrias are rare disorders of haem synthesis resulting from enzymatic deficiencies and characterized by the accumulation of porphyrins and their precursors. Unfortunately, delayed diagnosis is common with a mean lag time between symptoms and diagnosis of 15 years. 1 However, the diagnosis of acute porphyria can be rapidly established by measuring urinary porphobilinogen (PBG), and hence clinical awareness remains the key rate limiting step in effective management. Acute attacks should be treated with intravenous (IV) haem arginate, which can reduce morbidity and mortality, and all porphyrinogenic drugs should be avoided.2,3
Posterior reversible encephalopathy syndrome (PRES) is a disorder of usually reversible subcortical vasogenic brain oedema characterized by a myriad of neurological features which can include seizures and impaired consciousness. 4 In rare cases, PRES can be life-threatening. We present a case of PRES in hereditary coproporphyria (HCP) and propose that this complication may have been contributory to seizures and could have been avoided if early diagnosis had been made.
Case report
A 21-year-old female presented to the emergency department (ED) with a two-day history of myalgia, vomiting and generalized abdominal pain. Medical history included one unexplained seizure as a child and an ED presentation with presumptive viral gastroenteritis. There was no history of photosensitivity. She took no regular medications but had an intrauterine MIRENA placed five years prior to presentation. Examination revealed generalized tenderness only. She was diagnosed with a viral illness and discharged home.
Two days later, she re-presented to the ED with ongoing symptoms. She also described lower back pain that radiated to her thighs. Examination was unremarkable. Urine was negative for infection. Inflammatory markers and blood biochemistry were normal. Her symptoms were again attributed to a viral illness and she was discharged.
Three days later, she presented for a third time with ongoing myalgia and vomiting. Her abdominal pain had increased in severity, but examination of the abdomen remained unremarkable. She was clinically volume depleted with low jugular venous pressure, poor skin turgor and dry mucous membranes (there was no evidence of acute kidney injury but creatinine was 75 μmol/L compared with 50 μmol/L at baseline). She had a hyponatraemia of 125 mmol/L (reference interval: 135–145). She had low serum osmolality (249 mmol/kg), high urine osmolality (879 mmol/kg) but high urine sodium (137 mmol/L). Inflammatory markers, liver function and amylase were within normal limits. Plain radiographs of the chest and abdomen were normal.
She was referred to Internal Medicine but prior to transfer to a medical ward, she developed generalized seizures. At this juncture, porphyria was considered to be a possible unifying diagnosis. She was administered lorazepam and levetirectam and transferred to the intensive care unit (ICU). Computed tomography (CT) of the head and abdomen were normal. She had fluctuating hypertension in the ED and ICU (systolic blood pressure: 141–171 mmHg; diastolic blood pressure: 81–125 mmHg).
Raised urine PBG/creatinine ratio of 12:4 μmol/mmol (reference interval < 1:5) confirmed an acute porphyria, and IV haem arginate was subsequently commenced. The urine sample was collected on day 7 (from the onset of the illness). Urinary porphyrin/creatinine ratio (673 nmol/mmoL, reference interval <35) and faecal porphyrins (2430 μmol/kg dry weight, reference interval <200) were markedly elevated with metabolite profiles showing raised faecal CIII:CI ratio which was consistent with acute HCP. A novel missense variant in the coproporphyrinogen oxidase (CPOX) gene c.863T > G (p.Leu288Trp) predicted to be deleterious by in silico analysis was subsequently found. Her seizures ceased and her sodium and blood pressure normalized. She had a delirium that improved over three days. There was no report of headache or visual disturbance.
She received a further course of IV haem arginate for ongoing mild abdominal pain on the medical ward. A Montreal cognitive assessment (MOCA) showed unexplained significant impairment at 18/30. Magnetic resonance imaging (MRI) scan revealed symmetrical signal abnormalities and swelling in the parietal and occipital lobes consistent with PRES (Figure 1). Repeat MOCA four weeks later was 26/30 and repeat MRI scan six weeks later showed improvement consistent with PRES.
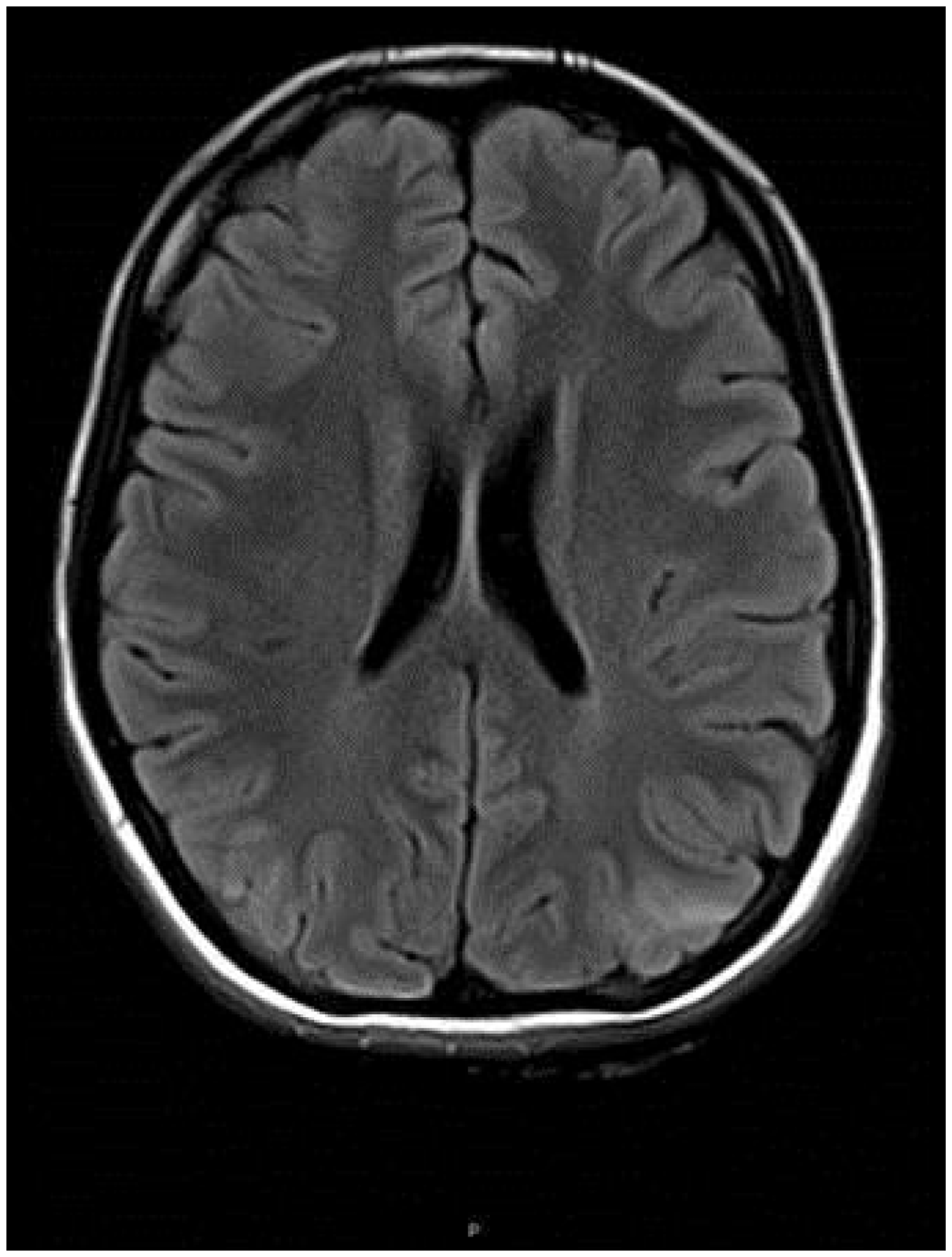
Parieto-occipital swelling showing multiple non-enhancing white matter lesions, consistent with PRES.
The aetiology of this acute porphyria attack was likely multifactorial with potential contributions from an initial viral illness, secondary caloric deprivation, dehydration and hyponatremia. No drug precipitant has been identified. We postulate that untreated hypertension played a key role in the development of PRES. Our patient has had one further mild attack.
Cascade family testing has so far identified three other family members with the mutation. Her sister has had several confirmed acute attacks characterized by pain in the abdomen, back and peripheries. Her mother has also had one confirmed acute attack. A maternal female cousin has had two acute attacks precipitated by caloric deprivation.
Discussion
HCP is the rarest of the three autosomal dominant acute porphyrias and is characterized by deficient activity of the mitochondrial enzyme CPOX. 5 It is inherited in an autosomal dominant manner and has low penetrance; approximately 90% of affected individuals never experience an acute attack. 6 This family is therefore highly unusual in that all of those who have been shown to have inherited the mutation have had documented acute attacks (confirmed by raised PBG and urinary porphyrins in the setting of acute neurovisceral symptoms); the novel mutation currently has 100% penetrance.
The clinical presentation of HCP can vary, but acute symptoms are typically neurovisceral. 2 Severe abdominal pain is the most frequent symptom although back and extremity pain can coexist. Neurological features include neuropathies and seizures. Photosensitive skin changes and psychiatric symptoms can occur in HCP but are less common. 2 Quiescent phases of the disease are interspersed with attacks which are usually precipitated by medication, inadequate caloric intake, intercurrent illness or hormonal changes. 2
Hyponatraemia frequently complicates acute attacks, with features often attributed to the syndrome of inappropriate anti diuretic hormone secretion (SIADH). SIADH, however, is a diagnosis of exclusion. Criteria to confirm SIADH include low serum sodium, high urine osmolality relative to plasma and high urine sodium. There should also be no evidence of hypovolaemia and normal pituitary, adrenal, thyroid, cardiac and liver function. The patient was clinically volume depleted, thus SIADH could not be substantiated. Hyponatraemia in this patient was likely multifactorial, with contributory factors including sodium loss related to vomiting and replacement with salt-poor fluid due to poor oral intake. The urine was also collected after the patient had started IV fluid. The patient, however, was on codeine and paracetamol which would increase antidiuretic hormone (ADH) secretion and potentiate ADH action, respectively.
Urinary PBG can quickly confirm an acute attack and hence timely availability of this test is crucial to establish the diagnosis of acute porphyria. It is recommended in best practice guidelines that urgent, quantitative PBG testing should be available within 24 h. 7
In our patient, the urinary PBG on day 7 was unequivocally elevated but to a lesser degree than is considered usual in acute attacks where PBG elevations in the order of 10 times the upper limit of normal are commonly found. 8 However, the extent of PBG elevation to confirm an acute attack is not well defined and there may be significant differences in PBG elevation between AIP and HCP. In a cohort of 260 AIP and 25 HCP mutation-positive patients, the median PBG of the AIP group was 2.5-fold higher than the HCP group. 9
The mechanism causing the elevation of PBG in AIP and HCP might provide an explanation for this difference. In AIP, the defect is at hydroxymethylbilane synthase which is the immediate enzyme converting PBG to hydroxymethylbilane. Hence, accumulation of PBG in AIP is significant. In contrast, the enzyme defect of HCP is more downstream in the haem synthesis pathway where coproporphyrinogen III is accumulated due to the defective CPOX. In essence, the mechanism leading to PBG elevation in HCP is later and more indirect than in AIP. Observations have also shown that the PBG is often negative in the latent phase of HCP and elevation may also be more transient when compared with AIP.7,8,10 A 2-fold increase from baseline may be enough to indicate an acute exacerbation in the HCP subtype.11,12
Quantitative analysis of porphyrins, with metabolite profiling in urine and faeces, is required to distinguish the three types of acute porphyrias. Genotyping confirms the diagnosis and enables cascade family screening. Genetic testing of the proband, and their families, is essential to identify pre-symptomatic carriers and prevent overt disease. Prognosis is good if the condition is identified early.
PRES is a clinico-radiological diagnosis with MRI typically showing bilateral subcortical oedema in the parieto-occipital region. 4 PRES is associated with a wide range of diseases, conditions and medications, but the pathophysiology remains incompletely understood.13–15 One central theory is that rapidly rising or fluctuating blood pressure may compromise cerebral blood flow autoregulation leading to hyperperfusion and endothelial damage. 4 The subsequent injury to the blood–brain barrier allows the interstitial extravasation of plasma and molecules with posterior brain regions potentially more vulnerable due to relatively poor sympathetic innervation. 4
Our patient was noted to have fluctuating hypertension, and hence it is plausible that this contributed to her PRES. Hyponatremia can also cause fluid shifts within the brain, but this is usually a consequence of acute (<48 h) hyponatremia. 16 In a chronic setting (>48 h), the effect is likely to be mild or absent. It is unclear whether seizures were secondary to PRES or a direct consequence of the acute porphyric attack in this case. There has been one other reported case of PRES in HCP, which also occurred in the context of hypertension. 17 However, this patient had no stool metabolite testing to confirm HCP and no CPOX gene mutation analysis. She also had unexplained very high inflammatory markers which do not usually occur in acute porphyria.
Treatment of PRES is supportive and focuses on identification and removal of the underlying precipitant. Delayed recognition can lead to cerebral ischemia, haemorrhage or herniation resulting in permanent neurological damage, and even death. 14 Novel treatment modalities for acute porphyrias include small interfering RNA, targeted at the enzyme ALA synthase, which may offer promise for the long-term control of these disorders. 18
This case highlights the importance of early diagnosis in acute porphyrias. An inexpensive and rapid screening test is available (urinary PBG) and prompt treatment can prevent complications. It is also possible that early treatment of hypertension may prevent or mitigate the development of PRES, but this remains speculative. Regardless, the role of early clinical suspicion and screening by physicians is integral to the prevention of complications. It is possible that PRES and a prolonged ICU stay could have been avoided in this young woman were porphyria considered during her first presentation to the ED. Given that urinary PBG is a relatively cheap and non-invasive test, we recommend a low threshold for including it in the work-up of unexplained abdominal pain especially in those with recurrent presentations. This may, however, have resource implications for detection of an otherwise rare condition.
Footnotes
Acknowledgements
We would like to thank Dr John Denton for providing the MRI images.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not required. The patient in the case report has provided written consent.
Guarantor
CT.
Contributorship
CT was responsible for the medical management of the case and conceived the case report. DL and CT performed the literature review and DL wrote the first draft. WKS, CS, AR and CF performed the diagnostic investigations, provided the final text pertaining to those investigations and provided the DNA sequence image. All authors reviewed and edited the manuscript and approved the final version of the manuscript.